

Abstract
The chronic accumulation of amyloid beta (Aβ) peptides is thought to underlie much of the pathology of Alzheimer’s disease (AD), and transgenic mice overexpressing Aβ show both behavioral defects and impairments in hippocampal synaptic transmission. In the present study, we examined excitatory transmission at the Schaffer collateral synapse in acute hippocampal slices from APPSwe/PS-1A246E transgenic mice to determine whether the synaptic impairment in these mice is due to a reduction in the activity-independent synaptic gain, or to a change in the activity-dependent synaptic dynamics. We observed a strong reduction in synaptic transmission in slices from APPSwe/PS-1A246E mice compared to those from their wildtype littermates. However, there was no resolvable change in the synaptic dynamics observed in response to either simple or complex stimulus trains. We conclude that the chronic accumulation of Aβ impairs synaptic transmission through a reduction in the synaptic gain, while preserving the synaptic dynamics.
Keywords: hippocampus, synaptic, glutamate, Alzheimer’s, amyloid
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease that leads to severe cognitive impairments, most notably memory loss (reviewed in [16]). These impairments are thought to be due to a presenilin (PS)-dependent increase in the production of beta-amyloid peptides (Aβ) from the amyloid precursor protein (APP) in several brain regions, including the hippocampus. Transgenic mice with mutations in APP and/or PS-1 that promote Aβ production have been generated as animal models for AD. These mice show hippocampal accumulation of Aβ, synaptic dysfunction, and impaired performance of hippocampus-dependent behavioral tasks (reviewed in [20]). This has led to a working model for the early stages of AD in which the accumulation of Aβ leads to synaptic defects that impair hippocampal function and cause deficits in learning and memory.
The mechanisms by which Aβ affects hippocampal synaptic transmission remain poorly understood, and both presynaptic [1,10,22] and postsynaptic [8,9] effects of Aβ have been reported. One challenge in understanding how these synaptic impairments lead to cognitive defects is that their functional consequences also remain poorly understood. The size of a given excitatory postsynaptic potential (EPSP) depends mainly on two parameters: the synaptic gain, and the synaptic dynamics (reviewed in [21]). The synaptic dynamics are generated by activity-dependent short-term synaptic plasticity induced during the afferent stimulus train, and are mainly determined by factors influencing the probability of transmitter release [5,12,18,19]. The synaptic gain is a scaling factor that determines the mean EPSP amplitude independent of synaptic dynamics, and is determined by a combination of the number of active synapses and the number of postsynaptic receptors.
Synaptic dynamics are coarsely assessed using two afferent stimuli in rapid succession to elicit paired pulse facilitation (PPF). Mouse models for AD have little if any difference in PPF [4,6,8,11,17; but see 10], suggesting that Aβ has little effect on the release probability. Surprisingly, however, recent studies have suggested that endogenous Aβ acutely increases the release probability [1,13] leading to a profound change in synaptic dynamics [1]. One possible resolution of these divergent results is that endogenous Aβ alters synaptic dynamics through a mechanism that is poorly assessed by PPF, so that mouse models for AD express an impairment in dynamics that has gone undetected.
To assess this possibility, we examined synaptic transmission in hippocampal slices (500μM thick) from 9-12 month old wildtype or transgenic mice using the prion promoter to drive expression of human variants of PS-1 (A246E) and APP (K670N and M671L) associated with familial AD [2]. At this age, APPSwe/PS-1A246E mice have increased Aβ load, amyloid plaques, and defects in hippocampus-dependent behaviors [3,14].
Materials and Methods
Hippocampal slice preparation, extracellular recordings, and data analysis were performed as described more extensively in [12]. Briefly, mice were deeply anesthetized with isoflurane. The hippocampi were removed by dissection, and sliced using a Vibratome in cooled (3-5°C) solution containing (in mM): 110 choline Cl, 26.2 NaHCO3, 11 glucose, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.0 NaH2PO4, 1.3 Na ascorbate, bubbled with 95% O2-5% CO2. Recordings were performed in artificial cerebrospinal fluid containing (in mM): 119 NaCl, 26.2 NaHCO3, 11 glucose, 2.5 KCl, 4 CaCl2, 4 MgSO4, 1.0 NaH2PO4, bubbled with 95% O2-5% CO2, as well as 50-100 μM D-2-amino-5-phosonovalerate (APV) and 25-50 μM picrotoxin to block NMDA and GABAA receptors respectively. Area CA3 was microdissected to prevent recurrent excitation, and the temperature was maintained at 32-35°C.
