

Abstract
C-alkyl amidine analogs of asymmetric Nω,Nω -dimethyl-L-arginine are dual-targeted inhibitors of both human DDAH-1 and nitric oxide (NO) synthase, and provide a promising scaffold for developing therapeutics to control NO overproduction in a variety of pathologies including septic shock and some cancers. Using a two-part clickchemistry-mediated activity probe, a homologated series of C-alkyl amidines are ranked for their ability to inhibit DDAH-1 within cultured HEK 293T cells. N5-(1-iminopentyl)-L-ornithine was determined to be the most potent compound in vitro (Kd = 7 μM) as well as in cultured cells, and the binding conformation and covalent reversible mode of inhibition was investigated by comparison of interactions made with DDAH-1 and a catalytically inactive C274S variant as gauged by Xray crystallography and isothermal titration calorimetry. By interrupting the ability of the inhibitor to form a covalent bond, the contribution of this interaction can be estimated. These results suggest further stabilization of the covalent adduct as a promising strategy for lead optimization in the design of effective reagents to block NO synthesis.
Keywords: DDAH, nitric oxide, inhibitors, covalentreversible, activity probe
Introduction
Dimethylarginine dimethylaminohydrolase-1 (DDAH-1) catalyzes the hydrolysis of asymmetric Nω, Nω -dimethyl-L-arginine (ADMA, 1), an endogenous inhibitor of nitric oxide (NO) synthase. As a consequence, DDAH plays a key role in the complex, multifaceted, regulation of NO synthesis and in pathological conditions arising from irregularities in NO concentrations.[1]
The effect of NO on tissues is complicated and seemingly contradictory.[2] Diminished NO production is often associated with pathologies, like endothelial dysfunction and cardiovascular risk.[1a, 3] The role of NO in tumor growth is also complicated.[4] In some cases, it appears that tumor growth is inversely correlated with NO production,[5] whereas other studies suggest that NO concentrations may promote tumor growth[6] or vascularization.[2] Although the issue is complex, there is sufficient evidence to suggest that agents that reduce NO production in cancer cells are worth investigation.[7] This includes the study of inhibitors of human DDAH-1, which will increase the level of natural NO synthase inhibitors and indirectly reduce NO levels in selected tissues.
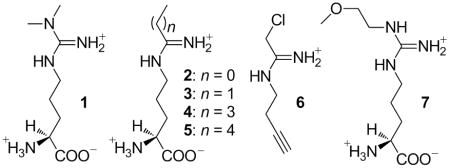
Targeted polypharmacology has been an effective strategy for anticancer agents,[8] and dual-targeted DDAH-1/NO synthase inhibitors may be particularly effective as anti-tumor agents, keeping in mind the potential cardiovascular risks of elevated concentrations of 1.[9] A series of C-alkyl amidine analogs of L-arginine (2 - 5) were shown to be dual-targeted inhibitors of both NO synthase and human DDAH-1.[10] For example, N5-(1-iminopropyl)-L-ornithine (3) inhibits neuronal NO synthase with a Ki of 3 μM and DDAH-1 with a Ki of 52 μM.[10a] Kinetic analysis indicates that 3 is a rapidly reversible competitive inhibitior of DDAH-1, but a high resolution (1.9 Å) X-ray crystallographic analysis of the DDAH-1•3 complex shows that a covalent bond is formed between the active site Cys 274 and the inhibitor.[10a] Analogs with longer alkyl substitutions have increased potency for DDAH-1 in vitro, with N5-(1-iminopentyl)-L-ornithine (4) showing the most potent inhibition of DDAH-1 (Ki = 7.5 μM) while still retaining potency for inhibition of NO synthase (Ki = 20 μM).
These inhibitors’ covalent reversible mode of inhibition may have particular advantages for drug design. However, it is unknown whether the analogs bearing longer alkyl substitutions (3 - 5) are transported, like 2, through the y+ cationic amino acid transporter,[11] or if they become less bioavailable as their structures diverge from L-arginine. The binding pose of these larger compounds is also of interest for developing more useful inhibitors, and determining the contribution of their reversible covalent bond to potency. To explore these questions more fully, we report here the use of a cell-permeable activity-based probe (6)[12] to rank the potency of 2 - 5 for inhibition of DDAH-1 within intact HEK293T cells. The binding interactions and the contribution of reversible covalent bond formation between DDAH-1 and the most potent bioavailable inhibitor in this series are then characterized using site-directed mutagenesis of the active-site Cys, combined with X-ray crystallography and isothermal titration calorimetry. This study provides a better understanding of how C-alkyl amidines inhibit DDAH-1 in cells, and will facilitate the future design of therapeutic reagents to impact NO production.
