

Abstract
Core pathologies of Alzheimer's disease (AD) are aggregated amyloid-β peptides (Aβ) and tau, and the latter is also characteristic of diverse neurodegenerative tauopathies. These amyloid lesions provoke microglial activation, and recent neuroimaging technologies have enabled visualization of this response in living brains using radioligands for the peripheral benzodiazepine receptor also known as the 18 kDa translocator protein (TSPO). Here, we elucidated contributions of Aβ and tau deposits to in vivo TSPO signals in pursuit of mechanistic and diagnostic significance of TSPO imaging in AD and other tauopathies. A new antibody to human TSPO revealed induction of TSPO-positive microgliosis by tau fibrils in tauopathy brains. Emergence of TSPO signals before occurrence of brain atrophy and thioflavin-S-positive tau amyloidosis was also demonstrated in living mice transgenic for mutant tau by positron emission tomography (PET) with two classes of TSPO radioligands, [11C]AC-5216 and [18F]fluoroethoxy-DAA1106. Meanwhile, only modest TSPO elevation was observed in aged mice modeling Aβ plaque deposition, despite the notably enhanced in vivo binding of amyloid radiotracer, [11C]Pittsburgh Compound-B, to plaques. In these animals, [11C]AC-5216 yielded better TSPO contrasts than [18F]fluoroethoxy-DAA1106, supporting the possibility of capturing early neurotoxicity with high-performance TSPO probes. Furthermore, an additional line of mice modeling intraneuronal Aβ accumulation displayed elevated TSPO signals following noticeable neuronal loss, unlike TSPO upregulation heralding massive neuronal death in tauopathy model mice. Our data corroborate the utility of TSPO-PET imaging as a biomarker for tau-triggered toxicity, and as a complement to amyloid scans for diagnostic assessment of tauopathies with and without Aβ pathologies.
Introduction
Hallmark fibrillar pathologies in Alzheimer's disease (AD) are classified into senile plaques and neurofibrillary tangles (NFTs) composed of self-polymerized amyloid-β peptide (Aβ) and tau proteins, respectively. As initiators of a cascade-like neuronal deterioration, Aβ and tau fibrils have been targeted by developments of diagnostic and therapeutic agents, and in vivo detection of the Aβ plaque formation in brains of AD patients and amyloid precursor protein (APP) transgenic (Tg) mice has been enabled by positron emission tomography (PET) with [11C]Pittsburgh Compound-B ([11C]PIB) and other amyloid-binding radiochemicals (Klunk et al., 2004; Maeda et al., 2007). Meanwhile, imaging techniques for assessing amyloid-induced neurotoxicities remain to be established. A large number of studies documented the informativeness of volumetric magnetic resonance imaging (MRI) and metabolic PET with [18F]fluoro-deoxy-glucose ([18F]FDG) in quantifying neuronal damages consequent to brain amyloidoses, but these assays do not necessarily reflect the degrees of ongoing neurotoxic insults.
It is well known that amyloid deposition triggers microglial and astroglial activation in AD brains (McGeer and McGeer, 1995). Moreover, beneficial outcomes of Aβ and tau immunizations in humans and mouse models have highlighted crucial roles of immunocompetent glia in the protection of neurons against amyloid toxicities (Dodel et al., 2003; Asuni et al., 2007). By contrast, our previous study of tau Tg mice demonstrated mitigation of tau-induced neurodegeneration by attenuating neuroinflammation with an immunosuppressant (Yoshiyama et al., 2007). These contradictory functions of activated glia in pathological conditions have elevated the significance of neuroimaging systems capable of monitoring toxic conversion of glia in response to amyloidogenesis, toward the aim of therapeutically regulating glial activities at a desirable level and mode.
Upregulation of 18 kDa translocator protein (TSPO), also known as peripheral benzodiazepine receptor, in activated glia is of diagnostic importance in neurological diseases (Diorio et al., 1991), and several different PET radioligands for this molecule are now available or in development. Elevated TSPO levels in living AD brains were initially detected by [11C]PK11195 (Cagnin et al., 2001), and compounds with improved blood–brain barrier permeability and affinity for TSPO, including [11C]DAA1106 (Maeda et al., 2004) and [18F]fluoroethoxy-DAA1106 ([18F]FEDAA1106) (Zhang et al., 2004), were developed and applied to neuroimaging of AD patients (Yasuno et al., 2008). Autoradiographic assays of model mice using [18F]FEDAA1106 combined with immunohistochemistry have also indicated that microglial TSPO expression is linked to toxic injuries of neurons and may herald neuronal death (Yoshiyama et al., 2007; Ji et al., 2008). Furthermore, we have developed a novel class of TSPO ligands, [11C]AC-5216 (Zhang et al., 2007) and its analogs (Yanamoto et al., 2009a,b), exhibiting faster kinetics in the brain than DAA1106 families. These probes potentially produce high-contrast TSPO images and are thus likely to allow assessment of neurotoxic insults from an early pathogenic stage.
The present study examined the interplay between fibrillar Aβ and tau amyloid and TSPO-expressing microglia and in vivo detectability of Aβ- and tau-associated microgliosis by PET imaging of APP and tau Tg mice with [11C]AC-5216 and [18F]FEDAA1106. TSPO-PET was also supplemented by imaging of plaques with [11C]PIB and immunostaining of postmortem human and mouse brains.
Materials and Methods
Neuropathological analyses of TSPO-positive glia in postmortem human brains.
Postmortem human brains were obtained from autopsies for patients with AD, Pick's disease, progressive supranuclear palsy (PSP), and nonneurological conditions at the University of Pennsylvania Center for Neurodegenerative Disease Research, as described previously (Trojanowski, 2008). Tissues were fixed in 10% neutral buffered formalin and embedded in paraffin blocks. Representative 6-μm-thick sections were stained with a newly developed rabbit polyclonal antibody (NP157) against a synthetic peptide spanning residues 157–169 of human TSPO (DNHGWRGGRRLPE). Sections were also labeled with a fluorescent amyloid dye, (E,E)-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy) styrylbenzene (FSB; Dojindo Laboratories), as described previously (Sato et al., 2004; Higuchi et al., 2005). Similarly, rabbit polyclonal antibodies against mouse TSPO (NP155) (Ji et al., 2008) and ionized calcium binding adapter molecule-1 (Iba-1; Wako Pure Chemicals), mouse monoclonal antibodies against phosphorylated tau proteins (AT8; Endogen), NeuN (clone A60; Millipore Bioscience Research Reagents/Millipore) and general (clone 6E10; Covance) and N-terminally truncated, pyroglutamylated (AβN3pE; clone 1A10; Immuno-Biological Laboratories) Aβ peptides, and rat monoclonal antibody against glial fibrillary acidic protein (GFAP) (2.2B10; Zymed/Invitrogen) were used for multicolor fluorescence microscopy.
Animals.
The animals were maintained and handled in accordance with recommendations of the National Institutes of Health and institutional guidelines at the National Institute of Radiological Sciences. All animal experiments conducted here were approved by the Animal Ethics Committee of the National Institute of Radiological Sciences.
Tg mice (PS19 line) overexpressing the P301S mutant human tau under the control of mouse prion promoter (Yoshiyama et al., 2007) were created on a B6C3H/F1 background (Yoshiyama et al., 2007), and mice aged from 3 to 15 months were used for the experiments. Tg mice termed APP23, which overexpress the Swedish doubly mutant APP751 under the control of a neuron-specific Thy-1 promoter element (Sturchler-Pierrat et al., 1997), were maintained on a C57BL/6J background, and mice aged from 12 to 26 months were examined. Tg mice overexpressing human APP with the E693Δ mutation under the control of mouse prion promoter were generated on a B6C3H/F1 background, and were backcrossed with C57BL/6J mice at least 10 times (Tomiyama et al., 2010). Non-Tg (nTg) littermates were also used as wild-type controls. Tg and nTg offspring were identified by PCR assays of tail DNA.
Generation of MRI template.
A 12-month-old C57BL/6J mouse was lethally anesthetized by pentobarbital. The mouse head was embedded in 3% aqueous agarose and scanned by 9.4-Tesla Bruker AVANCE 400WB imaging spectrometer (Bruker BioSpin), as described previously (Higuchi et al., 2005). Coronal T2-weighted MR images were acquired by three-dimensional (3D) fast spin-echo sequence with the following imaging parameters: echo time = 5.5 ms, repetition time = 3000 ms, RARE factor = 32, field of view (FOV) = 20 × 20 × 25 mm3, matrix dimensions = 256 × 512 × 60, and nominal resolution = 78 μm × 39 μm × 417 μm. The MRI data were used as an anatomical template for subsequent PET studies.
Radiochemical synthesis.
