

Abstract
Atypical hemolytic uremic syndrome (aHUS) associates with complement alternative pathway defects in over 50% of cases. Mutations in factor H (fH) are most common, usually point mutations affecting complement surface regulation and sometimes null mutations in heterozygosity. The latter are difficult to identify; although consistently low plasma fH concentration is suggestive, definitive proof has required the demonstration that the mutant sequence does not express in vitro. Here, novel reagents and assays that distinguish and individually quantify the common fH-Y402H polymorphic variants were used to identify alleles of the CFH gene resulting in low or no (‘null’) expression of full-length fH, but normal or increased expression of the alternative splice product FHL-1, also detected in these assays. Their use in an aHUS cohort identified three Y402H heterozygotes with low or absent fH-H402 but normal or increased FHL-1 levels. Novel mutations in heterozygosis explained the null phenotype in two cases, confirmed by family studies in one. In the third case, family studies showed that a known mutation was present on the Y allele; the cause of the reduced expression of H allele was not found, although data suggested altered fH/FHL-1 splicing. In each family, inheritance of “low expression” or “null” alleles for fH strongly associated with aHUS. These assays provide a rapid means to identify fH expression defects in aHUS without resorting to gene sequencing or expression analysis.
Introduction
Hemolytic uremic syndrome (HUS), characterized by the triad of thrombocytopenia, microangiopathic hemolytic anaemia and acute renal failure, is one of the commonest causes of renal failure in children (1). When not associated with diarrhoeal illness, or when recurrent, the disease is considered atypical (aHUS), accounting for less than 10% of all HUS cases. aHUS has a poor prognosis; it is fatal in up to 25% in the acute phase and 50% of survivors require ongoing renal replacement therapy (2). Numerous environmental precipitants of aHUS have been described, including infections (3, 4), tumours (5), pregnancy (6), drugs (7), and metabolic syndromes (8). In some families, both autosomal recessive and autosomal dominant inheritance modes were seen (9, 10). Research over the last decade has identified mutations in genes encoding complement regulators or components in 50% of aHUS cases; these include factor H (fH) (reviewed in 11), membrane cofactor protein (12), factor I (13), C3 (14) and factor B (15), provoking the suggestion that aHUS is a disease caused by dysregulation of the alternative pathway of complement (15). Mutations in the gene encoding fH (CFH) are the most frequent association with aHUS, with over 100 different mutations identified (16). The genetic basis of about half of aHUS cases in all cohorts remains undefined, provoking a search for other causative factors.
FH, a 150-kDa serum glycoprotein, regulates the alternative pathway (AP) of complement by acting as cofactor for factor I-mediated proteolytic inactivation of C3b, competing with factor B for C3b binding, and accelerating decay of the C3 convertase (11). FH is the key fluid-phase regulator of the AP, but also regulates AP activation on host cells and exposed basement membranes by binding glycosaminoglycans (GAGs) through its C-terminal domain (short consensus repeats (SCRs) 19 and 20) (17). The complement regulatory domain (SCRs 1–4) then provides regulation on the surface. Mutations in CFH have been described in multiple cohorts (collated on www.fh-hus.org) and account for some 30% of aHUS cases (16). The vast majority are heterozygous, either premature stop codons or single amino acid changes. Incomplete penetrance has been described in all series, suggesting that aHUS is multi-factorial, resulting from a combination of environmental triggers that injure endothelial cells, activate complement and precipitate disease in genetically susceptible individuals (18). Most fH mutations associated with aHUS are in the C-terminal SCRs and cause decreased binding of fH to GAGs on endothelial cells and basement membranes (19). This will cause impaired regulation of AP amplification at these sites, while fluid phase regulation is unimpaired. In a minority of aHUS cases, null mutations are found, resulting in heterozygous or, rarely, homozygous deficiency of fH (20). Although patients with null mutations in heterozygosity will usually have low plasma levels of fH (20), the large variability in fH concentrations in normal individuals makes it difficult or impossible to identify cases simply by measuring fH levels in plasma. Definitive proof that a particular mutant is null has previously required gene sequencing and the demonstration that the mutant cDNA, transfected into an appropriate cell line, failed to make fH protein (21, 22). Methods for measuring expression from individual fH alleles would facilitate identification and assignation of null alleles without the need for laborious cloning and expression.
