

Abstract
Autosomal recessive polycystic kidney disease (ARPKD) is a developmental disorder that mainly affects the kidneys and the biliary tract. Affected patients often have massively enlarged cystic kidneys as well as congenital hepatic fibrosis (CHF) characterized by dilated bile ducts and associated peribiliary fibrosis. This review will examine what is known about ARPKD‐associated liver disease and will highlight areas of ongoing research into its pathogenesis and potential treatment. Clin Trans Sci 2011; Volume 4: 460–465
Keywords: liver disease, pediatrics, cirrhosis
Introduction
Autosomal recessive polycystic kidney disease (ARPKD) is a rare genetic disorder that causes cystic kidneys in the developing fetus in utero and frequently results in pulmonary insufficiency after birth. Along with renal manifestations, affected patients also have abnormal biliary development with dilated bile ductules and peribiliary fibrosis, collectively termed as congenital hepatic fibrosis (CHF). ARPKD results from mutations in the PKHD1 gene, which encodes fibrocystin/polyductin (FC/PD), a protein located primarily in the primary cilia and apical surfaces of biliary and renal tubular epithelial cells. Little is known about the mechanism whereby mutations in FC/PD lead to ductal plate malformation and fibrosis is evident even in newborn infants.
Clinical Features
ARPKD is estimated to occur at a frequency of 1 in 20,000 live births, affecting all ethnic groups, including Caucasians, Africans, and Asians. 1 , 2 Approximately, half of the patients present in the neonatal period with enlarged, cystic kidneys and pulmonary hypoplasia, 3 30–50% of afflicted neonates have pulmonary insufficiency severe enough to result in death. 4 ARPKD patients who survive the neonatal period, however, have a good long‐term prognosis, with some surviving into their sixth decade. 5 Most patients will eventually develop systemic hypertension and many will develop chronic renal insufficiency that requires kidney transplantation. A total of 29–68% of patients also develop portal hypertension due to progressive liver fibrosis, with bleeding from esophageal varices contributing significantly to the morbidity and mortality of the disease. 1 , 5 A subset of patients develops recurrent or persistent bacterial cholangitis due to dilated bile ducts and stagnant bile flow. Overall, 7% of patients surviving the neonatal period will eventually require liver transplantation for complications of portal hypertension or cholangitis. 2 There is also an increased incidence of liver tumors, particularly cholangiocarcinomas, in ARPKD patients. 6 , 7
In contrast, ADPKD occurs at a much higher frequency of 1:400–1:1,000. 8 The majority of ADPKD patients present in adulthood, although there have been rare infantile cases. Kidney cysts that eventually progress to kidney failure are the main clinical feature of ADPKD. Less commonly, patients can also develop liver and pancreatic cysts. Rarely, patients may develop massive polycystic liver disease that requires surgical resection. There have been case reports of ADPKD patients with liver disease characterized by ductal plate malformation as seen in ARPKD 9 , 10 , 11 as well as patients who develop portal hypertension secondary to compression effect of the portal vein from extensive liver cysts. 12
Pathologic Findings
Liver disease in ARPKD is the result of a developmental defect. During normal intrahepatic biliary development, bipotential hepatoblasts adjacent to the portal mesenchyme begin to express biliary specific cytokeratins and form a single‐layer ring called the ductal plate. This ring of cells then becomes bilayered and undergoes remodeling to give rise to bile ducts. The part of the ductal plate that does not become bile ducts undergoes regression starting at the hilum and progressing to the liver periphery. 13 In ARPKD, there is a failure of ductal plate remodeling, with persistence of embryological bile duct structures embryological bile duct structures (Fig. 1); these eventually become massively dilated. The dilated bile ducts can go on to become macroscopic cysts that are in connection to the intrahepatic bile ducts and can be detected by imaging modalities. Associated portal veins are often abnormal, demonstrating an increased number of smaller portal vein branches. This pattern is thought to be intrinsic to the disease rather than reactive since the abnormal venous structures can be seen prior to the onset of significant portal hypertension. 14 There is often a significant amount of fibrosis in the portal tract even at birth, and as these affected children age, the amount of fibrosis increases, frequently resulting in hepatomegaly and portal hypertension. Interestingly and for unclear reasons, ARPKD‐affected livers often demonstrate proportionally larger left lobes compared to the right lobes. 7
Figure 1.
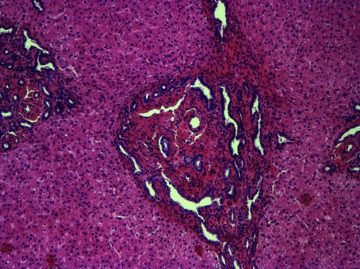
Liver explant specimen from a patient with ARPKD/CHF. The bile ducts are in a ring‐like pattern surrounding the portal vein. The portal tract is expanded with dilated bile ducts surrounded by fibrous tissue. The liver parenchyma is well preserved. This is typical of ductal plate malformation. H&E100× magnification.