Preparation of [11C]CH3I for labeling and radiosynthesis of [11C]AC-5216 [N-benzyl-N-ethyl-2-(7-11C-methyl-8-oxo-2-phenyl-7,8-dihydro-9H-purin-9-yl) acetamide] were performed as described previously (Zhang et al., 2007). Briefly, [11C]CO2 was bubbled into 0.04 m LiAlH4 in anhydrous tetrahydrofuran (300 μl). After evaporation of tetrahydrofuran, the remaining complex was treated with 57% hydroiodic acid (300 μl). [11C]CH3I was transferred under helium gas flow with heating into a reaction vessel containing 0.6 mg of precursor compound, 4 μl of aqueous NaOH (0.5 N), and 300 μl of anhydrous dimethylformamide cooled to −10 to −15°C. After radioactivity reached a plateau, the reaction vessel was warmed to 30°C and maintained at this temperature for 3 min. The reaction was terminated by adding a solution of CH3CN and H2O (6:4, 500 μl), and the radioactive mixture was applied to a semipreparative HPLC system. HPLC purification was completed by the use of a Capcell Pack column (ϕ10 mm × 250 mm; Shiseido) with a mobile phase (CH3CN:H2O = 6:4) at a flow rate of 6.0 ml/min. Radioactive fraction corresponding to the desired product was collected in a sterile flask, evaporated to dryness in vacuo, redissolved in 10 ml of sterile saline, and passed through a 0.22 μm filter (Millipore) for analysis and animal experiments. The radiochemical purity of the resultant compound was >99%, and its specific radioactivity was 199 ± 72 GBq/μmol at the end of synthesis.
[18F]FEDAA1106 [N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F] fluoroethoxy-5-methoxybenzyl)acetamide] was radiosynthesized using its desmethyl precursor, DAA1123, generously provided by Taisho Pharmaceutical, as described previously in detail (Zhang et al., 2004). The radiochemical purity of the end product exceeded 95%, and its specific radioactivity was 255.5 ± 14.8 GBq/μmol (mean ± SD) at the end of synthesis. [11C]PIB [N-[11C]methyl-2-(4-methylaminophenyl)-6-hydroxybenzothiazole] for amyloid imaging was synthesized as described previously (Maeda et al., 2007). The radiochemical purity of [11C]PIB was >97%, and its specific radioactivity was 169 ± 37.3 GBq/μmol at the end of synthesis.
In vitro [11C]AC-5216 autoradiography and immunohistochemical and histochemical examinations of tau Tg mouse brains.
PS19 mice at 3–17 months of age (n = 21), APPE6993Δ Tg mice at 22–33 months of age (n = 9), and age-matched nTg mice on B6C3H/F1 (n = 15) and C57BL/6 (n = 5) backgrounds deeply anesthetized by excessive dose of pentobarbital (200 mg/kg, i.p), were transcardially perfused with PBS, and then brains were removed. The tissues were fixed with 4% paraformaldehyde overnight, cryoprotected with 20% sucrose in phosphate buffer. Frozen brain samples were cut into 10-μm-thick coronal slices by cryotome (HM560; Carl Zeiss), and the slices were mounted on slide glass (Matsunami Glass) and stored at 4°C pending assays. These sections were reacted with 1 nm [11C]AC-5216 in 50 mm Tris-HCl buffer, pH7.4, at room temperature for 1 h, washed with ice-cold Tris-HCl buffer for 2 min twice, warmly blow-dried, and contacted to an imaging plate (BAS-MS; Fuji Film) for 2 h. The imaging plate data were scanned with a BAS5000 system (Fuji Film).
Slices adjacent to those used for autoradiography were immunostained with mouse monoclonal antibody (AT-8) against phosphorylated tau proteins and chemically stained with a common amyloid dye, thioflavin-S (Waldeck) (Yoshiyama et al., 2007). All stained sections were examined with an all-in-one microscope/digital camera (BZ-9000; Keyence). Additional adjacent sections were also doubly immunostained with mouse monoclonal antibody against NeuN and rabbit polyclonal antibody against mouse TSPO (NP155). Areas with AT8 and NeuN immunoreactivities and intensity of TSPO immunoreactivity were estimated in each hippocampal section by Photoshop 7.0 software (Adobe System).
Ex vivo [11C]AC-5216 autoradiography and histochemical examinations of APP and tau Tg mice.
Ex vivo autoradiographic assessments of regional brain radioactivities were conducted for APP23 and PS19 mice. Animals were given injections of [11C]AC-5216 (37 MBq) into the tail vein under anesthesia with 1.5% (v/v) isoflurane in air (2 ml/min flow rate) and were killed by decapitation at 30 min after the tracer injection. Brains were immediately removed and frozen in powdered dry ice. Frozen brain tissue was coronally cut into 20-μm-thick sections using a HM560 cryotome (Carl Zeiss). Autoradiographic signals were acquired and mapped as in in vitro assays.
Following attenuation of radioactivity, brain sections of APP23 mice used for autoradiographic experiments were fixed with 4% paraformaldehyde in PBS for 24 h and stained with 0.01% FSB in 50% ethanol (Sato et al., 2004; Higuchi et al., 2005).
Small animal PET imaging.
All PET scans were performed using a microPET Focus 220 animal scanner designed for rodents and small monkeys, which provides 95 transaxial slices 2.0 mm (center-to-center) apart, a 19.0 cm transaxial FOV, and a 7.6 cm axial FOV (Tai et al., 2005). Before the scans, mice were anesthetized with 1.5% (v/v) isoflurane, and a 30-gauge needle connected to a 1 ml polypropylene syringe via a length of polyethylene tubing was inserted into the tail vein. A dynamic emission scan in 3D acquisition mode immediately after intravenous injection of [11C]AC-5216 (38.5 ± 10.1 MBq; specific radioactivity, 140 ± 44.4 at injection) or [18F]FEDAA1106 (30.3 ± 9.29 MBq; specific radioactivity, 197 ± 48 at injection) was performed for 60 or 90 min. All list-mode data were sorted into 3D sinograms, which were then Fourier rebinned into 2D sinograms (frames, 10 × 1, 8 × 5, and 1 × 10 min). Summation images in three time frames (30 min each) after radioligand injection were reconstructed with maximum a posteriori (MAP) method, and dynamic images were reconstructed with filtered back-projection using a 0.5 mm Hanning's filter. Volumes of interest (VOIs) were placed on the hippocampus, entorhinal cortex, and striatum using PMOD image analysis software (PMOD Group) with reference to the MRI template. The tracer uptake in each VOI was estimated as percentage of injection dose per unit volume (%ID/ml). We also quantified binding potential (BPND; ratio at equilibrium of specifically bound radioligand to that of nondisplaceable radioligand in tissue) and distribution volume ratio (DVR; target-to-reference ratio of the distribution volume) for [11C]AC-5216 and [18F]FEDAA1106 in the hippocampus on the basis of a simplified reference tissue model (Lammertsma et al., 1996) and Logan's noninvasive method (Logan et al., 1996), respectively, by using the striatal data as references.
Kinetic properties of [11C]AC-5216 and [18F]FEDAA1106 were analyzed in the same PS19 mice aged 11 months (age = 10.7 ± 0.12 months; n = 8), and thereafter compared with those in age-matched nTg mice (age = 11.7 ± 0.77 months; n = 6). For the purpose of clarifying the effects of age on the levels of [11C]AC-5216 accumulation in tau Tg mouse brains, the test group was expanded to cover ages ranging from 3 to 14 months, and a total of 45 [11C]AC-5216-PET examinations were performed for 13 nTg and 22 PS19 mice, including 11 individuals receiving two or three scans 2–3 months apart.
We also analyzed the TSPO levels in APP23 mice aged from 23 to 29 months (age = 25.1 ± 1.3 months; n = 9), APPE693Δ mice at 25.4 months of age (n = 3), and age-matched nTg mice (age = 25.4 ± 2.60 months; n = 5) by the comparative use of [11C]AC-5216 and [18F]FEDAA1106. [11C]PIB-PET scans were also performed for 12 APP23 mice undergoing [11C]AC-5216-PET measurements as described previously in detail (Maeda et al., 2007). Briefly, emission scans were acquired for 60 min in 3D list mode immediately after the intravenous injection of [11C]PIB (30.3 ± 5.5 MBq).
PS19, APP23, and nTg mice prepared for PET analyses were distinct from those used for postmortem in vitro autoradiographic and neuropathological assays, while all APPE693Δ mice undergoing PET scans were incorporated in the postmortem study group.
Statistical analyses.
Statistical examinations in the present study were performed using Prism version 5 software (GraphPad Software). For group comparisons of pharmacokinetic variables, we performed either t-statistic or ANOVA followed by post hoc Bonferroni's test for multiple comparison. Correlations between two variables were evaluated by Pearson product-moment correlation coefficient. Partial correlation coefficient analysis was conducted with SPSS software (SPSS).
Results
Localization of TSPO-immunoreactive microglia in AD and non-AD tauopathy brains
We initially investigated associations of TSPO immunoreactivity with Aβ and tau lesions in autopsied human brains. In AD patients, diffuse plaques, which were only faintly labeled with FSB, were enveloped by a small number of Iba-1-positive microglia, which were negative for TSPO (Fig. 1A–D). By contrast, numerous microglia positive for Iba-1 and TSPO were in close contact with neuritic plaques intensely labeled with FSB (Fig. 1E–H). Localization of TSPO signals to astrocytes was ruled out by double immunostaining for TSPO and GFAP (Fig. 1I,J). Microglia weakly labeled with anti-Iba-1 antibody were also present in control brains, but did not exhibit TSPO immunoreactivity (Fig. 1K,L). Iba-1 and TSPO signals were abundantly detected in brains of patients with Pick's disease and PSP enriched with AT8-positive NFTs, Pick's bodies, and neuropil threads (Fig. 1M–P). Thus, microglia expressing TSPO were recruited to fibrillar tau inclusions but not Aβ deposits unaccompanied by tau pathologies.
Figure 1.