Results and Discussion
Ranking Inhibitor Potency Within HEK 293T Cells
We evaluated inhibitor potency within cultured cells by using a click-chemistry mediated activity probe, N-but-3-ynyl-2-chloroacetamidine (6). Previously, 6 was developed as a selective in vivo activity probe that labels the active fraction of DDAH-1 in intact mammalian cells.[12] Labeling can be blocked by the presence of bioavailable inhibitors, providing a technique to determine inhibitor potency within a cell.[12] Herein we adopt this methodology to evaluate a series of inhibitors (2 - 5). Briefly, human DDAH-1 bearing an N-terminal myc-tag was expressed in HEK 293T cells by transient transfection. Inhibitors were added into the growth medium of the cells in separate wells at a fixed concentration, followed by a short incubation. Then 6 was added to covalently label the uninhibited fraction of DDAH-1. Cells were then washed, harvested and lysed. The resulting lysate was incubated with biotin-PEO3-azide and catalysts for the azide/alkyne cycloaddition click reaction to attach biotin to the fraction of DDAH-1 previously labeled by 6.[12–13] SDS-PAGE then resolved the crude protein mixture. A two-color Western blot assay was used to detect DDAH-1 expression levels and the biotin-labeling signals by using rabbit anti-myc and mouse antibiotin primary antibodies in combination with two corresponding near-infrared dye-labeled secondary antibodies (anti-rabbit, em: 680 nm; anti-mouse, em: 800 nm) (Figure 1).[12, 14] The response to the biotin tag signal for each inhibitor was normalized for the expression levels of myc-DDAH-1. In order to compare in vivo inhibition potency among inhibitors, each normalized response was converted to percent activity relative to a no-inhibitor control (100 %) and a no-probe (6) control (0 %).
Figure 1.
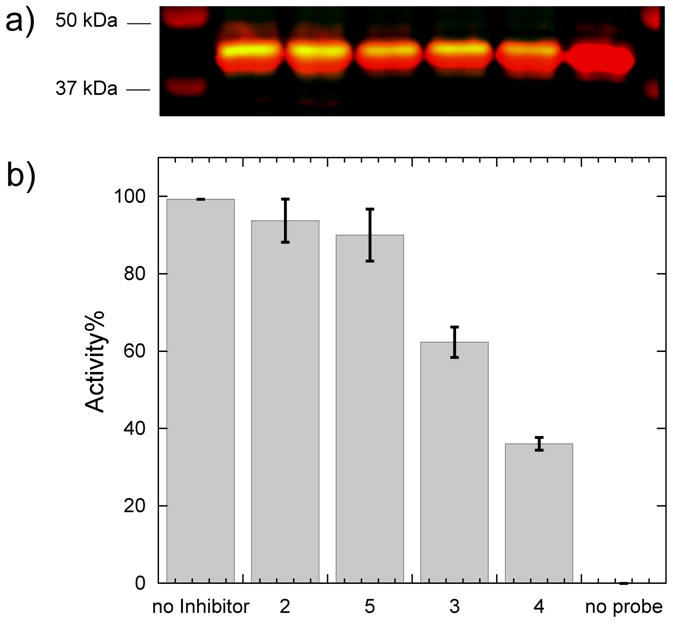
Ranking of 2 – 5 for potency of DDAH-1 inhibition in cells. a) Two-color Western blot reflecting the presence of myc (red) and biotin (green) tags after labeling of overexpressed human DDAH-1 in HEK 293T cells in the presence of inhibitors (350 μM each). Left from right: molecular weight markers, no-inhibitor control, 2, 5, 3, 4, no-probe control. b) Fluorescence intensities for the biotin-derived signal are calculated for every inhibitor and converted to Activity (%) by normalizing each intensity to the no-inhibitor control as 100 % and no probe (6) as 0 %. Data are the mean ± standard error for replicate experiments (n > 3).
These results indicate that treatment with the shortest C-alkyl amidine 2 does not significantly inhibit cellular DDAH-1 under these conditions. Extending the C-alkyl substitution of 2 by one methylene group to form 3, increases inhibitor potency. Extending the substituent by an additional two methylenes to form 4 results in an additional increase in potency. However extension by one more methylene group (5) results in a loss of ability to inhibit cellular DDAH-1. The general trend of increasing inhibitor potency in cells, 2 < 5 < 3 < 4, is consistent with that measured in vitro[10a] and gives the same rank order. The same trend seen both in vitro and in cells suggests a similar uptake of C-alkyl amidine scaffold inhibitors with different length of alkyl substituents, despite their divergence from the structure of L-arginine. Therefore, N5-(1-iminopentyl)-L-ornithine (4), is determined to be the most potent bioavailable DDAH-1 inhibitor in this series, and was selected for a more detailed structural and functional characterization. Specifically, a comparison of the interactions of 4 with wild-type and C274S DDAH-1 are of interest to better understand the binding conformation and the contribution of reversible covalent bond formation.
Structural Characterization of 4 complexed with wild-type and C274S DDAH-1
Crystals of the wild-type and C274S mutants of DDAH-1, complexed with 4 were essentially isomorphous with the previously studied 3 complex,[10a] and the X-ray structures were readily solved using molecular replacement methods. Complexes of 4 with both wild-type DDAH-1 and with the C274S mutant diffracted to 2.5 Å resolution. Statistics for the crystallographic refinement of the complexes are presented in Table 1. The electron density maps were readily interpreted and allowed a clear view of the binding of 4 to each protein.
Table 1.