Caroli Disease vs. Caroli Syndrome
The relationship between ARPKD, Caroli disease and Caroli syndrome is often confused. Jaques Caroli was a French physician who first described congenital dilation of the intrahepatic bile duct in 1958. Caroli disease refers to isolated dilation of the intrahepatic biliary tree that can be segmental. Caroli syndrome describes dilated bile ducts along with fibrosis surrounding the portal tracts, and it is most commonly associated with ARPKD. Caroli disease is much more rare, and can occur sporadically or be inherited in an autosomal‐dominant fashion, while Caroli syndrome is inherited in an autosomal recessive fashion.
In addition to ARPKD, there are several other diseases that cause CHF, including Bardet–Biedl syndrome, Meckel Gruber syndrome, Joubert syndrome, nephronophthisis, orofacial digital syndrome, congenital defect of glycosylation type 1b, and rarely, the autosomal‐dominant form of polycystic kidney disease (ADPKD). Since the identification of the genes involved in these conditions, the term Caroli syndrome has been gradually replaced by the name of the specific syndrome associated with the gene defect.
Genetic Defect
Polycystic kidney and hepatic disease 1 (PKHD1), the defective gene in ARPKD that encodes the protein FC/PD, is located on chromosome 6p12 and is one of the largest human genes, with at least 86 exons. The longest open reading frame consists of 66 exons (exon 2–67), encoding a protein with a molecular weight of 447 kD. 15 It is a large receptor‐like protein with the majority of the protein residing in the extracellular domain, with a single transmembrane region and a short cytoplasmic tail. 16 There are multiple alternatively spliced transcripts, including those without the transmembrane domain, presumably a secreted form. However, the functional significance of the various splice forms is not known. It is also important to note that all of the clinically relevant mutations have been described within the exons in the open reading frame; the function of the additional exons is also not known. 17
PKHD1 transcript expression is limited to the kidneys, liver, pancreas, and lungs, with the highest expression in the developing kidneys by RT‐PCR and northern blotting. 16 There is no mRNA expression in brain, heart, spleen, colon, thymus, and skeletal muscle in the adult mouse tissue. However, during mouse development, there is wide expression of PKHD1 in the neural tube, gut, bronchi, and vascular system prior to the appearance in the meatanephric primordium and liver by in situ hybridization and immunostaining. The function of FC/PD during this stage of development is unknown, although presumptively plays a role in the morphogenesis of primary tubules. 18
Within kidney tubular cells, FC/PD expression is predominantly located on the primary cilia of polarized epithelial cells and colocalizes with polycystin‐2, one of the proteins responsible for ADPKD by immunostaining. 18 It also interacts genetically with Pkd1, 19 the gene encoding polycystin‐1, a second protein associated with ADPKD. Together, these proteins are thought to be important in tubulogenesis and maintenance of the ductal structure. 20
FC/PD undergoes Notch‐like proteolytic processing via a γ‐secretase dependent mechanism. The released cytoplasmic tail then translocates to the nucleus, although its direct downstream target is not known. 21 , 22 Mutations in ARPKD have been found throughout the gene, without clustering to a specific region. There is a clear genotype–phenotype correlation with patients having two truncating mutations being the most severely affected, and generally dying in the perinatal period. 23 , 24 In our clinical experience, however, affected members of the family, presumably carrying the same mutation, can sometimes have markedly different clinical manifestations with regard to the severity of their liver and kidney disease.
Primary Cilia
Mutations in FC/PD seem to act at the level of the primary cilia. Primary cilia are nonmotile structures possessed by most mammalian cell types. They arise from basal bodies or centrosomes, and are formed by nine doublets of microtubules. Primary cilia are sensory organelles that detect various extracellular stimuli, depending on the cell type, and have been highly conserved through evolution. 25 , 26 In addition to ARPKD, there are several other genetic disorders that have been described to result from defective proteins associated with the primary cilia, and result in abnormally formed cilia, or ciliopathies. Most of them, including Bardet–Biedl syndrome, Meckel Gruber syndrome, Joubert Syndrome, nephronophthisis, and orofacial digital syndrome, are associated with cystic kidneys as well as hepatic ductal plate malformations. 27 , 28
There are multiple cell types in the liver that possess primary cilia, including portal fibroblasts (PF) and hepatic stellate cells (HSC), the predominant precursor cell types that cause fibrosis during liver injury. In contrast, hepatocytes do not have primary cilia and do not appear to express FC/PD (our unpublished finding). Masyuk et al. demonstrated by immunofluoresence staining that FC/PD localizes to the primary cilia in normal rat cholangiocytes and the primary cilia appears malformed in polycystic kidney (PCK) rat cholangiocytes by electron microscopy. 29 , 30
Experimental Models
Animal models have enhanced our ability to study the mechanism of disease in ARPKD. There are several animal models of ARPKD, some of which are orthologs of human ARPKD and some are phenotypically similar but not orthologous models.