The Y402H polymorphism of fH is strongly linked to age-related macular degeneration (23). In Caucasians, the allele frequency (Y:H) is approximately 2.5:1 in healthy individuals; hence, over 40% of Caucasians are Y402H heterozygous. This polymorphism therefore represents a useful “marker” for individual CFH alleles. We have previously reported a monoclonal antibody (mAb) specific to fH-H402 (24). Here we describe production of a mAb specific to fH-Y402 and the development of assays for independent quantification of the Y402H variants. Although the Y402H polymorphism has no apparent direct link to aHUS, application of the new assays to aHUS families enabled us to identify, characterize and confirm new CFH alleles associated with low or no expression of full-length fH, but normal or increased expression of the alternative splice product of the CFH gene, FHL-1. We show that these low/no expression alleles for fH conferred strong predisposition to aHUS. These novel tools will not only help identify the molecular basis of disease in patients with aHUS, but also aid prediction of risk in their relatives.
Results
Variant-specific mAb permit independent measurement of CFH allele products
The fH-H402 specific mAb was described previously (24); for this study a mAb specific for the Y402 variant was needed. From ten fusions, two mAb were obtained that selectively bound fH-Y402; one IgG1 isotype, the other IgM, designated MBI-6 and MBI-8 respectively. The IgG1 mAb MBI-6 was expanded and purified; specificity for fH-Y402 was confirmed in ELISA (Fig. 1A) and western blot (Fig. 1B), confirming it reacted exclusively with fH-Y402. Dot blotting of plasma from donors of known Y402H polymorphic status confirmed specificity for fH-Y402 (Fig. 1C). The fH-H402-specific mAb MBI-7 is included as an additional control and to demonstrate the utility of these mAb for independently identifying the presence of these two isoforms of fH. Affinities of MBI-6 for fH-Y402 and fH-H402 were assessed by surface plasmon resonance; the mAb failed to bind fH-H402 but bound fH-Y402 with a KD=15.2 nM (χ2=8.96), confirming the high affinity and absolute specificity of the mAb.
FIGURE 1. MAb MBI-6 specifically detects fH-Y402.

A. ELISA with each fH variant directly immobilised on plate; OX-24 detected both Y402 (closed squares) and H402 (closed circles), whereas MBI-6 detected only Y402 (open squares) and failed to detect H402 (open circles).
B. Western blot showing the detection of fH variants using Y402-specific (MBI-6; left) and H402-specific (MBI-7; right) mAb. The same amount of each protein was loaded in each lane. MBI-6 detected only the Y402 variant and MBI-7 only the H402 variant.
C. Dot blot in which 5μl, 10μl or 20μl aliquots (from left to right) of 10-fold diluted plasma from YY402 and HH402 homozygous donors were spotted on the membrane and probed with either Y402-specific (MBI-6; top), non-selective (OX-24; middle) or H402-specific (MBI-7; bottom) mAb.
ELISA for quantification of each variant in plasma were developed using the variant-specific antibodies. Both the variant-specific and total fH assays measure the combined levels of fH and FHL-1, the latter an alternative splice product of the CFH gene comprising the first seven SCRs of fH and thus sharing the Y402H polymorphism (in SCR7), but neither assay detects any of the reported fH-related proteins (FHR-1, -2, -3, -4, -5). The plasma concentrations of fH and FHL-1 correlate in individual donors with FHL-1 concentrations 10 – 50 fold lower than fH in normal individuals (11, 25).
The calculated assay detection limit was 0.01mg/l and the working range 0.02 – 0.3mg/l. Assay performance was assessed by taking multiple measures from independently diluted aliquots of the same samples. Within-assay precision was 1.3% to 15.5% across the working range with an average of 5.5% for fH-Y402 measurement, and 1.4% to 16.8% with an average of 5.6% for fH-H402 measurement. Between-assay precision was 6.7% to 17.6% with an average of 11.9% for fH-Y402 measurement and 5.1% to 12.2% with an average of 9.7% for fH-H402 measurement.