Crystallographic Data
| DDAH-1•4 | C274S•4 | |
|---|---|---|
| PDB code | XXXX | XXXX |
| Space group | P21 | P21 |
| Cell constants | a=47.0, b=80.9, c=74.1 Å, β=90.1° | a=47.2, b=79.6, c=73.6 Å, β=89.6° |
| Resolution (Å) | 20.-2.5 (2.54–2.50)[a] | 20.-2.5 (2.54–2.50) |
| Rmerge (%) | 0.140 (0.575) | 0.100 (0.477) |
| <I/σI> | 5.9 (2.0) | 9.2 (2.5) |
| Completeness (%) | 97.4 (99.8) | 92.0 (95.8) |
| Unique reflections | 18,983 | 18,125 |
| Redundancy | 3.4 | 3.1 |
| Wavelength | 1.5418 | 1.0 |
| # of molecules per asymmetric unit | 2 | 2 |
| # of residues | 548 | 550 |
| # of protein atoms | 4184 | 4089 |
| # of ligand atoms | 30 | 30 |
| # of solvent atoms | 49 | 137 |
| Rworking | 0.212 | 0.228 |
| Rfree | 0.308 | 0.305 |
| Average B factor for protein atoms (Å2) | 37.2 | 38.8 |
| Average B factor for ligand atoms (Å2) | 37.4 | 27.7 |
| Average B factor for solvent atoms (Å) | 32.5 | 31.6 |
| rms deviation from ideality | ||
| bonds (Å) | 0.008 | 0.007 |
| angles (°) | 1.195 | 1.433 |
| Ramachandran plot | ||
| % of residues in favored region | 93.7 | 91.9 |
| % of residues in additional allowed region | 6.3 | 8.1 |
Values in parentheses correspond to highest resolution shell
Figure 2a) shows a portion of the wild-type complex with an omit electron density map superimposed. The Cys 274 sulfur and 4 were omitted from the phases to reduce bias in this map. It is clear that there is a covalent bond, with strong continuous electron density between C274 and Cζ of the inhibitor. Furthermore, the inhibitor has tetrahedral geometry at Cζ, with distinct electron density for each of the four substituent groups bonded there. Therefore, the amidine moiety of 4 forms a covalent bond with Cys 274 in the inhibited complex and does not appear to have a different mode of inhibition than the structurally related inhibitor 3.[10a]
Figure 2.

Electron density for 4. a) Omit density for the complex with wild type DDAH-1; There is continuous density from C274 to the tetrahedral inhibitor. b) Omit density for 4 in the C274S mutant. The inhibitor is trigonal at Cζ and the inhibitor is clearly not covalently attached to the enzyme.
The amino acid portion of 4 makes non-covalent, but specific contacts with the protein (Figure 3). There is an ion pair, or a pair of hydrogen bonds, between the inhibitor carboxylate and Arg 145 of the protein (2.3 Å). The α-amino acid group of the inhibitor is strongly anchored in a pseudo-tetrahedral receptor pocket. It ion pairs, or donates a hydrogen bond to the side chain carboxylate of Asp 73 (2.5 Å). The α-amino acid group also donates hydrogen bonds to the carbonyl oxygens of Leu 30 (3.0 Å) and V268 (3.2 Å). In the covalent complex, Nε and Nω (on either side of Cζ) donated hydrogen bonds to the two oxygens of the D79 side chain. The distances are 2.9 and 3.1 Å respectively. The alkyl substituent of 4 fits into a pocket within the active site, and makes several hydrophobic contacts. For example, the terminal methyl group of the C-alkyl substituent is roughly centered in a hole formed by the Cδ 1 of Leu 271, Cβ of His 173, and the Cα of Gly 129.
Figure 3.

Comparison of 4 and 3 binding to DDAH-1. In this divergent (wall-eyed) stereo image, the protein is shown with green carbon backbone. 4 is shown as cyan sticks, while 3 is shown as black bonds. Hydrogen bonds are shown as black dashed lines.
The orientation of 4 in the DDAH-1 active site is very similar to what was seen previously with the DDAH-1•3 complex.[10a] We superimposed these two complexes and found an rms deviation of the corresponding Cαs to be 0.248 Å. Figure 3 shows the relative positions of the two inhibitors sitting in the active site of the DDAH-1•4 complex. 4, shown in green stick bonds, has a four carbon substituent compared to the two carbon substituent of the otherwise identical 3. The amino acid portion of the inhibitors is essentially identical up to and including Cζ, which is covalently bonded in the complexes to C274. The main difference is that the longer C-alkyl substituent of 4 is folded more snugly against the face of H173 than is the smaller substituent of 3. This added van der Waals contact likely contributes to tighter binding, but other explanations are also plausible.
Figure 2b) shows omit density for the model of 4 binding to the C247S mutant. There is no density between Ser 274 and the inhibitor; indeed the Oγ is 4.0 Å from Cζ of 4. The inhibitor has trigonal geometry at Cζ, as expected if no new bond is formed to the sp2 hybridized inhibitor. The interactions of the amino acid portion of the inhibitor are very similar to that seen in the covalent complex (Figure 3, 4). That is, ion pairs are formed between the carboxylate and Arg 145 (2.8 Å), between the α-amino and D73 (2.6 Å) and hydrogen bonds are formed to the carbonyls of L30 (2.7 Å), and V268 (2.3 Å). The interactions between the enzyme and the Nε and Nω of the inhibitor are quite different in the noncovalent complex. Nω donates a hydrogen bond (2.5 Å) to the side chain of D79, but Nε is rotated around and donates to the carbonyl oxygen of D269 (2.4 Å). The C-alkyl chain of the inhibitor lies in the same general area as seen for the covalent derivative, but there are displacements since the inhibitor is not pulled so deeply into the binding pocket.