Stimuli were delivered with a bipolar electrode placed in stratum radiatum, and extracellular fEPSPs were recorded. Fiber volley amplitude was calculated after subtraction of the stimulus artifact by exponential fitting to the decay of the artifact. Field EPSP (fEPSP) slopes were measured using the 1-2 milliseconds of rising phase. All experiments were performed blind to animal genotype, which was determined using standard PCR of genomic DNA. Data were compared using the Student’s t-test or the Rank-Sum test as appropriate, and evaluated based on the number of animals rather than the number of slices to prevent errors associated with larger variability across animals than across slices. All data are presented as mean ± SEM. The OHSU Institutional Animal Care and Use Committee approved all procedures for animal care according to NIH guidelines.
Results
To compare synaptic output across genotypes, we constructed input-output functions while varying the stimulus strength, using the amplitude of the fiber volley to measure axonal activation and the initial slope of the fEPSP to assess the postsynaptic response (Figure 1A). The fiber volley amplitude was linearly related to the fEPSP over a range of stimulus intensities (Figure 1C), with the slope of this linear relationship defining the synaptic strength (Figure 1D). The synaptic strength of APPSwe/PS-1A246E mice was strongly reduced relative to wildtype mice (Figure 1B-D; wildtype mice, 2.2 ±0.4 mV/msec fEPSP/mV fiber volley, n= 9; APPSwe/PS-1A246E mice, 1.0 ±0.2 mV/msec fEPSP/mV fiber volley, n= 9; P<0.01). Thus, synaptic transmission in APPSwe/PS-1A246E mice is impaired, as in other transgenic mouse models for AD.
Figure 1. Synaptic transmission is impaired in hippocampal slices from APPSwe/PS-1A246E mice relative to those from wildtype mice.
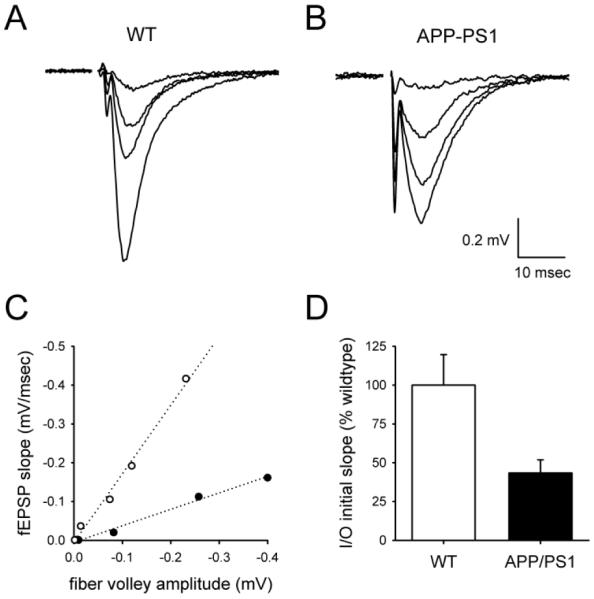
fEPSPs were generated in response to a range of stimulus strengths, as shown for representative experiments in hippocampal slices from wildtype (A) and APPSwe/PS-1A246E (B) mice. Axonal recruitment required to elicit the same sized fEPSP in APPSwe/PS-1A246E mice was larger, as assessed by the fiber volley (arrows in A and B). (C) The synaptic input/output function for each experiment was linear using low-intensity stimuli, as shown using the data from A (open circles) and B (filled circles). (D) The slope of this linear function was smaller in APPSwe/PS-1A246E mice than in wildtype mice, indicating that comparable axonal recruitment leads to a smaller synaptic output.
Despite the reduction of synaptic strength, both wildtype and APPSwe/PS-1A246E mice showed PPF in response to paired stimuli with a 100 msec interstimulus interval (Figure 2A), with no significant difference in PPF across genotypes (Figure 2B; wildtype mice, 1.52±0.12, n= 9; APPSwe/PS-1A246E mice, 1.60±0.08, n= 9; P>0.5). To assess slower forms of short-term plasticity such as frequency facilitation (FF; see [23]), we examined the enhancement of synaptic transmission that takes place during a 30 second stimulus train at 1 Hz. This stimulus paradigm robustly elicited FF (Figure 2C) that was not resolvably different between genotypes (Figure 2D; wildtype mice, 1.43 ± 0.11, n= 9; APPSwe/PS-1A246E mice, 1.39 ±0.10, n= 9; P>0.7).
Figure 2. Paired-pulse facilitation and frequency facilitation are normal in hippocampal slices from APPSwe/PS-1A246E mice.

fEPSPs in hippocampal slices from both wildtype and APPSwe/PS-1A246E mice exhibit robust PPF that was similar across genotypes, as shown in representative experiments (A) and population data (B). Frequency facilitation was elicited using a 1 Hz train of 30 stimuli, and was clearly evident in individual experiments when comparing averages of the first and last 3 fEPSPs (black traces in C) during the train (grey traces in C). Frequency facilitation was similar across genotypes (D).