The best‐characterized model is the PCK rat that arose from a spontaneous mutation in Sprague‐Dawley rats. The mutated gene on a region of rat chromosome 9 was later found to be the ortholog of the PKHD1 gene, with a splicing mutation IVS35–2A⇒T. 16 PCK rats have abnormally shaped primary cilia on the cholangiocytes by electron microscopy. Similar to the human disease, this rat model has both renal and hepatic involvement. Kidney cysts are noted at 3 weeks of age while dilated intrahepatic bile ducts and expanded portal tracts are present at birth. 31 , 32 Masyuk et al. characterized the segmental dilation and focal budding features of biliary tree in the PCK rats by three‐dimensional reconstruction. 30 As the animal ages, there is worsening of kidney cysts, biliary dilation, and hepatic fibrosis. The biliary dilation can eventually progress to large cysts that are disconnected from the rest of the biliary tree as shown by microimaging. 30 These rats do not have pancreatic abnormalities.
There are several orthologous PKHD1 mice, including those with target deletions in exon 40, exon 2, exon 3–4, and exon 4. 19 , 33 , 34 , 35 All of these animals have liver disease that is characterized by ductal plate malformation, but not all have kidney involvement. Some of the models have pancreatic cysts as well.
In addition to the PKHD1 orthologs, several other murine models have been used to study ARPKD (Table 1), including cpk, bpk, orpk, and inv mice, which have mutations in cystin, Bicc1, Inversin, and Polaris, respectively. 36 , 37 , 38 All of these mice have ciliopathies with cystic kidneys and biliary dysgenesis that are inherited in an autosomal recessive fashion.
Table 1.
Murine models of ARPKD.
| Model | Gene | Protein | Liver involvement | Kidney | Pancreas |
|---|---|---|---|---|---|
| Pkhd1 | Exon 40 | Yes (E16.5) | Normal | ||
| Exon 2 | Yes (1 month) | Yes, in females only, 9 months | Yes | ||
| Exon 3–4 | Yes | Yes | Yes (33.3% at 9 months) | ||
| Exon 4 | Yes (2 weeks) | Normal | Yes | ||
| cpk | Cys1 | Cystin | Yes | Yes | |
| bpk | Bicc1 | Bicaudal | Yes | Yes | |
| Inv | Invs | Inversin | Yes, early onset cholestasis | Yes | |
| orpk | TqN737Rpw | Polaris | Yes | Yes |
Factors implicated in ARPKD‐associated congenital hepatic fibrosis
In ARPKD, tubular epithelial cells are thought to be hyperproliferative 30 , 31 with increased apoptosis. 39 The roles of various growth factors and other soluble mediators that contribute to this phenotype have been explored in detail in recent years.
Calcium influx
Intracellular calcium is important in maintaining many important cellular processes, such as cell proliferation, gene expression, cell differentiation, and apoptosis. Primary cilia on renal tubular epithelial cells and cholangiocytes are thought to act as mechanosensors. 40 , 41 When the primary cilia are bent from shear force, there is calcium influx followed by release of calcium from intracellular stores. Polycystin 1 and polycystin 2, the defective proteins in ADPKD, are thought to function together to mediate the mechanotransduction from primary cilia to the intracellular calcium response. 40 , 42 Since FC is found in a complex with polycystin1 and polycystin 2 in the primary cilia, it is thought to play a role in the mechanotransduction process as well.
In cholangiocytes isolated from PCK rats, there is decreased intracellular calcium, 43 which is thought to be associated with cellular hyperproliferation. 8 , 44 Gardilone et al. showed that transient receptor potential vanilloid 4 (TRPV4), a calcium entry channel, is overexpressed in the cholangiocytes of PCK rats as well as in the ARPKD patients. 44 , 45 However, the overexpressed TRPV4 in PCK cholangiocytes are mainly localized intracellularly rather than localizing to the primary cilia as in the normal cholangiocytes. By activating TRPV4 with activators, there is restoration of intracellular calcium as well as increased AKT activity and decreased ERK activity. This increase in intracellular calcium is associated with decreased proliferation as well as decreased cystogenesis in cholangiocytes isolated from PCK rats. However, in in vivo experiments where young PCK rats were administered TRPV4 activator daily for 8 weeks, the TRPV4 activator was only effective in reducing kidney cysts and kidney fibrosis but had no significant effect on fibrocystic disease of the liver at the dose administered.
Cyclic AMP
Along with calcium, cyclic AMP (cAMP), an intracellular second messenger, is an important regulator of proliferation in different cell types. cAMP is thought to promote cystogenesis by stimulating chloride‐driven fluid secretion, and is increased in the cholangiocytes of PCK rats. Masyuk et al. demonstrated that in animals treated with Octreotide, a somatostatin analog that inhibits cAMP, there is a decrease in hepatic cyst volume, fibrotic score, and mitotic indices. 46 A vasopressin V2 receptor (VPV2) antagonist has also been shown to lower renal cAMP and inhibits renal disease progression in PCK rats. 47 However, the VPV2 receptor is not expressed in the liver and the antagonist does not improve fibrocystic liver disease in PCK rats. 48
cAMP has two downstream effectors, Epac and protein kinase A (PKA). cAMP binds to the regulatory unit of PKA and causes release of the catalytic unit, which then regulates phosphorylation of various proteins, including MEK and ERK, which are important intracellular signaling molecules in the MAP kinase pathway, regulating cell growth. Banales et al. showed that Epac activation causes increase in proliferation of both normal and PCK cholangiocytes while PKA causes increased proliferation in PCK cholangiocytes but decreased proliferation in normal cholangiocytes. 43 This group also showed that there is decreased intracellular calcium in PCK cholangiocytes, and that restoration of intracellular calcium inhibits both baseline proliferation of PCK cholangiocytes as well as PKA‐associated proliferation.