Figure 4.

Binding of 4 to C274S DDAH-1. In this divergent stereo image, the protein is shown with green carbon atoms, and the inhibitor in cyan. Hydrogen bonds are shown as black dashed lines.
The C274S•4 complex can be superimposed on the covalent DDAH-1•4 complex with an rms deviation over the Cαs of 0.349 Å. Figure 5 shows the relative positions of the covalent and non-covalent ligands in the wild-type active site. The covalent inhibitor is shown as a stick model with green carbon atoms, while the non-covalent inhibitor has magenta carbons. The amino acid portions of the ligand conformations are nearly identical, but the amidine group is oriented differently. Without covalent bond formation, Cζ of the inhibitor moves out to a van der Waals distance from the side chain of 274, that is about 4 Å. The Nε and Nω atoms make strong, but differing interactions with the protein. Presumably, the C-alkyl group is freer to take up a low energy conformation with respect to the enzyme active site. It may therefore be that this non-covalent bonding mode represents the reversible binding form of the inhibitor (see below).
Figure 5.

Comparison of 4 complexed to wild type and C274S DDAH-1. In this divergent stereo image, the protein is shown with green carbon backbone. 4 in the wild type is shown as cyan sticks, while 4 in the mutant is shown in magenta. Hydrogen bonds are shown as black dashed lines; for clarity, only bonds to the C-alkyl amidine moiety are shown and those to the amino acid portion of the inhibitor have been omitted.
The amidine and the C-alkyl substituent of 4 in the noncovalent C274S•4 complex adopt a bent conformation that is strikingly similar to that seen with DDAH-1 and 7, an Nω - substituted arginine inhibitor of DDAH-1.[15] It is unclear why wildtype DDAH-1 is able to make a covalent bond with 3 and 4, and not 7, but the length of the terminal alkyl substituent is not apparently the determining factor.
Dissociation constants of 4 with wild-type and C274S DDAH- 1
Previously, IC50 values were used to calculate a Ki value (7.5 ± 04 μM) for 4.[10a] However, this methodology has potential pitfalls for accurately calculating dissociation constants and cannot be used to characterize ligand interactions with the non-cataytic C274S mutant.[16] Therefore, isothermal titration calorimetry (ITC) was used to determine the overall dissociation constants for 4 and its interactions with both wild-type and C274S DDAH-1.
Differences in heat changes after sequential injections were monitored upon titration of 4 into wild-type DDAH-1 until no significant differences were observed (Figure 6). In order to obtain an overall dissociation constant, these differences were fitted using a single-site binding model to obtain the parameters: Ka = 1.4 ± 0.2 ×105 M−1, ΔH° = − 5580 ± 180 cal/mol, ΔS° = 4.79 cal/mol/deg. Binding is exothermic and is favored by negative enthalpy and positive entropy terms. This ITC experiment gives an overall dissociation constant (Kd,overall) of 7 ± 1 μM that encompasses both non-covalent and covalent complexes (see below). This value matches quite well with the Ki,calc derived earlier from the IC50 value.[10a]
Figure 6.

Isothermal titration calorimetry of the wild-type DDAH-1•4 complex. Top panel shows raw data output for titration of 4 into DDAH-1 at 25° C. Bottom panel shows integrated values from the top panel, as best fit to a single-site binding model to give: Ka = 1.4 ± 0.2 × 105 M−1, ΔH° = − 5580 ± 180 cal/mol, ΔS° = 4.79 cal/mol/deg, n = 0.6. See experimental section for details.
The same experiment was repeated using 4 but instead substituting the C274S mutant of DDAH-1 to obtain the parameters: Ka = 3.6 ± 0.2 ×105 M−1, ΔH° = − 5660 ± 100 cal/mol, ΔS° = 1.85 cal/mol/deg (Figure 7). The resulting dissociation constant is 28 ± 2 μM, which is only 4-fold higher than that observed for the covalent complex. The increase in Kd stems not from a change in enthalpy (these values are within error of each other), but rather from the entropy term, which is 2.6-fold less positive for the C274S mutant. The larger ΔS° observed with wild-type DDAH-1 may, in part, reflect displacement of water molecules upon binding 4 that are otherwise retained in the C274S complex (e.g. Wat 23), although other interpretations are possible.
Figure 7.

Isothermal titration calorimetry of the C274S•4 complex. Top panel shows raw data output for titration of 4 into C274S DDAH-1 at 25° C. Bottom panel shows integrated values from the top panel, as best fit to a single-site binding model to give: Ka = 3.6 ± 0.2 × 105 M−1, ΔH° = − 5660 ± 100 cal/mol, ΔS° = 1.85 cal/mol/deg, n = 1. See experimental section for details.