While these experiments demonstrate that APPSwe/PS-1A246E mice have normal dynamics at a few frequencies of activation, CA3 pyramidal cells in vivo fire over a range of frequencies that spans ~3 orders of magnitude [7]. To assess synaptic dynamics more comprehensively, we examined the fEPSPs generated by a complex stimulus train based on in vivo single unit recordings of CA3 pyramidal cells (as in [12]; Figure 3A, top). The fEPSPs during these trains were highly variable, but the pattern of fEPSPs during the train was similar across genotypes (Figure 3A). To assess the synaptic dynamics more quantitatively, the fEPSPs during each spike of the train were normalized according to a baseline period preceding the train, and averaged together across mice of the same genotype. The average fEPSPs for each spike during the train in wildtype mice were plotted against the corresponding fEPSPs for each spike during the train in APPSwe/PS-1A246E mice. The two sets of fEPSPs were highly correlated along the line of identity (Figure 3B; wildtype mice, n= 9; APPSwe/PS-1A246E mice, n= 9; r2=0.90).
Figure 3. Synaptic dynamics in response to a complex spike train are normal in hippocampal slices from APPSwe/PS-1A246E mice.

(A) A stimulus train derived from in vivo recordings (top) was used to elicit fEPSPs (bottom) in response to a broad range of stimulus frequencies in both wildtype (open symbols) and APPSwe/PS-1A246E (filled symbols) mice. fEPSP slopes are normalized to a control period preceding delivery of the complex stimulus train, which corrects for differences in synaptic gain across genotypes. (B) Each normalized fEPSP during the stimulus train in slices from wildtype mice was compared to the corresponding normalized fEPSP during the same stimulus train in slices from APPSwe/PS-1A246E mice. The entire set of fEPSPs during the train was highly correlated across genotypes (r2=0.90). (C) The ratio of normalized fEPSP slopes in slices from APPSwe/PS-1A246E mice over that of wildtype mice was calculated for each fEPSP during the train, and plotted against the ISI preceding that fEPSP (filled symbols). There was no significant effect of genotype observed at any ISI seen during the train. The same ratio was calculated using data from a previous study in which the same stimulus train was applied in the presence and absence of the GABAB receptor agonist baclofen to engage presynaptic inhibition, confirming the sensitivity of the analysis ([12]; grey symbols). Error bars for the ratios in (C) were calculated by resampling of each term in the ratio 10,000 times using the measured variance, then measuring the variance of the ratio across resamplings and converting to standard error.
To ensure that there was no effect of genotype on fEPSPs at a small subset of the full frequency range in the train, we examined the ratio of each fEPSP during the train in APPSwe/PS-1A246E mice over the corresponding fEPSP during the train in wildtype mice. This ratio was independent of the interspike interval (ISI) immediately preceding each fEPSP, for all fEPSPs in the train (Figure 3C, filled symbols). For comparison, a similar analysis was performed on data from prior experiments that used the same stimulus train to examine the effects of the GABAB receptor agonist baclofen on synaptic dynamics [12]. The effect of baclofen was easily resolvable (Figure 3C, grey symbols) at a level of basal synaptic inhibition (~60%) comparable to the defect in I/O curve seen in APP/PS-1 mice here. We conclude that synaptic dynamics are unaffected in APPSwe/PS-1A246E mice over a broad range of firing frequencies.
Discussion
We found that synaptic transmission in APPSwe/PS-1A246E mice is profoundly impaired at an age when Aβ accumulation is substantial, due to a change in synaptic gain and not synaptic dynamics. These findings are consistent with the literature using PPF to assess synaptic dynamics in other mouse lines that overexpress Aβ [4,6,8,11,17; but see 10]. One report found that fatigue in the response to prolonged 100 Hz stimulation is modestly enhanced in mice overexpressing Aβ [22], but it remains unclear whether this fatigue is synaptic or due to a loss of axonal reliability during high-frequency stimulation [15]. Our own stimulus trains were limited to a maximal frequency of 50 Hz to avoid this complication, so we cannot exclude the possibility that a synaptic defect could be uncovered in APPSwe/PS-1A246E mice during prolonged high-frequency firing. However, such a defect would likely have little effect in vivo because CA3 pyramidal cells do not express such prolonged bursts during hippocampus-dependent behavioral tasks [7].
A reduction in synaptic gain can be explained by two possible synaptic defects in APPSwe/PS-1A246E mice: an impairment in postsynaptic receptor function, or a reduction in the density of active synapses. From a functional perspective, the former represents a graded change in synaptic gain applied to all synapses, while the latter represents an all-or-none change in synaptic gain applied to only a subset of synapses. In contrast, the absence of an effect on synaptic dynamics is difficult to reconcile with a change in the release probability, as the expression of short-term plasticity interacts with the release probability (reviewed in [23]). The use of complex spike trains as a method for probing several forms of plasticity at once provides a particularly high resolving power for detecting such interactions.