Epidermal growth factor
Epidermal growth factor (EGF) plays a major role in epithelial growth and differentiation. It binds to the EGF receptor and activates the mitogen‐activated protein kinases (MAPKs), which are serine/threonine specific kinases. The MAPK cascade consists of three protein kinases, an MAPK kinase kinase, an MAPK kinase (MAPK/ERK kinase, or MEK), and an MAPK (extracellular signal‐regulated kinase or ERK). Primary cholangiocytes isolated from a recessive murine model of PCK disease (BPK mouse) demonstrate exaggerated proliferation in response to EGF. 49 Similar responses were found in PCK cholangiocytes, with increased downstream activity of MEK5, which subsequently led to increased phosphorylation of ERK5. 50 Sato et al. administered Gefitinib, an EGF receptor tyrosine kinase inhibitor, to PCK rats and found that it led to improved fibrocystic liver disease, but no significant effect on cystic renal disease. 48 In vitro, Gefitinib also resulted in decreased cyst formation and proliferation of PCK cholangiocytes.
AKT/mTOR
Akt is a serine/threonine protein kinase that plays a key role in cell proliferation, apoptosis, transcription, and cell migration. It is activated by phosphoinositide 3‐kinase (PI3K), and is an important upstream regulator of mTOR (mammalian target of rapamycin). In ADPKD, aberrant activation of the AKT/mTOR pathway is thought to contribute to the disease, 51 , 52 and inhibition of mTOR by sirolimus results in improvement in cystic kidney and liver disease in ADPKD animal models. 53 , 54 , 55 , 56 , 57 Furthermore, sirolimus has been anecdotally shown to decrease native kidney and liver cystic volume of ADPKD patients who have undergone renal transplant and taken sirolimus as immunosuppression. 56 , 58 However, in recent prospective clinical trials of ADPKD patients, the results are mixed. 59 , 60 In one study, sirolimus did not halt cystic kidney growth while in another study, there was a decrease in the rate of kidney cystic growth without significant effects on the progression of renal impairment.
In ARPKD, the AKT/mTOR pathway has also been shown to be overexpressed in kidney tubular epithelial cells. 61 However, sirolimus treatment fails to improve fibrocystic liver and kidney disease in the PCK rat. 62
Renin‐angiotensin system
The renin‐angiotensin system (RAS), in addition to being a circulating hormone system, has also been recognized as a tissue‐based system that mediates fibrosis in several organs, including heart, kidney, vessels and liver. 63 , 64 , 65 , 66 , 67 Angiotensinogen is a zymogen that is cleaved by renin into angiotensin I. Angiotensin I is then converted to angiotensin II by angiotensin‐converting enzyme (ACE), and it is the major bioactive product of RAS. In ARPKD, the systemic level of angiotensin I and II has found to be unchanged. 68 However, there is increased intrarenal expression of renin, ACE, and angiotensin II; 69 there is also increased expression of ACE and angiotensin II in the portal tract by immunostaining. 70 Jia et al. demonstrated that PCK rats treated with an ACE inhibitor, Lisinopril, have an improvement in collecting duct cysts and decreased ERK activities in the kidneys. 71 There is no reported result on the effect of chronic ACE inhibition in fibrocystic liver disease associated with ARPKD.
Others
Recently, Pioglitazone, a PPAR‐γ agonist, was found to be effective in decreasing hepatic fibrosis and cystic lesions as well as cystic kidney disease in PCK rats. 72 PPAR‐γ is a nuclear receptor that is activated by naturally occurring fatty acids and fatty acid derivatives. It has a variety of functions, including in adipocyte differentiation and the inflammatory response of endothelial cells as well as in renal fibrosis and hepatic regeneration. 73 , 74 , 75 Yoshihara et al. propose that Pioglitazone improves fibrocystic liver disease in PCK rats by inhibiting ERK and TGF‐β without affecting the mTOR pathway, while in the kidneys, it inhibits both ERK and mTOR.