Comparison of the interactions of 4 with wild-type and C274S DDAH-1 proteins can give an estimation of the contribution of the covalent bond to inhibitor potency. For the wild-type protein, structural and functional characterization supports a reversible covalent mode of inhibition. This is represented by three states in rapid equilibrium: the unbound enzyme and inhibitor, a non-covalent enzyme•inhibitor complex, and the covalent adduct observed in the structural models (Equation 1). The resulting Kd,overall is a combination of Kd1 for formation of the non-covalent complex, and Kd2 for conversion to the covalent complex (Equation 2).
| Eq.(1) |
| Eq.(2) |
In contrast, no covalent bond is observed in the structural model of C274S•4, so its dissociation constant represents a simple equilibrium between bound and free ligand. Because of the conservative nature of the Ser for Cys substitution, we considered this complex to be an approximation of the noncovalent E•I complex with wild-type DDAH-1. Therefore, by substituting Kd1 with the Kd measured for the C274S•4 complex, the contribution of the covalent bond formation (1/Kd2) can be determined to favor bond formation by a factor of 3. Therefore, covalent bond formation is not a major contributing factor for the potency of these C-alkyl amidine inhibitors. Its minor contribution is consistent with the rapid onset and reversibility of inhibition. It is also consistent with structural data that show the majority of ligand-protein interactions as being the same in both complexes, with only the covalent bond and some relatively non-specific interactions differing in the wild-type DDAH-1•4 complex.
Typically, covalent reversible inhibitors can be quite potent. Two well-known classes are peptide aldehydes and peptide nitriles, both capable of inhibiting various hydrolases that use Cys as an active-site nucleophile.[17] The covalent adducts of these inhibitors have been proposed to mimic reaction transition states, thereby converting the stabilization energy usually used during catalytic turnover into binding energy to increase potency. For one example, peptide nitriles were shown to be potent, reversible inhibitors of papain.[18] These inhibitors make a covalent thioimidate bond with the active site Cys,[19] mimicking the covalent acyl-intermediate observed during catalytic turnover, yet have only a short lifetime that leads to rapid reversibility of inhibition.[20] The C-alkyl amidines appear to use a similar mechanism, reacting with the active-site Cys of DDAH-1 to form a tetrahedral adduct similar to that found on the normal catalytic pathway. However, the potency of these inhibitors is moderate, with values only 24-fold lower than the KM for substrate (1, 170 μM).[21] Although C-alkyl amidines can favorably form covalent tetrahedral adducts with DDAH-1, they appear to be unable to take advantage of the stabilization energy normally afforded to reaction intermediates or transition states. One likely missed interaction is that between the terminal dimethylamine nitrogen in 1 and the active-site His173 that serves as a general acid in the reaction.[22] The position corresponding to the substrate’s leaving group nitrogen is substituted in 4 with a methylene, which is incapable of making the same interactions. Instead, this C-alkyl substituent makes weaker nonspecific van der Waals interactions with the face of the His173 ring. This analysis suggests that alteration of the inhibitor’s structure to capture this or similar interactions would likely further shift the equilibrium to the covalent complex and result in increased potency.
Conclusion
C-Alkyl amidines are covalent reversible inhibitors of human DDAH-1. They have relatively modest (low μM) Ki values, but are still one of the most potent classes of human DDAH-1 inhibitors reported to date.[10d, 23] A click-chemistry mediated two-part activity probe demonstrates that these compounds can inhibit DDAH-1 within cultured cells, despite their divergence from the structure of L-arginine. N5-(1-iminopentyl)-L-ornithine (4) is determined to be the most potent compound in a homologated series as assayed in vitro and in cultured cells. Structural determination of 4 complexed with wild-type and C274S DDAH-1, along with ITC experiments are used to characterize it’s specific binding interactions and to dissect the contribution of the covalent bond to potency. Here, covalent bond formation is favorable, but only contributes 4-fold to overall potency. This minor contribution is likely due to the inability of the tetrahedral covalent inhibitor adduct to recapitulate some of the key binding interactions normally made by substrates during turnover. Targeting these interactions is suggested for optimizing the potency of this class of compounds as bioavailable DDAH-1 inhibitors in the development of future NO-blocking therapeutics.
Experimental Section
Materials
N5-(1-Iminioethyl)-L-ornithine (2) was purchased from Calbiochem (San Diego, CA). N5-(1-Iminopropyl)-, N5-(1-iminopentyl)-, and N5-(1-iminohexyl)-L-ornithine (3, 4, and 5, respectively) were synthesized previously.[10a] N-But-3-ynyl-2-chloro-acetamidine (6) and biotin–PEO3-azide were previously synthesized.[11] Unless specified otherwise, all other chemicals were from the Sigma Aldrich Chemical Co. (St. Louis, MO).