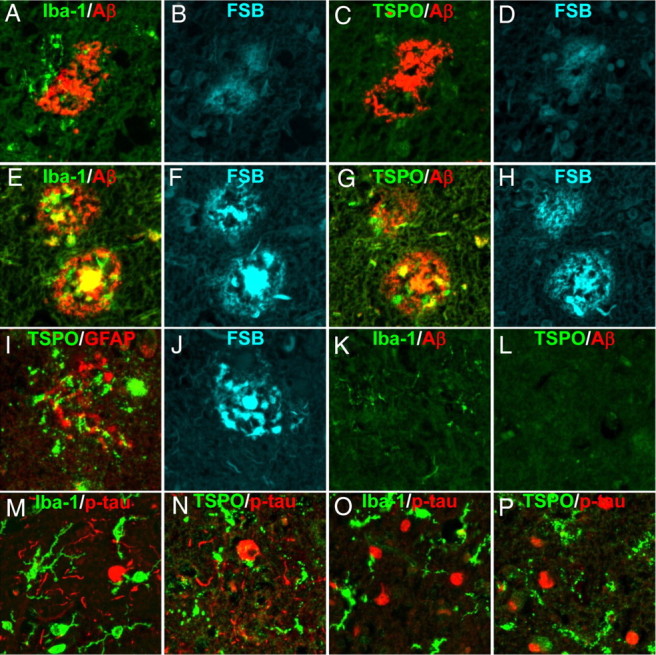
Emergence of TSPO-expressing microglia in AD and non-AD tauopathy brains as assessed by fluorescence microscopy. A–D, Triple labeling of a diffuse plaque in a frontal cortex section of AD brain (A, B) and its adjacent slice (C, D). The plaque deposition enriched with AβN3pE (red in A, C) was weakly stained with FSB (B, D), and induced recruitment of a few Iba-1-immunoreactive microglia (green in A), which were negative for TSPO (green in C). E–H, Double labeling of neuritic plaques in a hippocampal section of AD brain (E, F) and its adjacent slice (G, H). The AβN3pE-rich plaques (red in E, G) contained dense amyloid cores and dystrophic neurites strongly stained with FSB (F, H), and were surrounded by Iba-1 (green in E) and TSPO (green in G) immunoreactivities. I, J, Triple staining of a neuritic plaque in AD hippocampus, showing intense FSB labeling (J) and no overlaps between TSPO (green in I) and GFAP (red in I) immunoreactivities. K, L, Double immunostaining of frontal cortex sections generated from a nondemented control brain sample. Resident microglia displayed faint Iba-1 (green in K) but no TSPO (green in L) signals. AβN3pE-positive lesions were barely detectable (red in K, L). M–P, Two-channel fluorescence microscopic views of frontal cortex sections generated from PSP (M, N) and Pick's disease (O, P) brains. Anti-phospho-tau antibody, AT8, illuminated numerous neuropil threads, NFTs (red in M, N) and Pick's bodies (red in O, P). Iba-1 (green in M, O) and TSPO (green in N, P) immunoreactivities were present in cells with nearly the same morphology, which were characterized as putative microglia.
In vivo PET imaging of TSPO in nTg mouse brains by [11C]AC-5216
Before analyses of Tg mice, pharmacokinetic profiles of intravenously administered [11C]AC-5216 in brains of nTg mice aged 7–11 months were assessed by in vivo PET (supplemental Fig. 1A,C, available at www.jneurosci.org as supplemental material). Images generated by averaging dynamic data from 30 to 90 min indicated that retention of [11C]AC-5216 was highest in the pons/medulla, followed by the cerebellum (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Radiosignals were relatively low in the hippocampus and neocortex and lowest in the striatum (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). [11C]AC-5216 accumulation in the pons/medulla and cerebellum was markedly abolished by pretreatment with a nonradioactive TSPO ligand, PK11195 (5 mg/kg, i.v.), at 2 min before radiotracer injection (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). The uptake of [11C]AC-5216 in all brain regions peaked at 90 s after injection, and thereafter the decay-corrected radioactivity in the striatum, mostly derived from free and nonspecifically bound tracers, decreased with a half-life of ∼25 min (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). The initial uptake of [11C]AC-5216 in all regions was significantly modified by pretreatment with PK11195 [F(25,375) = 12.7, p < 0.05 (pons/medulla); F(25,375) = 9.4, p < 0.001 (hippocampus); F(25,375) = 11.3, p < 0.001 (striatum) for interaction between treatment and time], which is attributable to the blockade of TSPO in blood cells and peripheral organs and consequent elevation of plasma radiotracer concentration. Meanwhile, the retention of [11C]AC-5216 at 60–90 min was significantly suppressed by PK11195 in the pons/medulla (p < 0.05 by Bonferroni's test) but not in the hippocampus and striatum (p > 0.05 by Bonferroni's test).
In vivo PET imaging of neuroinflammation in PS19 mice
MAP reconstruction of microPET data provided high-resolution images illustrating localization of [11C]AC-5216 binding to TSPO in 11-month-old PS19 (Fig. 2A) and nTg (Fig. 2B) mouse brains. Levels of [11C]AC-5216 signals in the hippocampus and entorhinal cortex of PS19 mice were higher than those of age-matched nTg mice, while there was no overt difference in the striatal radiosignal intensity between these two genotypes. Similar to PET findings, ex vivo autoradiographic mapping of [11C]AC-5216 demonstrated that radiotracers intensely accumulated in the hippocampus and entorhinal cortex of PS19 mice relative to nTg controls, which displayed radioactivities primarily in ventricular areas, at 30 min after being intravenously administered (Fig. 2C). These data were in line with the spatial extent of tau-induced neuroinflammation and neurodegeneration in PS19 mice clarified in our previous in vitro autoradiographic and in vivo MRI experiments (Yoshiyama et al., 2007).
Figure 2.

A–C, Induction of neuroinflammatory TSPO signals in PS19 mice detected by in vivo PET (A, B) and ex vivo autoradiographic (C) imaging with [11C]AC-5216. A, B, Orthogonal views of TSPO distributions in brains of 11-month-old nTg (A) and PS19 Tg (B) mice scanned by PET at 30–90 min after intravenous injection of [11C]AC-5216. Coronal images (top) were generated to include the striatum (left; bregma +0.5 mm) and dorsal (middle; bregma −2.2 mm) and ventral (right; bregma −3.6 mm) hippocampi, and sagittal (bottom left) and horizontal (bottom right) slices were constructed at 1.7 mm lateral to the midline and 3.0 mm ventral to the bregma, respectively. PET maps are superimposed on the MRI anatomical template. C, Ex vivo autoradiographic mapping of TSPO in brains of 13-month-old nTg mouse, 11-month-old PS19 mouse with marginal neurodegenerative changes, and 11- and 14-month-old PS19 mice with massive neuronal loss in the hippocampus and entorhinal cortex. Brain tissues were collected at 30 min after intravenous injection of [11C]AC-5216, and coronal sections were generated to contain the hippocampus (bregma −2.8 mm).
Time-radioactivity curves for intracranial kinetics of [11C]AC-5216 also supported elevated TSPO levels in tau-rich regions of PS19 mice (n = 8) compared with nTg controls (n = 6) (Fig. 3A,B) in contrast to the similarity of striatal dynamic data between Tg and nTg mice (Fig. 3C). The uptake and retention of [11C]AC-5216 estimated as %ID/ml in the striatum of PS19 mice were comparable to those of nTg mice without correction for body weight. Indeed, the area under the curve for PS19 striatum did not differ from nTg control value (t = 0.22, p > 0.05), despite a notable difference in body weight between these two genotype groups (23.6 g vs 36.8 g, t = 4.6, p < 0.001). In consideration of changes in body weight disproportionate to brain weight, we presumed %ID/ml to be more suitable than body mass-corrected index for quantifying distribution of this radioligand in the brain.
Figure 3.

Kinetics of intravenously administered [11C]AC-5216 in brains of 11-month-old nTg (open symbols; n = 6) and PS19 Tg (closed symbols; n = 8) mice. A–C, Time-radioactivity curves in the hippocampus (A), entorhinal cortex (B), and striatum (C) obtained from dynamic PET scans. D, E, Target-to-cerebellum ratio of radioactivities in the hippocampus (D) and entorhinal cortex (E). F, BPND values for radioligand binding to TSPO in the hippocampus and entorhinal cortex calculated by simplified reference tissue model. **p < 0.01 and ***p < 0.001 by Bonferroni's multiple comparison after ANOVA. Error bars represent SE.
The difference in retention of [11C]AC-5216 in the hippocampus and entorhinal cortex between PS19 and nTg mice was more explicitly displayed by plotting target-to-striatum ratios of the radioactivity over the scan time [F(1,12) = 24.9, p < 0.001 (hippocampus); F(1,12) = 30.5, p < 0.001 (entorhinal cortex) for main effect of genotype] (Fig. 3D,E). We also determined BPND for [11C]AC-5216 based on simplified reference tissue model using striatal data as references (Fig. 3F). BPND values were significantly increased in the hippocampus and entorhinal cortex of PS19 mice compared with nTg mice (F(1,24) = 39.9, p < 0.001 for main effect of genotype).