Proliferating bile duct epithelium has been shown to be a major source of connective tissue growth factor (CTGF). 76 In ARPKD, there is increased expression of CTGF in the portal area and this may be a contributor to liver fibrogenesis in this disease as well. 77
Liver Fibrosis and ARPKD
Despite the various factors implicated in the cyst formation in ARPKD, there has been no clear connection between the development of hepatic cysts and the development of hepatic fibrosis. In the liver, there are two major myofibroblastic cell types, HSC and PF. HSC reside in the sinusoids while PF reside in the periportal area. In chronic liver injury, these cells may undergo myofibroblastic differentiation to become myofibroblasts, responsible for collagen production in the fibrotic liver. Myofibroblasts from both cell types acquire α‐smooth muscle actin (α‐SMA) during activation. HSC have been well studied, and found to be involved in many models of liver fibrosis. 78 , 79 PF have recently come under the spotlight as an important population of myofibroblastic precursors, and shown to be particularly important in biliary fibrosis. 80 , 81 By immunohistochemical staining, PF appear to be the major myofibroblastic precursor in ARPKD/CHF (our unpublished finding). There is evidence that interactions between cholangiocytes and PF are important in the progression of liver fibrosis 82 , 83 and these interactions may be critical in the pathogenesis of hepatic fibrosis in this disease.
Future Directions
Much progress has been made in understanding the pathogenesis of fibrocystic kidney and liver disease of ARPKD over the past 10 years, since the mutated gene was identified. Most studies on ARPKD‐associated CHF to date have focused on the hyperproliferative properties of cholangiocytes. However, the morbidity of the fibrocystic liver disease mainly results from progressive portal fibrosis leading to significant portal hypertension. There is very little known about the mechanism whereby FC/PD mutations and ciliopathies lead to progressive liver fibrosis. It will be critical in the future to develop a better understanding of the role of abnormal FC/PD in the myofibroblastic differentiation of PF so that future therapy may direct at halting and possibly reverse the fibrosis process. Antifibrosis therapy has the potential to not only improve hepatic manifestations of ARPKD, it may also improve renal function since prior therapy for PCK disease aimed at decreasing renal cystic volume has failed to stop the deterioration of kidney function.
References
- 1. Dias NF, Lanzarini V, Onuchic LF, Koch VH. Clinical aspects of autosomal recessive polycystic kidney disease. J Bras Nefrol. 2010; 32(3): 263–267. [PubMed] [Google Scholar]
- 2. Guay‐Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003; 111(5 Pt 1): 1072–1080. [DOI] [PubMed] [Google Scholar]
- 3. Gunay‐Aygun M, Tuchman M, Font‐Montgomery E, Lukose L, Edwards H, Garcia A, Ausavarat S, Ziegler SG, Piwnica‐Worms K, Bryant J, et al PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol Genet Metab. 2010; 99(2): 160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zerres K, Mucher G, Becker J, Steinkamm C, Rudnik‐Schoneborn S, Heikkila P, Rapola J, Salonen R, Germino GG, Onuchic L, et al Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet. 1998; 76(2): 137–144. [PubMed] [Google Scholar]
- 5. Zerres K, Rudnik‐Schoneborn S, Steinkamm C, Becker J, Mucher G. Autosomal recessive polycystic kidney disease. J Mol Med. 1998; 76(5): 303–309. [DOI] [PubMed] [Google Scholar]
- 6. Fonck C, Chauveau D, Gagnadoux MF, Pirson Y, Grunfeld JP. Autosomal recessive polycystic kidney disease in adulthood. Nephrol Dial Transplant. 2001; 16(8): 1648–1652. [DOI] [PubMed] [Google Scholar]
- 7. Turkbey B, Ocak I, Daryanani K, Font‐Montgomery E, Lukose L, Bryant J, Tuchman M, Mohan P, Heller T, Gahl WA, et al Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). Pediatr Radiol. 2009; 39(2): 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006; 2(1): 40–55; quiz55. [DOI] [PubMed] [Google Scholar]
- 9. Kanaheswari Y, Hamzaini AH, Wong SW. Congenital hepatic fibrosis in a child with autosomal dominant polycystic kidney disease. Med J Malaysia. 2008; 63(3): 251–253. [PubMed] [Google Scholar]
- 10. Lipschitz B, Berdon WE, Defelice AR, Levy J. Association of congenital hepatic fibrosis with autosomal dominant polycystic kidney disease. Report of a family with review of literature. Pediatr Radiol. 1993; 23(2): 131–133. [DOI] [PubMed] [Google Scholar]
- 11. Cobben JM, Breuning MH, Schoots C, ten Kate LP, Zerres K. Congenital hepatic fibrosis in autosomal‐dominant polycystic kidney disease. Kidney Int. 1990; 38(5): 880–885. [DOI] [PubMed] [Google Scholar]
- 12. Misra A, Loyalka P, Alva F. Portal hypertension due to extensive hepatic cysts in autosomal dominant polycystic kidney disease. South Med J. 1999; 92(6): 626–627. [DOI] [PubMed] [Google Scholar]