Ranking inhibitor potency within HEK 293T cells
HEK 293T cells were seeded in a 6 well polystyrene plate using complete growth medium containing DMEM with 10 % FBS (Invitrogen, Carlsbad, CA) and grown to 50 - 60 % confluency. pEF6a-hDDAH-1[12] was transfected into HEK 293T cells using lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 24 h, spent medium was removed and cells were washed with 1 mL and then 0.5 mL PBS (pH = 7.2, Invitrogen). Inhibitors 2, 3, 4 and 5 (350 μM each) were added to the growth medium of cells in culture (1 mL) and incubated for 30 min at 37° C in a CO2 incubator before subsequent addition of the activity-probe 6 (110 μM), followed by an additional 10 min incubation. After these treatments, cells were washed twice with PBS (1 mL) at pH 7.4 and harvested by suspension in PBS (1 mL) and centrifugation at 74 × g for 5 min at 4° C. Cell pellets were stored at -80° C. Frozen cell pellets were lysed and labeled with Biotin-PEO3-azide as described earlier.[12] Two-color western blot detection was used to detect the expression levels of myc-DDAH-1 and the response to the biotin tag as described previously.[12] Images were scanned using an Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln, NE) at the core DNA Facility (University of Texas, Austin). Integrated fluorescence intensities were taken for both 680 nm and 800 nm channels. The 680 nm value (the response to myc tag, displayed in red) was used for normalization and the resulting fluorescence intensities for the response to biotin (displayed in green) was converted to a percent activity value for each inhibitor as described above. Four experimental replicates of each inhibitor were completed to obtain an average and standard error, both plotted using Kaleidagraph (Synergy Software, Reading, PA).
Preparation of wild-type and C274S human DDAH-1
For wild-type human DDAH-1, an N-terminal His6-tagged expression plasmid with a reengineered N-terminus (pET28a-hDDAH-1re) was used to avoid N-terminal nonenzymatic gluconoylation as described earlier.[12]
The C274S site directed mutant was made using the protocol outlined in the QuikChange Site-Directed Mutagenesis Kit (Stragene, La Jolla, CA). Briefly, the forward primer 5’-GGATGGGCTGCTCACCTCCTGCTCAGTTTTAATTA-3 ’ (S274 underlined) and reverse primer 5’-TAATTAAAACTGAGCAGGAGGTGAGCAGCCCATCC-3’ (S274 underlined) were used with PfuTurbo DNA polymerase to amplify the C274S gene by 12 cycles of PCR (30s denaturation at 95 °C, 30s annealing at 55°C, and 6.5 minute extension at 68°C) using the pET28a-hDDAH-1 plasmid[21] as a template. This template does not carry the same modifications found in pET28a-hDDAH-1re. After DpnI treatment, the PCR product was transformed directly into E. coli strain DH5α. Plasmid DNA was then purified from transformants and sequenced (DNA core facility, Austin, TX) to confirm the C274S mutation. The pET28a-C274S-hDDAH-1 plasmid was then transformed into E. Coli strain Rosetta 2 (DE3) for expression.
There were small differences between the protein preparation procedures used for crystallography and those for calorimetry. For crystallography, wild-type DDAH-1 was expressed from plasmid pET28a-hDDAH-1 in E. coli strain Rosetta 2 (DE3) (Novagen, Gibbstown, NJ), under the dual selection of kanamycin and chloramphenicol. Purification of the C274S variant instead substituted the pET28a-C274S-hDDAH-1 plasmid. Each variant was purified using the same procedure. Briefly, transformants were grown in Terrific Broth media (Difco, Franklin Lakes, NJ) at 37° C to an OD600 of 0.7; protein overexpression was initiated by addition of 1 mM IPTG. Cells were grown for another three hours post induction, and were harvested by centrifugation at 4000 × g. Cells were resuspended in Lysis buffer (25 mM Tris-HCl, pH 8, 250 mM NaCl) and lysed in a French Pressure cells three times at 1000 PSI. Cell debris was removed by centrifugation at 30,000 × g for 1 h and the crude cell lysate passed though a column containing 3 mL of Ni-NTA His Bind resin that was previously equilibrated with Lysis buffer. The column was washed with 500 mL of a 25 mM Tris-HCl, pH 8, 250 mM NaCl, 10 mM imidazole buffer and the protein was eluted with a linear gradient from 10 – 250 mM imidazole. Protein-containing fractions were pooled, dialyzed against a 10 mM Tris-HCl 50 mM KCl buffer, and concentrated using an Amicon Ultra-15 centrifugal filter (10,000 molecular weight cutoff) (Millipore, Billerica, MA). The recombinant wild-type or C274S DDAH-1 was further purified by gel filtration using a HiLoad 16/60 Superdex S-200 prep grade column (120mL) and an AKTA FPLC system (GE Healthcare, Piscataway, NJ). The column was preequilibrated with a 10 mM Tris-HCl, pH 8, 50 mM KCl buffer and was run at a flow rate of 1 mL/min. Protein-containing fractions were pooled, concentrated to 15 – 17 mg/mL, and stored at 4° C.
For the calorimetry experiments, wild-type DDAH was expressed from the pET28a-hDDAH-1re plasmid, and the C274S variant from pET28a-C274S-hDDAH-1. Each was overexpressed and purified according to the procedure described elsewhere (final pH 7.0).[10a] The purified proteins were analyzed by ESI-MS (Analytical Core Facility, College of Pharmacy, The University of Texas) and gave the masses expected (all in Da) both for wild type (MWcalc = 33,311; MWexptl = 33,304) and the C274S variant (MWcalc = 33,429; MWexptl = 33,423; C274S also contains a minor +177 Da peak due to N-terminal α-gluconoylation[24]), respectively. The MW of the C274S variant is slightly larger than wild-type because the differences in the N-terminal sequence outweigh the Ser for Cys substitution. Theoretical protein molecular weights were calculated based on their amino acid sequences without the first Met (http://workbench.sdsc.edu/) [25].