All eight PS19 mice undergoing [11C]AC-5216-PET received an additional scan with [18F]FEDAA1106 (Fig. 4). Retention of both [11C]AC-5216 (Fig. 4A) and [18F]FEDAA1106 (Fig. 4B) in the hippocampus exceeded that in the striatum. However, slow clearance of unbound and nonspecifically bound [18F]FEDAA1106 from brain tissues hampered high-contrast detection of specific binding sites (Fig. 4B). BPND for [18F]FEDAA1106 in the hippocampus was intimately correlated with that for [11C]AC-5216 (r2 = 0.71, p < 0.01 by Pearson's correlation coefficient), but the detection power of [18F]FEDAA1106 for TSPO increase was approximately half that of [11C]AC-5216, judging from the regression slope in the correlation plot (Fig. 4C).
Figure 4.
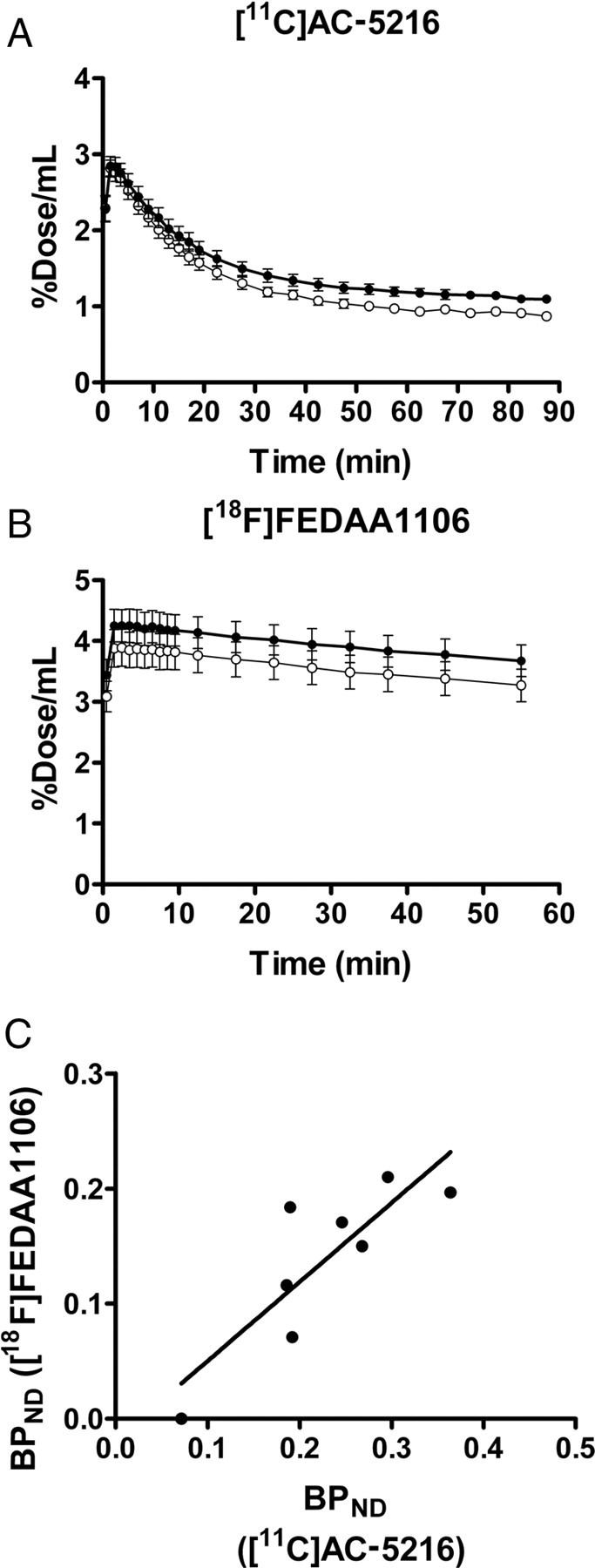
Comparison of brain kinetics and performance in detecting neuroinflammatory changes between two TSPO ligands, [11C]AC-5216 and [18F]FEDAA1106, in 11-month-old PS19 Tg mice (n = 8). A, B, Hippocampal (closed circles) and striatal (open circles) time-radioactivity curves in mice following intravenous injection of [11C]AC-5216 (A) and [18F]FEDAA1106 (B). Error bars represent SE. C, Scatterplot of hippocampal BPND for [18F]FEDAA1106 against that for [11C]AC-5216 in each individual. Significant correlation between these BPND estimates was observed (r2 = 0.71, p < 0.01 by F test), while [11C]AC-5216 yielded larger values and better detectability of subtle inflammations than did [18F]FEDAA1106. Solid line indicates the regression for all mice.
Progressive increase of in vivo TSPO signals and their neuropathological correlates in PS19 mice
In an expanded group of PS19 mice, in vivo radiolabeling of TSPO with [11C]AC-5216 was quantified in the hippocampus and entorhinal cortex of PS19 and nTg mice at different ages (Fig. 5A). Increase of radiotracer retention was also noticeable in the medial frontal cortex, while no overt changes were found in the striatum (Fig. 5A). Quantitative measurements of ligand binding in the hippocampus demonstrated no change in BPND at 3 months of age and significant increase of this index at 7 and 11 months of age relative to age-matched nTg controls (F(2,34) = 11.2, p < 0.001 for interaction between age and genotype; p < 0.01 and p < 0.001 at 7 and 11 months of age, respectively, by Bonferroni's test) (Fig. 5B). In view of a known finding that PS19 mice exhibit MRI-detectable hippocampal atrophy from 9 months of age (Yoshiyama et al., 2007), this result indicates that TSPO elevates antecedent to measurable neuronal loss at an in vivo level. Age-related intensification of TSPO signals in the hippocampus of PS19 mice was also supported by the scatterplot of BPND for [11C]AC-5216 against age (Fig. 5C), with these two parameters showing a significant correlation with each other (r2 = 0.40, p < 0.001 by Pearson's correlation coefficient). Notably, a longitudinal series of PET scans of hippocampal BPND in nine PS19 mice revealed that progression of TSPO upregulation is individually assessable (Fig. 5C, red symbols). Notwithstanding the statistical significance of its correlation with age, hippocampal BPND values displayed a large variance among individuals at the same age (Fig. 5C), and thus the level of TSPO expression in each PS19 mouse was not simply predictable by its age.
Figure 5.
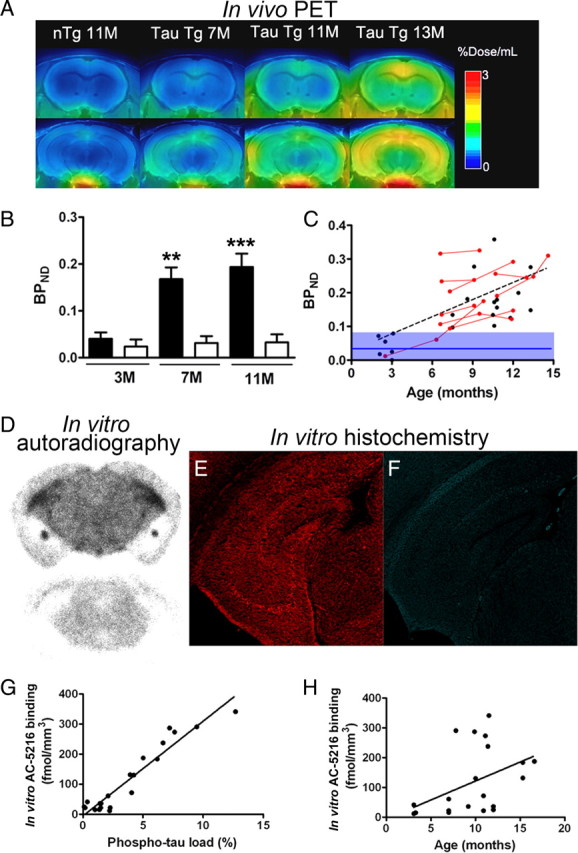
Progressive increase of PET and autoradiographic [11C]AC-5216 signals in the hippocampus of PS19 mice as a function of age and phosphorylated tau load. A, Coronal PET images containing the striatum (top) and hippocampus (bottom) in 11-month-old nTg and 7-, 11-, and 13-month-old PS19 Tg mice. Images were generated by averaging dynamic scan data at 30–90 min after radiotracer injection. B, BPND for [11C]AC-5216 in the hippocampus of nTg (open columns) and PS19 Tg (closed columns) mice at 3, 7, and 11 months of age. **p < 0.01 and ***p < 0.001 by Bonferroni's multiple comparison after ANOVA. Error bars represent SE. C, Scatterplot of hippocampal BPND for [11C]AC-5216 against age in PS19 Tg mice (n = 28; mean age ± SD = 8.9 ± 3.4 months; age range, 2.2–14.6 months). Red symbols represent data from a longitudinal analysis of the same individuals (n = 9), and the blue line and purple area indicate the mean ± SD of hippocampal BPND values in nTg mice (n = 17; mean age ± SD = 7.5 ± 3.6 months; age range, 2–12 months). The increase of BPND in PS19 Tg mice became noticeable at ∼6 months of age, and was thereafter augmented in a significant correlation with age (r2 = 0.40, p < 0.001 by Pearson's correlation coefficient). The broken line denotes the regression in PS19 Tg mice. D, Representative in vitro autoradiograms showing total (top section) and nonspecific (bottom section) binding of [11C]AC-5216 in coronal brain slices obtained from a 12-month-old PS19 Tg mouse. Marked signal intensification was observed in the hippocampus, which exhibited severe atrophy. E, F, Double fluorescence labeling of the section shown in D with anti-phospho-tau antibody, AT8 (E), and amyloid dye, thioflavin-S (F). Numerous AT8-positive lesions lacking thioflavin-S positivity were present, and were conceived to be relatively immature intraneuronal amyloid fibrils. G, H, Scatterplots of autoradiographic [11C]AC-5216 binding in the hippocampus of PS19 mice (n = 21) against AT8-positive phospho-tau load (G; expressed as percentage of total hippocampal area) and age (H). The solid lines represent regression.