- 13. Lemaigre FP. Development of the biliary tract. Mech Dev. 2003; 120(1): 81–87. [DOI] [PubMed] [Google Scholar]
- 14. Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation”. Hepatology. 1992; 16(4): 1069–1083. [DOI] [PubMed] [Google Scholar]
- 15. Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R et al PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin‐like plexin‐transcription‐factor domains and parallel beta‐helix 1 repeats. Am J Hum Genet. 2002; 70(5): 1305–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, et al The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor‐like protein. Nat Genet. 2002; 30(3): 259–269. [DOI] [PubMed] [Google Scholar]
- 17. Bergmann C, Frank V, Kupper F, Schmidt C, Senderek J, Zerres K. Functional analysis of PKHD1 splicing in autosomal recessive polycystic kidney disease. J Hum Genet. 2006; 51(9): 788–793. [DOI] [PubMed] [Google Scholar]
- 18. Zhang MZ, Mai W, Li C, Cho SY, Hao C, Moeckel G, Zhao, R , Kim, I , Wang J, Xiong H, et al PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci USA. 2004; 101(8): 2311–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garcia‐Gonzalez MA, Menezes LF, Piontek KB, Kaimori J, Huso DL, Watnick T, Onuchic LF, Guay‐Woodford LM, Germino GG. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum Mol Genet. 2007; 16(16): 1940–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mai W, Chen D, Ding T, Kim I, Park S, Cho SY, Chu JS, Liang D, Wang N, Wu D et al Inhibition of Pkhd1 impairs tubulomorphogenesis of cultured IMCD cells. Mol Biol Cell. 2005; 16(9): 4398–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaimori JY, Nagasawa Y, Menezes LF, Garcia‐Gonzalez MA, Deng J, Imai E, Onuchic LF, Guay‐Woodford LM, Germino GG. Polyductin undergoes notch‐like processing and regulated release from primary cilia. Hum Mol Genet. 2007; 16(8): 942–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hiesberger T, Gourley E, Erickson A, Koulen P, Ward CJ, Masyuk TV, Larusso NF, Harris PC, Igarashi P. Proteolytic cleavage and nuclear translocation of fibrocystin is regulated by intracellular Ca2+ and activation of protein kinase C. J Biol Chem. 2006; 281(45): 34357–34364. [DOI] [PubMed] [Google Scholar]
- 23. Furu L, Onuchic LF, Gharavi A, Hou X, Esquivel EL, Nagasawa Y, Bergmann C, Senderek J, Avner E, Zerres K, et al Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J Am Soc Nephrol. 2003; 14(8): 2004–2014. [DOI] [PubMed] [Google Scholar]
- 24. Bergmann C, Senderek J, Sedlacek B, Pegiazoglou I, Puglia P, Eggermann T, Rudnik‐Schoneborn, S , Furu L, Onuchic LF, De Baca M et al Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J Am Soc Nephrol. 2003; 14(1): 76–89. [DOI] [PubMed] [Google Scholar]
- 25. Hildebrandt F, Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005; 6(12): 928–940. [DOI] [PubMed] [Google Scholar]
- 26. Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009; 111(3): 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009; 60: 321–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kolb RJ, Nauli SM. Ciliary dysfunction in polycystic kidney disease: an emerging model with polarizing potential. Front Biosci. 2008; 13: 4451–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Masyuk TV, Huang BQ, Ward CJ, Masyuk AI, Yuan D, Splinter PL, Punyashthiti R, Ritman EL, Torres VE, Harris PC, et al Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003; 125(5): 1303–1310. [DOI] [PubMed] [Google Scholar]
- 30. Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, Wang X, Harris PC, Larusso NF. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol. 2004; 165(5): 1719–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sanzen T, Harada K, Yasoshima M, Kawamura Y, Ishibashi M, Nakanuma Y. Polycystic kidney rat is a novel animal model of Caroli’s disease associated with congenital hepatic fibrosis. Am J Pathol. 2001; 158(5): 1605–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Katsuyama M, Masuyama T, Komura I, Hibino T, Takahashi H. Characterization of a novel polycystic kidney rat model with accompanying polycystic liver. Exp Anim. 2000; 49(1): 51–55. [DOI] [PubMed] [Google Scholar]
- 33. Nagasawa Y, Matthiesen S, Onuchic LF, Hou X, Bergmann C, Esquivel E, Senderek J, Ren Z, Zeltner R, Furu L, et al Identification and characterization of Pkhd1, the mouse orthologue of the human ARPKD gene. J Am Soc Nephrol. 2002; 13(9): 2246–2258. [DOI] [PubMed] [Google Scholar]
- 34. Woollard JR, Punyashtiti R, Richardson S, Masyuk TV, Whelan S, Huang BQ, Lager DJ, vanDeursen J, Torres VE, Gattone VH, et al A mouse model of autosomal recessive polycystic kidney disease with biliary duct and proximal tubule dilatation. Kidney Int. 2007; 72(3): 328–336. [DOI] [PubMed] [Google Scholar]
- 35. Gallagher AR, Esquivel EL, Briere TS, Tian X, Mitobe M, Menezes LF, Markowitz GS, Jain D, Onuchic LF, Somlo S. Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am J Pathol. 2008; 172(2): 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guay‐Woodford LM. Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol. 2003; 285(6): F1034–F1049. [DOI] [PubMed] [Google Scholar]