Crystallization of hDDAH-1 and C274S
The best quality DDAH-1 crystals were grown at 4° C by vapor diffusion using the sitting drop method from 0.1 M Tris-HCl pH 8.2, 20 % polyethylene glycol (PEG) 6000. Crystals of the C274S variant of DDAH-1 were grown at 4° C from 19–23 % PEG 6000, 0.1 M Tris-HCl, pH 8.0, using the hanging drop method.
The complex of DDAH-1 with 4 was formed by adding 1 μL of a 1mM stock solution of the inhibitor to a well containing preformed crystals in approximately 3 μL crystallization mother liquor and incubating overnight. For the purpose of determining the structure of C274S in complex with 4, a crystal was transferred to a reservoir containing 20 μL 5 mM 4 in artificial mother liquor (25 % PEG 6000, 0.1 M Tris-HCl, pH 8.0) and soaked overnight.
X-ray data collection and processing
The DDAH-1•4 and C274S•4 crystals were removed from their drops using a nylon loop, and flash frozen in liquid nitrogen. Prior to data collection, crystals were treated with cryoprotectant by transferring to 35 % PEG 6000, 0.1 M Tris-HCl, pH 8.0 for 1–5 seconds. Diffraction data from the DDAH-1•4 crystal were collected at the X-ray Crystallography Core Laboratory at the University of Texas Health Science Center at San Antonio. Data were collected at 100 K on a R-AXIS HTC image plate detector with X-rays generated by a Rigaku MicroMax-007HF rotating anode generator operated at 40 mV, 30 mA. Diffraction data from the C274S•4 crystal were collected at 100 K at the Advanced Light Source beamline 5.0.2 at the Lawrence Berkeley National Laboratory. Data were processed using the HKL2000 suite.[26]
Structure determination
The structures of both the human DDAH-1 and the C274S variant complexed with 4 were solved by molecular replacement with Phaser[27] using the structure of human DDAH-1[10a] (PDB accession code 3I2E) as the search model.
The complex models were adjusted by fitting to 2Fo-Fc and Fo-Fc maps using COOT.[28] The DDAH-1•4 and C274S•4 structures were subjected to several rounds of refinement using CNS[29] and PHENIX[30] followed by manual rebuilding of the model. 5% of the diffraction data were set aside throughout refinement for crossvalidation.[31] MolProbity[32] was used to determine areas of poor geometry and to make Ramachandran plots.
Atomic coordinates
Coordinates of the refined models of the human DDAH-1 and the C274S variant complexed with 4 have been deposited in the Protein Data Bank with entry codes XXXX and XXXX, respectively
Isothermal titration calorimetry of DDAH-1 variants
The binding of 4 to wild-type and C274S DDAH-1 was investigated using isothermal titration calorimetry. A MicroCal iTC200 microcalorimeter (MicroCal, Northampton, MA) was used to conduct the titrations, and data analysis was preformed using Origin 7.0. Briefly, 16 injections of 4 were titrated into the sample cell containing either wild-type or C274S DDAH-1 at 25°C. Inhibitor stock solutions were prepared using the outside final dialysis buffer from DDAH-1 purifications (50 mM KH2PO4, 10 % glycerol, pH 7.0). A stock solution of 4 (3.5 mM) was titrated into wild-type DDAH-1 (197.8 μM). A stock solution of 4 (3.09 mM) was titrated into C274S DDAH-1 (206.3 μM). Each titration curve was fitted to a single binding site model to determine the binding stoichiometry (n), association constant (KA), and changes to the thermodynamic parameters of enthalpy (ΔH°) and entropy (ΔS°) using the data fitting module in Origin software provided by the manufacturer.
Acknowledgments
We thank Alexander Taylor for assistance with data collection at the X-ray Crystallography Core Laboratory at the University of Texas Health Science Center at San Antonio. This work was supported by NIH grants AI 075509 (to J.D.R), GM 069754 (to W.F.) and GM 069754-S3 (to W.F.), by Robert A. Welch Foundation grants F1225 (to J.D.R.) and F1572 (to W.F.), and by the College of Natural Sciences support to the Center for Structural Biology. Portions of this research were conducted at the Advanced Light Source, a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The Berkeley Center for Structural Biology is supported in part by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences.
Contributor Information
Dr. Walter Fast, Email: WaltFast@mail.utexas.edu.
Dr. Jon D. Robertus, Email: jrobertus@cm.utexas.edu.