If the release probability is not affected in mouse models for AD that overexpress Aβ, how can this be reconciled with recent studies suggesting that endogenous Aβ rapidly changes the release probability? One possibility is that the acute increase in release probability induced by Aβ is homeostatically corrected, returning the release probability to its resting level in spite of the elevated Aβ. In terms of relevance to human AD, clinical detection of the disease is thought to be preceded by years of Aβ accumulation rather than a sudden acute change in Aβ levels.
In conclusion, the impairment in synaptic transmission seen in the APPSwe/PS-1 A246E mouse model for AD causes a reduced synaptic gain but does not change synaptic dynamics. This finding suggests that therapeutic treatments to compensate for these defects should optimally attempt to increase the synaptic gain.
Highlights.
We examined hippocampal synaptic function in mice overexpressing β-amyloids.
These mice had profoundly impaired synaptic output at Schaffer collateral synapses.
The impairment was due to a reduction in the gain of the synaptic output.
Activity-dependent short-term plasticity was normal over a wide frequency range.
Acknowledgements
APP/PS1 mice were a generous gift from Dr David Borchelt, John Hopkins University, Baltimore. Funding for this study was generously provided by the NIH (MH077120, AG026051).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 2009;12:1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- [2].Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- [3].Borchelt DR, Ratovitski T, Van Lare J, Lee MK, Gonzales VB, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice co-expressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- [4].Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- [5].Dobrunz LE, Stevens CF. Response of hippocampal synapses to natural stimulation patterns. Neuron. 1999;22:157–166. doi: 10.1016/s0896-6273(00)80687-x. [DOI] [PubMed] [Google Scholar]
- [6].Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, Seabrook GR. Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci. 2001;21:4691–4698. doi: 10.1523/JNEUROSCI.21-13-04691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Frerking M, Schulte J, Wiebe S, Staubli U. Spike timing in CA3 pyramidal cells during behavior: implications for synaptic transmission. J. Neurophysiol. 2005;94:1528–1540. doi: 10.1152/jn.00108.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Larson J, Lynch G, Games D, Seubert P. Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res. 1999;840:23–35. doi: 10.1016/s0006-8993(99)01698-4. [DOI] [PubMed] [Google Scholar]
- [11].Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- [12].Ohliger-Frerking P, Wiebe SP, Staubli U, Frerking M. GABA(B) receptor-mediated presynaptic inhibition has history-dependent effects on synaptic transmission during physiologically relevant spike trains. J. Neurosci. 2003;23:4809–4814. doi: 10.1523/JNEUROSCI.23-12-04809.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Parodi J, Sepúlveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG. Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J. Biol. Chem. 2010;285:2506–2514. doi: 10.1074/jbc.M109.030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Puoliväli J, Wang J, Heikkinen T, Heikkilä M, Tapiola T, van Groen T, Tanila H. Hippocampal Aβ42 levels correlate with spatial memory deficit in APP and PS1 double transgenic mice. Neurobiol. Dis. 2002;9:339–347. doi: 10.1006/nbdi.2002.0481. [DOI] [PubMed] [Google Scholar]
- [15].Raastad M, Shepherd GM. Single-axon action potentials in the rat hippocampal cortex. J Physiol. 2003;548:745–752. doi: 10.1113/jphysiol.2002.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008;2:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Trinchese F, Liu S, Battaglia F, Walter S, Mathews PM, Arancio O. Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 2004;55:801–814. doi: 10.1002/ana.20101. [DOI] [PubMed] [Google Scholar]
- [18].Tsodyks MV, Markram H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc. Natl. Acad. Sci. U.S.A. 1997;94:719–723. doi: 10.1073/pnas.94.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Varela JA, Sen K, Gibson J, Fost J, Abbott LF, Nelson SB. A quantitative description of short-term plasticity at excitatory synapses in layer 2/3 of rat primary visual cortex. J. Neurosci. 1997;17:7926–7940. doi: 10.1523/JNEUROSCI.17-20-07926.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 2004;44:181–93. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- [21].Zador AM, Dobrunz LE. Dynamic synapses in the cortex. Neuron. 1997;19:1–4. doi: 10.1016/s0896-6273(00)80341-4. [DOI] [PubMed] [Google Scholar]
- [22].Zhang H, Gong B, Liu S, Fa’ M, Ninan I, Staniszewski A, Arancio O. Synaptic fatigue is more pronounced in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer. Res. 2005;2:137–40. doi: 10.2174/1567205053585936. [DOI] [PubMed] [Google Scholar]
- [23].Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]