Next, we sought a neuropathological determinant of TSPO radiosignals by combined autoradiographic and histological assays using sliced brains of PS19 mice aged 3–17 months. In hippocampal sections displaying intense in vitro binding of [11C]AC-5216, phosphorylated tau immunoreactivity was markedly enhanced, while no or only several NFT-like amyloid inclusions were detected by thioflavin-S staining (Fig. 5D–F). Radiolabeling of TSPO with [11C]AC-5216 was linearly proportional to the amount of phospho-tau immunolabeling (Fig. 5G), and an excellent correlation between these two measures was observed according to partial correlation analysis (r2 = 0.95, p < 0.001). Radiotracer binding also showed a tendency to correlate with age (Fig. 5H), but the partial correlation was not statistically significant in these test samples (r2 = 0.32, p > 0.05). Hence, deposition of pathologically phosphorylated tau proteins rather than thioflavin-S-positive high-order tau assembly was the primary determinant of the increase in [11C]AC-5216-labeled TSPO, and interindividual variability in the onset and chronological rate of tau pathogenesis largely modified the course of neuroinflammatory changes during aging.
In vivo imaging of glial response to amyloid accumulation in APP23 mouse brains assessed by [11C]AC-5216 and [11C]PIB
Spatial mapping of [11C]AC-5216 retention in brains of 24-month-old nTg and APP23 mice illustrated the glial response to amyloid deposition in the neocortex and hippocampus (Fig. 6A). This observation was also supported by ex vivo (Fig. 6B) and in vitro (Fig. 6C) autoradiograms using [11C]AC-5216, and radiolabeling of TSPO was well colocalized with subsequent FSB staining of amyloid plaques in these regions (Fig. 6C). In accordance with our previous report (Ji et al., 2008), TSPO immunoreactivity was present primarily in astrocytes surrounding plaques (data not shown).
Figure 6.

PET and autoradiographic images of TSPO upregulation in old APP23 mice. A, Coronal PET images containing the striatum (top) and hippocampus (bottom) in 28-month-old nTg and 26-month-old APP23 Tg mice. Images were generated by averaging dynamic scan data at 30–60 min after [11C]AC-5216 injection. B, Ex vivo autoradiographic sections containing the hippocampus (at bregma −2.8 mm) in 27-month-old nTg and 30-month-old APP23 Tg mice. Brains were collected at 30 min after intravenous injection of [11C]AC-5216. C, Amyloidosis-associated in vitro autoradiographic [11C]AC-5216 signals in a 24-month-old APP23 Tg mouse brain section. The autoradiographic section (left) was subsequently stained with an amyloid dye, FSB (middle), and colocalization of radiotracer binding and plaque deposition was assessed in a merged image (right).
Time-radioactivity curves generated from dynamic [11C]AC-5216-PET data indicated lower levels of initial radiotracer uptake in all examined regions of APP23 mice (n = 9) than in nTg controls (n = 5) (Fig. 7A–C). Despite this difference, Tg mice exhibited augmented radioactivity retention relative to control mice in the hippocampus and neocortex beyond 45 min after tracer injection (Fig. 7A,B). Unlike these areas, radiosignals in the striatum of Tg mice beyond 60 min were nearly equivalent to those of controls (Fig. 7C), justifying the use of striatal data as reference. Indeed, the ratios of radioactivities to striatal values were significantly increased in the hippocampus (Fig. 7D) (F(1,12) = 5.3, p < 0.05 for main effect of genotype) and neocortex (Fig. 7E) (F(1,12) = 8.0, p < 0.05 for main effect of genotype) of APP23 mice compared with nTg mice. Although BPND values calculated by simplified reference tissue model were significantly increased in the neocortex and hippocampus of APP23 mice compared with nTg controls (Fig. 7F), the retention of [11C]AC-5216 signals in the striatum of aged nTg exceeded those in the hippocampus and neocortex, potentially resulting in inaccurate BPND estimates. Thus, we reexamined the radiotracer binding by calculating DVRs. Similar to BPND, values of DVR for [11C]AC-5216 in the hippocampus and neocortex of APP23 mice were significantly larger than those of nTg controls (data not shown; F(1,24) = 20.3, p < 0.001 for main effect of genotype), but were much smaller than those of aged tau Tg mice. In addition, PET scans of these animals with [18F]FEDAA1106 failed to detect differences in glial activities between the two genotype groups (n = 7; data not shown).
Figure 7.

Kinetics of intravenously administered [11C]AC-5216 in brains of 24-month-old nTg (open symbols; n = 5) and APP23 Tg (closed symbols; n = 9) mice. A–C, Time-radioactivity curves in the hippocampus (A), entorhinal cortex (B), and striatum (C) obtained from dynamic PET scans. D, E, Target-to-cerebellum ratio of radioactivities in the hippocampus (D) and entorhinal cortex (E). F, BPND values for radioligand binding to TSPO in the hippocampus and entorhinal cortex calculated by simplified reference tissue model. *p < 0.05 and **p < 0.01 by Bonferroni's multiple comparison after ANOVA. Error bars represent SE.
As in PS19 mice, hippocampal BPND for [11C]AC-5216 was significantly correlated with age in 15 APP23 mice at 12–27 months of age (Fig. 8A) (r2 = 0.29, p < 0.05 by Pearson's correlation coefficient), in agreement with progressive Aβ amyloidosis in these animals. Twelve of these Tg animals also received PET scans with [11C]PIB, and amyloid deposition in the hippocampus and neocortex was detected as increased BPND values, which were significantly greater than those for [11C]AC-5216 (Fig. 8B) (F(1,44) = 79.4, p < 0.001 for main effect of radioligand). Furthermore, there was no significant correlation between hippocampal BPND values for these two radioligands (Fig. 8C) (r2 = 0.19, p > 0.05 by Pearson's correlation coefficient), indicating that plaque deposition at a relatively early stage does not induce TSPO upregulation detectable in living brains.
Figure 8.
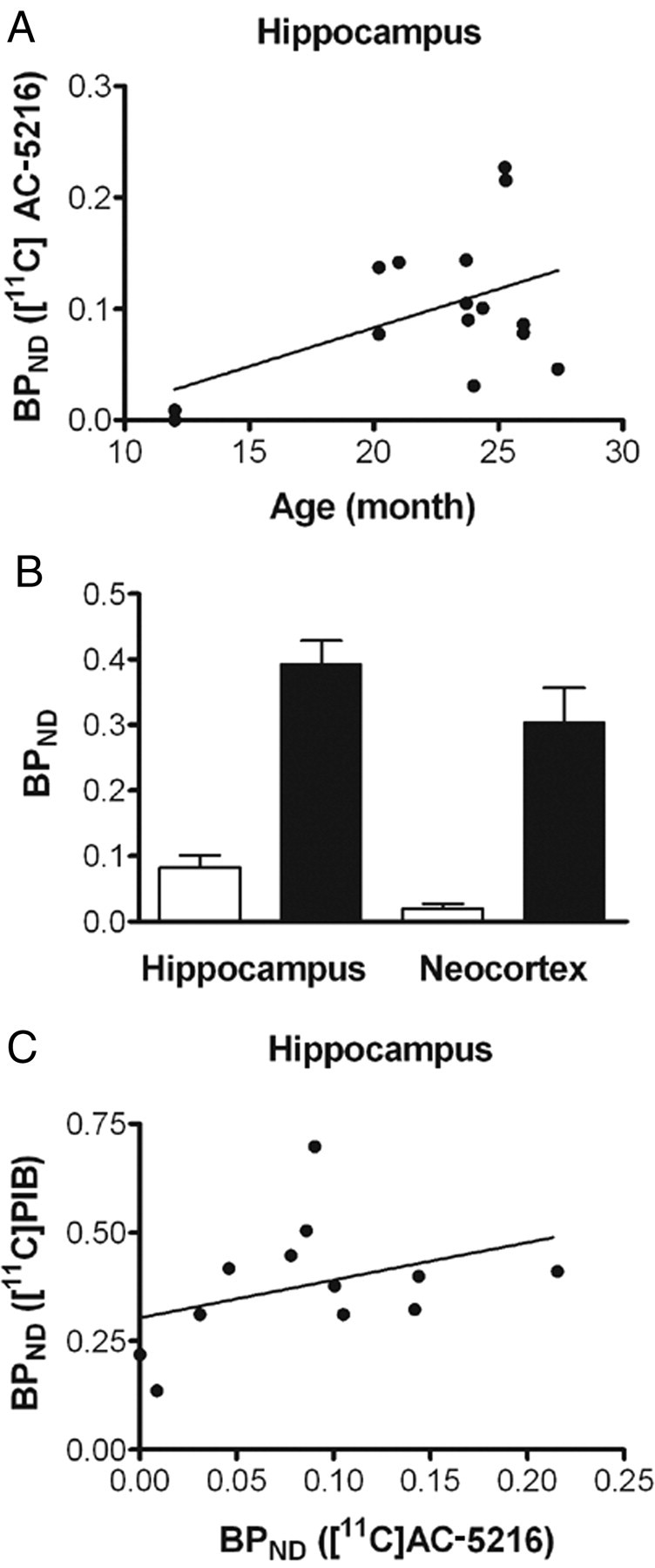
Association of TSPO signals with age-dependent deposition of Aβ amyloid in living brains of APP23 mice. A, Scatterplot of BPND for [11C]AC-5216 against age in the hippocampus of APP23 Tg mice (n = 15; mean age ± SD = 22.4 ± 4.9 months; age range, 12–28.4 months). The solid line represents regression. B, BPND of [11C]AC-5216 (open columns) and an amyloid radioligand, [11C]PIB (closed columns), in the hippocampus of APP23 Tg mice (n = 12; mean age ± SD = 22.6 ± 5.3 months; age range, 12–28.4 months). Error bars represent SE. C, Scatterplot of BPND for [11C]PIB against that for [11C]AC-5216 in the hippocampus of APP23 Tg mice (n = 12; mean age ± SD = 22.6 ± 5.3 months; age range, 12–28.4 months). The solid line represents regression.