References
- 1.a) Pope AJ, Karuppiah K, Kearns PN, Xia Y, Cardounel AJ. J Biol Chem. 2009 doi: 10.1074/jbc.M109.037036. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wadham C, Mangoni AA. Expert Opin Drug Metab Toxicol. 2009;5:303. doi: 10.1517/17425250902785172. [DOI] [PubMed] [Google Scholar]; c) Palm F, Onozato ML, Luo Z, Wilcox CS. Am J Physiol Heart Circ Physiol. 2007;293:H3227. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- 2.Jenkins DC, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC, Moncada S. Proc Natl Acad Sci U S A. 1995;92:4392. doi: 10.1073/pnas.92.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fliser D. Eur J Clin Invest. 2005;35:71. doi: 10.1111/j.1365-2362.2005.01457.x. [DOI] [PubMed] [Google Scholar]
- 4.Wink DA, Mitchell JB. Free Radic Biol Med. 2003;34:951. doi: 10.1016/s0891-5849(02)01362-x. [DOI] [PubMed] [Google Scholar]
- 5.Xie K, Fidler IJ. Cancer Metastasis Rev. 1998;17:55. doi: 10.1023/a:1005956721457. [DOI] [PubMed] [Google Scholar]
- 6.Thomsen LL, Miles DW. Cancer Metastasis Rev. 1998;17:107. doi: 10.1023/a:1005912906436. [DOI] [PubMed] [Google Scholar]
- 7.Sikora AG, Gelbard A, Davies MA, Sano D, Ekmekcioglu S, Kwon J, Hailemichael Y, Jayaraman P, Myers JN, Grimm EA, Overwijk WW. Clin Cancer Res. 2010;16:1834. doi: 10.1158/1078-0432.CCR-09-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roth BL, Sheffler DJ, Kroeze WK. Nat Rev Drug Discov. 2004;3:353. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- 9.a) Schade D, Kotthaus J, Clement B. Pharmacol Ther. 2010;126:279. doi: 10.1016/j.pharmthera.2010.02.005. [DOI] [PubMed] [Google Scholar]; b) Boger RH. Ann Med. 2006;38:126. doi: 10.1080/07853890500472151. [DOI] [PubMed] [Google Scholar]
- 10.a) Wang Y, Monzingo AF, Hu S, Schaller TH, Robertus JD, Fast W. Biochemistry. 2009;48:8624. doi: 10.1021/bi9007098. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Babu BR, Griffith OW. Curr Opin Chem Biol. 1998;2:491. doi: 10.1016/s1367-5931(98)80125-7. [DOI] [PubMed] [Google Scholar]; c) Bretscher LE, Li H, Poulos TL, Griffith OW. J Biol Chem. 2003;278:46789. doi: 10.1074/jbc.M306787200. [DOI] [PubMed] [Google Scholar]; d) Kotthaus J, Schade D, Muschick N, Beitz E, Clement B. Bioorg Med Chem. 2008;16:10205. doi: 10.1016/j.bmc.2008.10.058. [DOI] [PubMed] [Google Scholar]
- 11.Baydoun AR, Mann GE. Biochem Biophys Res Commun. 1994;200:726. doi: 10.1006/bbrc.1994.1511. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Hu S, Fast W. J Am Chem Soc. 2009;131:15096. doi: 10.1021/ja906432e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed Engl. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 14.Weldon S, Ambroz K, Schutz-Geschwender A, Olive DM. Anal Biochem. 2008;375:156. doi: 10.1016/j.ab.2007.11.035. [DOI] [PubMed] [Google Scholar]
- 15.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O’Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Nat Med. 2007;13:198. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 16.a) Copeland RA. Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. Wiley-Interscience; Hoboken, N.J: 2005. [PubMed] [Google Scholar]; b) Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 17.Wolfenden R. Annu Rev Biophys Bioeng. 1976;5:271. doi: 10.1146/annurev.bb.05.060176.001415. [DOI] [PubMed] [Google Scholar]
- 18.Sluyterman LA, Wijdenes J. Biochim Biophys Acta. 1973;302:95. doi: 10.1016/0005-2744(73)90012-0. [DOI] [PubMed] [Google Scholar]
- 19.Brisson JR, Carey PR, Storer AC. J Biol Chem. 1986;261:9087. [PubMed] [Google Scholar]
- 20.Liang TC, Abeles RH. Arch Biochem Biophys. 1987;252:626. doi: 10.1016/0003-9861(87)90068-3. [DOI] [PubMed] [Google Scholar]
- 21.Hong L, Fast W. J Biol Chem. 2007;282:34684. doi: 10.1074/jbc.M707231200. [DOI] [PubMed] [Google Scholar]
- 22.a) Stone EM, Costello AL, Tierney DL, Fast W. Biochemistry. 2006;45:5618. doi: 10.1021/bi052595m. [DOI] [PubMed] [Google Scholar]; b) Linsky TW, Monzingo AF, Stone EM, Robertus JD, Fast W. Chem Biol. 2008;15:467. doi: 10.1016/j.chembiol.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linsky TW, Fast W. In: Comprehensive Natural Products II: Chemistry and Biology. Mander L, Liu H-W, editors. Vol. 8. Elsevier; Oxford: 2010. p. 125. [Google Scholar]
- 24.Geoghegan KF, Dixon HB, Rosner PJ, Hoth LR, Lanzetti AJ, Borzilleri KA, Marr ES, Pezzullo LH, Martin LB, LeMotte PK, McColl AS, Kamath AV, Stroh JG. Anal Biochem. 1999;267:169. doi: 10.1006/abio.1998.2990. [DOI] [PubMed] [Google Scholar]
- 25.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Method Enzymol. 1997;27:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Cryst D Biol Crystallogr. 1998;54(Pt 5):905. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. Acta Crystallogr D Biol Crystallogr. 2002;58:1948. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 31.Brunger AT. Acta Cryst D Biol Crystallogr. 1993;49:24. doi: 10.1107/S0907444992007352. [DOI] [PubMed] [Google Scholar]
- 32.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. Nucleic Acids Res. 2007;35:W375. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]