- 37. Guay‐Woodford LM, Bryda EC, Christine B, Lindsey JR, Collier WR, Avner ED, D‘Eustachio P, Flaherty L. Evidence that two phenotypically distinct mouse PKD mutations, bpk and jcpk, are allelic. Kidney Int. 1996; 50(4): 1158–1165. [DOI] [PubMed] [Google Scholar]
- 38. Ibraghimov‐Beskrovnaya O, Bukanov N. Polycystic kidney diseases: from molecular discoveries to targeted therapeutic strategies. Cell Mol Life Sci. 2008; 65(4): 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Woo D. Apoptosis and loss of renal tissue in polycystic kidney diseases. N Engl J Med. 1995; 333(1): 18–25. [DOI] [PubMed] [Google Scholar]
- 40. Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001; 184(1): 71–79. [DOI] [PubMed] [Google Scholar]
- 41. Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006; 131(3): 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, et al Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003; 33(2): 129–137. [DOI] [PubMed] [Google Scholar]
- 43. Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD). Hepatology. 2009; 49(1): 160–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gradilone SA, Masyuk AI, Splinter PL, Banales JM, Huang BQ, Tietz PS, Masyuk TV, Larusso NF. Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion. Proc Natl Acad Sci USA. 2007; 104(48): 19138–19143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gradilone SA, Masyuk TV, Huang BQ, Banales JM, Lehmann GL, Radtke BN, Stroope A, Masyuk AI, Splinter PL, LaRusso NF Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology. 2010; 139(1): 304–314, e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 39,59‐cyclic monophosphate. Gastroenterology. 2007; 132(3): 1104–1116. [DOI] [PubMed] [Google Scholar]
- 47. Wang X, Gattone V 2nd, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC‐31260 and OPC‐41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005; 16(4): 846–851. [DOI] [PubMed] [Google Scholar]
- 48. Sato Y, Harada K, Furubo S, Kizawa K, Sanzen T, Yasoshima M, Ozaki S, Isse K, Sasaki M, Nakanuma Y. Inhibition of intrahepatic bile duct dilation of the polycystic kidney rat with a novel tyrosine kinase inhibitor gefitinib. Am J Pathol. 2006; 169(4): 1238–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nauta J, Sweeney WE, Rutledge JC, Avner ED. Biliary epithelial cells from mice with congenital polycystic kidney disease are hyperresponsive to epidermal growth factor. Pediatr Res. 1995; 37(6): 755–763. [DOI] [PubMed] [Google Scholar]
- 50. Sato Y, Harada K, Kizawa K, Sanzen T, Furubo S, Yasoshima M, Ozaki S, Ishibashi M, Nakanuma Y. Activation of the MEK5/ERK5 cascade is responsible for biliary dysgenesis in a rat model of Caroli’s disease. Am J Pathol. 2005; 166(1): 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mostov KE. mTOR is out of control in polycystic kidney disease. Proc Natl Acad Sci USA. 2006; 103(14): 5247–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer‐Zucker A, et al The mTOR pathway is regulated by polycystin‐1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA. 2006; 103(14): 5466–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zafar I, Ravichandran K, Belibi FA, Doctor RB, Edelstein CL. Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int. 2010; 78(8): 754–761. [DOI] [PubMed] [Google Scholar]
- 54. Wuthrich RP, Kistler AD, Serra AL. Impact of Mammalian target of rapamycin inhibition on autosomal‐dominant polycystic kidney disease. Transplant Proc. 2010; 42(9 Suppl): S44–S46. [DOI] [PubMed] [Google Scholar]
- 55. Belibi F, Ravichandran K, Zafar I, He Z, Edelstein CL. mTORC1/2 and rapamycin in female Han: SPRD rats with polycystic kidney disease. Am J Physiol Renal Physiol. 2011; 300(1): F236–F244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010; 21(3): 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, Tian X, Somlo S, Strazzabosco M. Mammalian target of rapamycin regulates vascular endothelial growth factor‐dependent liver cyst growth in polycystin‐2‐defective mice. Hepatology. 2010; 51(5): 1778–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol. 2008; 19(3): 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, et al Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363(9): 820–829. [DOI] [PubMed] [Google Scholar]
- 60. Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Horl WH, Obermuller N, et al Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363(9): 830–840. [DOI] [PubMed] [Google Scholar]
- 61. Fischer DC, Jacoby U, Pape L, Ward CJ, Kuwertz‐Broeking E, Renken C, Nizze H, Querfeld U, Rudolph B, Mueller‐Wiefel DE, et al Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol Dial Transplant. 2009; 24(6): 1819–1827. [DOI] [PubMed] [Google Scholar]