TSPO upregulation following neuronal loss in APPE693Δ mice
PET imaging of TSPO with [11C]AC-5216 was also performed in 25.4-month-old APPE693Δ mice, and radioligand retention was enhanced in the hippocampus and neocortex of these animals relative to age-matched controls (Fig. 9A). BPND values in the hippocampus of APPE693Δ mice (n = 3) was significantly greater than those of controls (n = 5) (p < 0.05 by t test). Postmortem assays demonstrated increased AT8 immunoreactivity in APPE693Δ mice compared with nTg mice (data not shown), while the intensity of phosphor-tau immunolabeling in these Tg mice was much weaker than that in PS19 mice. TSPO signals were concentrated in putative glial cells in the hippocampus of APPE693Δ mice (Fig. 9B), similar to PS19 mice (Yoshiyama et al., 2007; Ji et al., 2008). Based on quantification of hippocampal NeuN and TSPO immunoreactivities, APPE693Δ mice presented neuronal loss, but did not exhibit detectable TSPO immunolabeling before the occurrence of notable neuronal death (Fig. 9C). This was in contrast with PS19 mice, in which TSPO-positive gliosis preceded pronounced neuronal loss (Fig. 9C).
Figure 9.
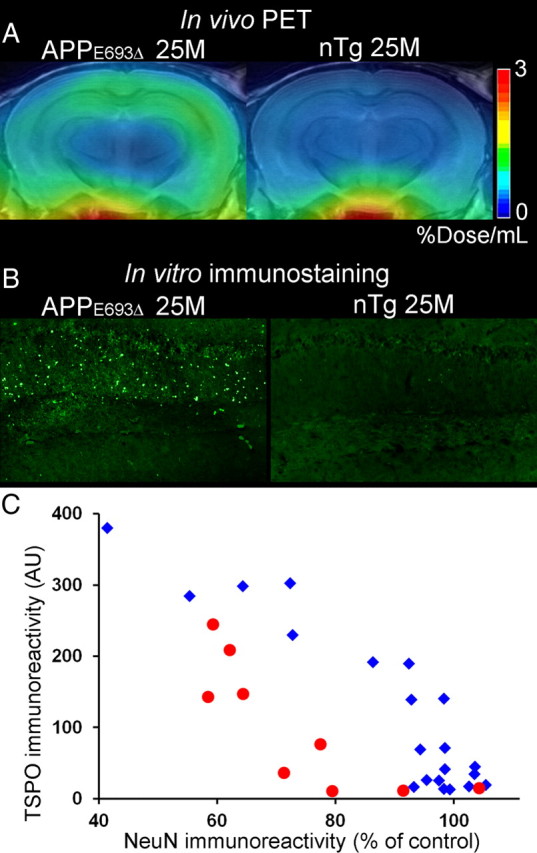
TSPO upregulation and neuronal loss in APPE693Δ and PS19 mice. A, Coronal PET images containing the hippocampus in 25.4-month-old APPE693Δ Tg (left) and 25-month-old nTg (right) mice. Images were generated by averaging dynamic scan data at 30–60 min after [11C]AC-5216 injection. B, Immunolabeling of TSPO with NP155 in the hippocampal CA1 region of 25.4-month-old APPE693Δ Tg (left; the same animal as shown in A) and 25-month-old nTg (right) mice. C, Intensity of TSPO immunolabeling (arbitrary values) plotted against NeuN immunoreactivity (percentage of area in controls matched by age with each individual) in APPE693Δ (red circles; n = 9; 22–33 months of age) and PS19 mice (blue rhombuses; n = 21; 3–17 months of age).
Discussion
Notwithstanding the abundance of evidence for the activation of inflammatory glia in many neurodegenerative disorders, the roles of these cells in the propagation of and/or protection against neurotoxic injuries in the disease state are yet to be determined. In view of the present data obtained by the use of a high-contrast imaging agent, [11C]AC-5216, and a newly generated antibody, microglial TSPO signals became detectable in living brains concomitant with accumulations of amyloid deposits as exemplified by fibrillar tau inclusions in neurons. Since TSPO immunoreactivities are associated with NFTs, neuropil threads, and plaque neurites rather than Aβ deposits, PET imaging of TSPO would provide a surrogate biomarker of tau-induced neuronal deteriorations in AD. This strategy also is applicable to non-AD tauopathies, in consideration of the numerous TSPO-positive microglia in PSP and Pick's disease brains and intense [11C]AC-5216 radiosignals in PET scans of PS19 mice.
In vivo TSPO imaging may not be as capable of capturing early AD pathologies as amyloid PET with [11C]PIB, because TSPO was not abundantly localized to AβN3pE-immunoreacitve diffuse plaques in AD brains, to which [11C]PIB can firmly bind (Maeda et al., 2007; Higuchi et al., 2010). However, [11C]AC-5216 could be more selective for proteinaceous aggregates that compromise neuronal survival like NFTs compared with plaques visualized by [11C]PIB and other specific tracers. In fact, there have been multiple reports documenting 20–40% of nondemented elderly subjects with increased levels of [11C]PIB retention in the brain (Mintun et al., 2006; Pike et al., 2007; Jack et al., 2009), a factor that might hinder the use of amyloid imaging agents for selecting individuals requiring disease-modifying and/or neuroprotective treatments. Likewise, radiotracers for fibrillar tau aggregates in preclinical development stages (Okamura et al., 2005; Higuchi, 2009; Higuchi et al., 2010; Maruyama et al., 2009) are potentially advantageous over TSPO ligands in differentiating between tau-positive and tau-negative frontotemporal lobar degenerations, while [11C]AC-5216 is likely to serve for visualizing neurotoxic events at a certain stage that PET with tau fibril agents may not cover. This notion is supported by our observation that thioflavin-S- and FSB-negative but AT8-positive phospho-tau inclusions were coexistent with autoradiographic [11C]AC-5216 signals in PS19 mice. It is accordingly possible that relatively immature tau assemblies poorly captured by β-sheet ligands damage neurons and recruit TSPO-expressing microglia in response to this damage. The neurotoxicities induced by low-order tau multimers has been implicated in disease through studies of tau Tg mouse models (Higuchi et al., 2002; Santacruz et al., 2005; Yoshiyama et al., 2007), and the consequent TSPO-positive microgliosis is in sharp contrast with the lack of TSPO upregulation in microglia responding to extracellular Aβ deposits except neuritic plaques. Although dense-cored plaques in APP23 were found to be surrounded by astrocytes expressing TSPO (Ji et al., 2008), such astrocytic TSPO signals were undetectable in postmortem AD brains (Fig. 1I), indicating the absence of tight links between nonneuritic plaques and TSPO-positive gliosis in human cases. The astrocytic TSPO expression was positively correlated with glial cell-derived neurotrophic factor immunoreactivity across diverse animal models including APP23 mice (Ji et al., 2008). By contrast, our auxiliary analysis has shown that activated astrocytes in AD brains barely displayed this neurotrophic signal (data not shown) and thus could aggressively act on neurons in synergy with TSPO-positive microglia, unlike those in APP23 mice. This distinction between species concerning glial characteristics is attributable to neurodegenerative tau pathologies, which exist in AD but are uncommon in APP Tg mice. Furthermore, the presence of neurotoxic inflammation antecedent to the NFT deposition would also be examined in living humans with prodromal tauopathies by the concomitant use of radioligands for TSPO and tau fibrils.
It is of additional importance that an increase in TSPO levels was observed in the hippocampus of PS19 mice at 7 months of age despite the lack of MRI-measurable regional atrophies at this stage (Yoshiyama et al., 2007), and our recent PET study with [18F]FEDAA1106 and [18F]FDG has also demonstrated precedence of TSPO increases to impairments of cerebral glucose metabolism in PS19 mice (Hattori S, Maeda J, Tokunaga M, Yu I, Ji B, Ono M, Maruyama M, Aoki I, Zhang MR, Fukumura T, Suhara T, Higuchi M, unpublished observations). These findings collectively imply advantages of TSPO-PET over morphometric and metabolic imaging in detecting incipient neurotoxicities induced by tau abnormalities. This notion is further supported by the present neuropathological data demonstrating emergence of TSPO-expressing glia antecedent to loss of NeuN signals in the hippocampus of PS19 mice, which is in distinction from increased TSPO levels apparently secondary to neuronal loss in APPE693Δ mice, despite neurotoxicities of intraneuronal oligomeric Aβ-inducing gliotic changes at relatively young ages (Tomiyama et al., 2010). It is, however, yet to be clarified with different experimental models whether TSPO-positive glia is specifically responsive to tau lesions, or whether intracellular accumulation of some other pathologically involved molecules, such as α-synuclein (Spillantini et al., 1997) and TDP-43 (Neumann et al., 2006) can be a strong inducer of glial TSPO upregulation at an early stage of neurodegeneration.