- 62. Renken C, Fischer DC, Kundt G, Gretz N, Haffner D. Inhibition of mTOR with sirolimus does not attenuate progression of liver and kidney disease in PCK rats. Nephrol Dial Transplant. 2011; 26(1): 92–100. [DOI] [PubMed] [Google Scholar]
- 63. Paizis G, Cooper ME, Schembri JM, Tikellis C, Burrell LM, Angus PW. Up‐regulation of components of the renin‐angiotensin system in the bile duct‐ligated rat liver. Gastroenterology. 2002; 123(5): 1667–1676. [DOI] [PubMed] [Google Scholar]
- 64. Bataller R, Gabele E, Parsons CJ, Morris T, Yang L, Schoonhoven R, Brenner DA, Rippe RA. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct‐ligated rats. Hepatology. 2005; 41(5): 1046–1055. [DOI] [PubMed] [Google Scholar]
- 65. Moreno M, Bataller R. Cytokines and renin‐angiotensin system signaling in hepatic fibrosis. Clin Liver Dis. 2008; 12(4): 825–852, ix. [DOI] [PubMed] [Google Scholar]
- 66. Yang L, Bataller R, Dulyx J, Coffman TM, Gines P, Rippe RA, Brenner DA. Attenuated hepatic inflammation and fibrosis in angiotensin type 1a receptor deficient mice. J Hepatol. 2005; 43(2): 317–323. [DOI] [PubMed] [Google Scholar]
- 67. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005; 115(2): 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Goto M, Hoxha N, Osman R, Dell KM. The renin‐angiotensin system and hypertension in autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2010; 25(12): 2449–2457. [DOI] [PubMed] [Google Scholar]
- 69. Loghman‐Adham M, Soto CE, Inagami T, Sotelo‐Avila C. Expression of components of the renin‐angiotensin system in autosomal recessive polycystic kidney disease. J Histochem Cytochem. 2005; 53(8): 979–988. [DOI] [PubMed] [Google Scholar]
- 70. Goto M, Hoxha N, Osman R, Wen J, Wells RG, Dell KM. Renin‐angiotensin system activation in congenital hepatic fibrosis in the PCK rat model of autosomal recessive polycystic kidney disease. J Pediatr Gastroenterol Nutr. 2010; 50(6): 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jia G, Kwon M, Liang HL, Mortensen J, Nilakantan V, Sweeney WE, Park F. Chronic treatment with lisinopril decreases proliferative and apoptotic pathways in autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2010; 25(6): 1139–1146. [DOI] [PubMed] [Google Scholar]
- 72. Yoshihara D, Kurahashi H, Morita M, Kugita M, Hiki Y, Aukema HM, Yamaguchi T, Calvet JP, Wallace DP, Nagao S. PPAR‐{gamma} agonist ameliorates kidney and liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. Am J Physiol Renal Physiol. 2011; 300(2): 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hamblin M, Chang L, Fan Y, Zhang J, Chen YE. PPARs and the cardiovascular system. Antioxid Redox Signal. 2009; 11(6): 1415–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yamamoto Y, Ono T, Dhar DK, Yamanoi A, Tachibana M, Tanaka T, Nagasue N. Role of peroxisome proliferator‐activated receptor‐gamma (PPARgamma) during liver regeneration in rats. J Gastroenterol Hepatol. 2008; 23(6): 930–937. [DOI] [PubMed] [Google Scholar]
- 75. Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, Kohno N, Yorioka N. PPAR‐gamma agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF‐beta. Lab Invest. 2009; 89(1): 47–58. [DOI] [PubMed] [Google Scholar]
- 76. Sedlaczek N, Jia JD, Bauer M, Herbst H, Ruehl M, Hahn EG, Schuppan D. Proliferating bile duct epithelial cells are a major source of connective tissue growth factor in rat biliary fibrosis. Am J Pathol. 2001; 158(4): 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ozaki S, Sato Y, Yasoshima M, Harada K, Nakanuma Y. Diffuse expression of heparan sulfate proteoglycan and connective tissue growth factor in fibrous septa with many mast cells relate to unresolving hepatic fibrosis of congenital hepatic fibrosis. Liver Int. 2005; 25(4): 817–828. [DOI] [PubMed] [Google Scholar]
- 78. Brenner DA. Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc. 2009; 120: 361–368. [PMC free article] [PubMed] [Google Scholar]
- 79. Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010; 7(8): 425–436. [DOI] [PubMed] [Google Scholar]
- 80. Dranoff JA, Wells RG. Portal fibroblasts: underappreciated mediators of biliary fibrosis. Hepatology. 2010; 51(4): 1438–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Beaussier M, Wendum D, Schiffer E, Dumont S, Rey C, Lienhart A, Housset C. Prominent contribution of portal mesenchymal cells to liver fibrosis in ischemic and obstructive cholestatic injuries. Lab Invest. 2007; 87(3): 292–303. [DOI] [PubMed] [Google Scholar]
- 82. Kruglov EA, Nathanson RA, Nguyen T, Dranoff JA. Secretion of MCP‐1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2006; 290(4): G765–G771. [DOI] [PubMed] [Google Scholar]
- 83. Baghdasaryan A, Claudel T, Kosters A, Gumhold J, Silbert D, Thuringer A, Leski K, Fickert P, Karpen SJ, Trauner M. Curcumin improves sclerosing cholangitis in Mdr2‐/‐ mice by inhibition of cholangiocyte inflammatory response and portal myofibroblast proliferation. Gut. 2010; 59(4): 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]