Research attempts to incorporate PET imaging with [11C]PK11195 in the clinical workup for AD have so far only proven its limited capability in discriminating afflicted patients from normal elderly (Cagnin et al., 2001; Edison et al., 2008; Okello et al., 2009; Wiley et al., 2009) and pathologically grading disease severity, but this shortcoming may reflect a relatively inefficient blood–brain barrier penetration and binding affinity of the radioligand (Maeda et al., 2004). A series of DAA1106 analogs, which yielded intense PET signals displaceable with nonradioactive TSPO ligands in the brains of rodents and primates (Maeda et al., 2004; Zhang et al., 2004; Venneti et al., 2007, 2008), have been reported to visualize increases of TSPO in AD brains (Yasuno et al., 2008), but these changes were not spatially linked to depositions of Aβ and tau fibrils. According to the small-animal PET data obtained here, [11C]AC-5216 enables PET mapping of glial TSPO with a higher contrast than does [18F]FEDAA1106 in AD model mice, benefiting from its prompt exit from the brain when unbound to the target-binding elements. Since Tg animals used here did not model the entire etiological pathway in the transition from aging to neurodegenerative disorders, the performance of [11C]AC-5216 in humans relative to [18F]FEDAA1106 is not fully predictable according to the current data, and will need to be evaluated by clinical PET assessments.
Despite myriad evidences for interplays between dying neurons and inflammatory gliosis (Block et al., 2007), little is known concerning molecular signals mediating the stimulation of TSPO-positive microglia by phosphorylated tau depositions antecedent to overt neuronal loss, as observed in the previous in vitro autoradiographic (Yoshiyama et al., 2007) and present in vivo PET analyses. Upregulations of cyclooxygenase-2 (cox-2) and interleukin-1β (IL-1β) were indicated to be early pathological alterations in PS19 and similar tau Tg mouse strains (Bellucci et al., 2004; Yoshiyama et al., 2007), and a study on neuronal cell cultures reported that IL-1β could induce cox-2 expression (Hoozemans et al., 2001). In these considerations, it is inferable that proinflammatory cytokines exemplified by IL-1β are responsible for tau-triggered neuronal deteriorations. In addition to these neuroinflammatory pathologies, PS19 mice were characterized as initially developing abnormal tau accumulations in presynaptic terminals of hippocampal neurons (Yoshiyama et al., 2007). A speculative hypothesis that synaptic tau lesions are inducers of an inflammatory cascade could be drawn by linking these facts, since disruptions of synaptic functionality and calcium homeostasis may locally give rise to activation of NF-κB (Meffert et al., 2003), a multifunctional component capable of provoking transcriptions of proinflammatory cytokines. Conversion of microglia from resident and /or neuroprotective forms to an aggressive mode could consequently occur, and could be detected as overexpression of TSPO (Ji et al., 2008). Presynaptic degeneration and microgliosis were also observed as early pathological alterations in APPE693Δ mice (Tomiyama et al., 2010), but were not concurrent with TSPO upregulation until the occurrence of neuronal death. As Aβ deposits in these animals were compartmentalized in endosomes, lysosomes, endoplasmic reticulum, and some other organelles (Nishitsuji et al., 2009), unlike cytoplasmic accumulation of tau proteins in PS19 mice (Yoshiyama et al., 2007), we presume that these Aβ species may initially provoke glial activation in a TSPO-negative mode distinct from tau-induced gliosis. TSPO can therefore be a biomarker for impending neuronal death accelerated by aggressive microglial response to tau lesions. This view may be in line with our recent observation of increased [18F]FEDAA1106 signals in a subset of asymptomatic individuals carrying the tau gene mutation causative of familial tauopathy (Miyoshi et al., 2010), and the sensitivity of capturing early neuroinflammatory changes in such cases would be enhanced by applying [11C]AC-5216 to clinical assays.
The tight interconnections between TSPO increase and tau-induced neurodegeneration as illustrated in the present work support the applicability of [11C]AC-5216-PET to nonclinical and clinical evaluations of emerging anti-amyloid treatments. Aβ immunization, for instance, triggers activation of TSPO-positive glial cells (Maeda et al., 2007), which may contain microglia unengaged in plaque removal and be prone to promoting neurotoxic tau pathogenesis. In vivo monitoring of TSPO levels would play a pivotal role in determining the safety margin of therapeutic agents counteracting the Aβ accumulation. Similarly, active and passive immunization strategies under development now against phosphorylated tau proteins (Asuni et al., 2007; Takeuchi et al., 2009) possibly stimulate a toxic conversion of microglia accompanied by a surge of TSPO, and this issue would be addressed with the aid of TSPO-PET scans of treated animal models.
To conclude, we describe here the properties of a biomarker for microglia that is a sensitive reporter of neurotoxic effects of tau inclusion formation in AD and related neurodegenerative tauopathies based on a new class of TSPO radioligands. As the kinetics of [11C]AC-5216 has been characterized in normal human subjects (Miyoshi et al., 2009), a side-by-side comparison would be enabled between courses of neuroinflammatory insults in humans and experimental models, toward the establishment of immunotherapeutic interventions in the disease pathogenesis and creation of better genetically engineered animals recapitulating the amyloidosis-gliosis intercontinuum in human diseases.
Footnotes
This work was supported in part by grants from the National Institute on Aging of the National Institutes of Health (AG10124 and AG17586 to J.Q.T. and V.M.-Y.L., respectively) and by Grants-in-Aid for Young Scientists (B) 20790047 (J.M.), Grants-in-Aid for the Molecular Imaging Program and Scientific Research on Priority Areas, and Research on Pathomechanisms of Brain Disorders Grant 20023036 (M.H.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank Taisho Pharmaceutical (Tokyo, Japan) for providing DAA1123. We also thank Takeharu Minamihisamatsu for technical assistance, staff of the Molecular Probe Group, National Institute of Radiological Sciences, for support with radiosynthesis, and Dr. Haruhisa Inoue of Kyoto University for critical discussion.
The authors declare no competing financial interests.
References
- Asuni et al., 2007.Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci et al., 2004.Bellucci A, Westwood AJ, Ingram E, Casamenti F, Goedert M, Spillantini MG. Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am J Pathol. 2004;165:1643–1652. doi: 10.1016/S0002-9440(10)63421-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block et al., 2007.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Cagnin et al., 2001.Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Diorio et al., 1991.Diorio D, Welner SA, Butterworth RF, Meaney MJ, Suranyi-Cadotte BE. Peripheral benzodiazepine binding sites in Alzheimer's disease frontal and temporal cortex. Neurobiol Aging. 1991;12:255–258. doi: 10.1016/0197-4580(91)90106-t. [DOI] [PubMed] [Google Scholar]
- Dodel et al., 2003.Dodel RC, Hampel H, Du Y. Immunotherapy for Alzheimer's disease. Lancet Neurol. 2003;2:215–220. doi: 10.1016/s1474-4422(03)00349-1. [DOI] [PubMed] [Google Scholar]
- Edison et al., 2008.Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ. Microglia, amyloid, and cognition in Alzheimer's disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Higuchi, 2009.Higuchi M. Visualization of brain amyloid and microglial activation in mouse models of Alzheimer's disease. Curr Alzheimer Res. 2009;6:137–143. doi: 10.2174/156720509787602906. [DOI] [PubMed] [Google Scholar]
- Higuchi et al., 2002.Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski J, Lee VM. Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron. 2002;35:433–446. doi: 10.1016/s0896-6273(02)00789-4. [DOI] [PubMed] [Google Scholar]
- Higuchi et al., 2005.Higuchi M, Iwata N, Matsuba Y, Sato K, Sasamoto K, Saido TC. 19F- and 1H-MRI detection of amyloid-β plaques in vivo. Nat Neurosci. 2005;8:527–533. doi: 10.1038/nn1422. [DOI] [PubMed] [Google Scholar]
- Higuchi et al., 2010.Higuchi M, Maeda J, Ji B, Maruyama M, Okauchi T, Tokunaga M, Ono M, Suhara T. In-vivo visualization of key molecular processes involved in Alzheimer's disease pathogenesis: insights from neuroimaging research in humans and rodent models. Biochim Biophys Acta. 2010;1802:373–388. doi: 10.1016/j.bbadis.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Hoozemans et al., 2001.Hoozemans JJ, Veerhuis R, Janssen I, Rozemuller AJ, Eikelenboom P. Interleukin-1β induced cyclooxygenase 2 expression and prostaglandin E2 secretion by human neuroblastoma cells: implications for Alzheimer's disease. Exp Gerontol. 2001;36:559–570. doi: 10.1016/s0531-5565(00)00226-6. [DOI] [PubMed] [Google Scholar]
- Jack et al., 2009.Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji et al., 2008.Ji B, Maeda J, Sawada M, Ono M, Okauchi T, Inaji M, Zhang MR, Suzuki K, Ando K, Staufenbiel M, Trojanowski JQ, Lee VM, Higuchi M, Suhara T. Imaging of peripheral benzodiazepine receptor expression as biomarkers of detrimental versus beneficial glial responses in mouse models of Alzheimer's and other CNS pathologies. J Neurosci. 2008;28:12255–12267. doi: 10.1523/JNEUROSCI.2312-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk et al., 2004.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Lammertsma et al., 1996.Lammertsma AA, Bench CJ, Hume SP, Osman S, Gunn K, Brooks DJ, Frackowiak RS. Comparison of methods for analysis of clinical [11C]raclopride studies. J Cereb Blood Flow Metab. 1996;16:42–52. doi: 10.1097/00004647-199601000-00005. [DOI] [PubMed] [Google Scholar]
- Logan et al., 1996.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- Maeda et al., 2004.Maeda J, Suhara T, Zhang MR, Okauchi T, Yasuno F, Ikoma Y, Inaji M, Nagai Y, Takano A, Obayashi S, Suzuki K. Novel peripheral benzodiazepine receptor ligand [11C]DAA1106 for PET: an imaging tool for glial cells in the brain. Synapse. 2004;52:283–291. doi: 10.1002/syn.20027. [DOI] [PubMed] [Google Scholar]
- Maeda et al., 2007.Maeda J, Ji B, Irie T, Tomiyama T, Maruyama M, Okauchi T, Staufenbiel M, Iwata N, Ono M, Saido TC, Suzuki K, Mori H, Higuchi M, Suhara T. Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer's disease enabled by positron emission tomography. J Neurosci. 2007;27:10957–10968. doi: 10.1523/JNEUROSCI.0673-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama et al., 2009.Maruyama M, Maeda J, Ji B, Zhang MR, Okauchi T, Ono M, Hattori S, Trojanowski JQ, Lee VMY, Fukumura T, Higuchi M, Suhara T. In-vivo optical and PET detections of fibrillar tau lesions in a mouse model of tauopathies. 2009 Abstract Viewer/Itinerary Planner. Program No. IC-P-009. Alzheimer's Association International Conference on Alzheimer's Disease 2009; July 2009; Chicago. 2009. [Google Scholar]
- McGeer and McGeer, 1995.McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- Meffert et al., 2003.Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-κB functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Mintun et al., 2006.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Miyoshi et al., 2009.Miyoshi M, Ito H, Arakawa R, Takahashi H, Takano H, Higuchi M, Okumura M, Otsuka T, Kodaka F, Sekine M, Sasaki T, Fujie S, Seki C, Maeda J, Nakao R, Zhang MR, Fukumura T, Matsumoto M, Suhara T. Quantitative analysis of peripheral benzodiazepine receptor in the human brain using PET with 11C-AC-5216. J Nucl Med. 2009;50:1095–1101. doi: 10.2967/jnumed.109.062554. [DOI] [PubMed] [Google Scholar]
- Miyoshi et al., 2010.Miyoshi M, Shinotoh H, Wszolek ZK, Strongosky AJ, Shimada H, Arakawa R, Higuchi M, Ikoma Y, Yasuno F, Fukushi K, Irie T, Ito H, Suhara T. In vivo detection of neuropathologic changes in presymptomatic MAPT mutation carriers: A PET and MRI study. Parkinsonism Relat Disord. 2010;16:404–408. doi: 10.1016/j.parkreldis.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Neumann et al., 2006.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Nishitsuji et al., 2009.Nishitsuji K, Tomiyama T, Ishibashi K, Ito K, Teraoka R, Lambert MP, Klein WL, Mori H. The E693Δ mutation in amyloid precursor protein increases intracellular accumulation of amyloid beta oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am J Pathol. 2009;174:957–969. doi: 10.2353/ajpath.2009.080480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura et al., 2005.Okamura N, Suemoto T, Furumoto S, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, Fujiwara H, Nemoto M, Maruyama M, Arai H, Yanai K, Sawada T, Kudo Y. Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer's disease. J Neurosci. 2005;25:10857–10862. doi: 10.1523/JNEUROSCI.1738-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okello et al., 2009.Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, Walker Z, Kennedy A, Fox N, Rossor M, Brooks DJ. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72:56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike et al., 2007.Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, Mathis CA, Klunk WE, Masters CL, Rowe CC. β-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- Santacruz et al., 2005.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato et al., 2004.Sato K, Higuchi M, Iwata N, Saido TC, Sasamoto K. Fluoro-substituted and 13C-labeled styrylbenzene derivatives for detecting brain amyloid plaques. Eur J Med Chem. 2004;39:573–578. doi: 10.1016/j.ejmech.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Spillantini et al., 1997.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat et al., 1997.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai et al., 2005.Tai YC, Ruangma A, Rowland D, Siegel S, Newport DF, Chow PL, Laforest R. Performance evaluation of the microPET focus: a third-generation microPET scanner dedicated to animal imaging. J Nucl Med. 2005;46:455–463. [PubMed] [Google Scholar]
- Takeuchi et al., 2009.Takeuchi H, Inoue H, Higuchi M, Tsukita K, Trojanowski JQ, Lee VMY, Ji B, Takahashi R, Suhara T. Development of therapies and monitoring for tauopathies with dementia. Soc Neurosci Abstr. 2009;35:236–16. [Google Scholar]
- Tomiyama et al., 2010.Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, Nishitsuji K, Ito K, Shimada H, Lambert MP, Klein WL, Mori H. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski, 2008.Trojanowski JQ. PENN neurodegenerative disease research—in the spirit of Benjamin Franklin. NeuroSignals. 2008;16:5–10. doi: 10.1159/000109753. [DOI] [PubMed] [Google Scholar]
- Venneti et al., 2007.Venneti S, Lopresti BJ, Wang G, Slagel SL, Mason NS, Mathis CA, Fischer ML, Larsen NJ, Mortimer AD, Hastings TG, Smith AD, Zigmond MJ, Suhara T, Higuchi M, Wiley CA. A comparison of the high-affinity peripheral benzodiazepine receptor ligands DAA1106 and (R)-PK11195 in rat models of neuroinflammation: implications for PET imaging of microglial activation. J Neurochem. 2007;102:2118–2131. doi: 10.1111/j.1471-4159.2007.04690.x. [DOI] [PubMed] [Google Scholar]
- Venneti et al., 2008.Venneti S, Wang G, Nguyen J, Wiley CA. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J Neuropathol Exp Neurol. 2008;67:1001–1010. doi: 10.1097/NEN.0b013e318188b204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley et al., 2009.Wiley CA, Lopresti BJ, Venneti S, Price J, Klunk WE, DeKosky ST, Mathis CA. Carbon 11-labeled Pittsburgh Compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch Neurol. 2009;66:60–67. doi: 10.1001/archneurol.2008.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamoto et al., 2009a.Yanamoto K, Kumata K, Yamasaki T, Odawara C, Kawamura K, Yui J, Hatori A, Suzuki K, Zhang MR. [18F]FEAC and [18F]FEDAC: Two novel positron emission tomography ligands for peripheral-type benzodiazepine receptor in the brain. Bioorg Med Chem Lett. 2009a;19:1707–1710. doi: 10.1016/j.bmcl.2009.01.093. [DOI] [PubMed] [Google Scholar]
- Yanamoto et al., 2009b.Yanamoto K, Yamasaki T, Kumata K, Yui J, Odawara C, Kawamura K, Hatori A, Inoue O, Yamaguchi M, Suzuki K, Zhang MR. Evaluation of N-benzyl-N-[11C]methyl-2- (7-methyl-8-oxo-2-phenyl-7,8-dihydro-9H-purin-9-yl)acetamide ([11C]DAC) as a novel translocator protein (18 kDa) radioligand in kainic acid-lesioned rat. Synapse. 2009b;63:961–971. doi: 10.1002/syn.20678. [DOI] [PubMed] [Google Scholar]
- Yasuno et al., 2008.Yasuno F, Ota M, Kosaka J, Ito H, Higuchi M, Doronbekov TK, Nozaki S, Fujimura Y, Koeda M, Asada T, Suhara T. Increased binding of peripheral benzodiazepine receptor in Alzheimer's disease measured by positron emission tomography with [11C]DAA1106. Biol Psychiatry. 2008;64:835–841. doi: 10.1016/j.biopsych.2008.04.021. [DOI] [PubMed] [Google Scholar]
- Yoshiyama et al., 2007.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2004.Zhang MR, Maeda J, Ogawa M, Noguchi J, Ito T, Yoshida Y, Okauchi T, Obayashi S, Suhara T, Suzuki K. Development of a new radioligand, N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoroethyl-5-methoxybenzyl)acetamide, for pet imaging of peripheral benzodiazepine receptor in primate brain. J Med Chem. 2004;47:2228–2235. doi: 10.1021/jm0304919. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2007.Zhang MR, Kumata K, Maeda J, Yanamoto K, Hatori A, Okada M, Higuchi M, Obayashi S, Suhara T, Suzuki K. 11C-AC-5216: a novel PET ligand for peripheral benzodiazepine receptors in the primate brain. J Nucl Med. 2007;48:1853–1861. doi: 10.2967/jnumed.107.043505. [DOI] [PubMed] [Google Scholar]