Measurement of fH Variants in Healthy Donors and aHUS Patients
To confirm assay performance and establish normal range (nr), concentrations of fH variants were measured by ELISA in 46 healthy control donors (mean age 42.2 ± 13.8 years, range 25-66 years; 63% female; 21 YY402, 5 HH402, 20 YH402; Y402:H402 allele ratio 0.67:0.33). Total fH was calculated both by summing the amounts of each variant and by using a non-selective fH assay. Values obtained by summing variant concentrations closely matched fH values obtained in the non-selective assay, confirming the validity of the variant-specific assays (for the whole cohort, 257.5±89.8mg/l from sum of assays and 263.3±69.4mg/l from total fH assay). The nr (mean ± 2sd) was 77.9 – 437.1 mg/l in the summed assay and 124.4 – 402mg/l in the total fH assay, illustrating the broad range in plasma fH levels in this normal population. Values for plasma fH in our assays are lower than those quoted in past studies, a consequence of re-calibration based upon accurate extinction coefficients calculated as described (24). Mean total fH was not different between the three groups (YY402, 267.2±66.7mg/l; YH402, 250.9±56.9mg/l; HH402, 214.7±81.5mg/l). In heterozygote controls, variant levels were not significantly different. Importantly, no heterozygote control had selective low or no expression of one allele.
From the Spanish aHUS cohort, 48 individuals heterozygous for the Y402H polymorphism were identified and tested in the Y402H variant specific assays (Fig. 2). Three unrelated individuals (Hus29, Hus90 and Hus169) were identified in whom expression of the H402 allele was very low compared to the Y402 allele. FH level in index case Hus90 was 109mg/l (Y402, 100mg/l; H402, 9mg/l), in case Hus169 was 73mg/l (Y402, 60mg/l; H402, 13mg/l) and in case Hus29 was 148mg/l (Y402, 130mg/l, H402, 18mg/l). Total fH level fell below the calculated nr (mean +/− 2SD) only for Hus169; in the others, total fH levels were in the lower quartile of the nr.
FIGURE 2. Measurement of Y402 and H402 variants of fH in aHUS patients heterozygous for the Y402H polymorphism.

Ratios of concentrations of fH-Y402 and fH-H402 were calculated for each individual and plotted on the graph. For the majority, the ratio was close to 1. For three unrelated individuals (Hus29, Hus90 and Hus169), the concentration of fH-H402 was very low compared to fH-Y402 in that individual, giving high ratios, apparent on the figure.
Index case Hus29 had been reported before as a carrier of the fH mutation R1210C, a known aHUS-associated mutation causing disulfide bonding of fH to albumin in plasma (19, 26); the albumin-bound protein is present in plasma in normal amounts, retains the capacity to inhibit fluid-phase complement activation but has impaired binding to surfaces. All exons in CFH had been sequenced in Hus29 and no other mutations found. Eight family members, all healthy, were available, of whom five carried the R1210C mutation (Fig. 3A). Father, aunt and paternal great-grandfather were R1210C carriers, Y402 homozygous with normal plasma fH levels, demonstrating that R1210C was on the Y402 allele. Paternal grandfather, a R1210C carrier, was Y402H heterozygous with normal plasma fH levels and similar levels of fH-H402 and fH-Y402. Mother, a H402 homozygote, did not carry the R1210C mutation and had low plasma fH levels. Both the index case and her sister were Y402H heterozygous, and carried the R1210C mutation; however, while the sister had normal plasma fH levels with similar amounts of each variant, the index case had low total fH and very low fH-H402 levels in plasma. Western blot of plasma fH in the index case and her sister using the fH-H402-specific mAb demonstrated differential expression of the two maternal H402 alleles, 12% of control levels in the index case by densitometry, and normal expression (90% of control) in her sister (Fig. 4). Because the R1210C mutation was on the Y402 allele, no high molecular weight albumin-fH complex was detected by this H402-specific mAb. Expression of the H402 allele of the alternative splice product of CFH, FHL-1, was increased five-fold (by densitometry) in the index case but not increased compared to controls in her sister (Fig. 4). Analysis of the promoter and intronic sequences within the gene has so far not revealed any genetic variation that could explain increased expression of FHL-1.
FIGURE 3. Family studies in aHUS pedigrees.

Circles represent females, squares males; closed symbols are aHUS cases, open symbols healthy relatives, crossed-through symbols represent deceased relatives. Numbers within a symbol provide the case number. Total fH (mg/l) and Y402H status, where available, are shown below symbols; concentrations of Y402 and H402 variants in heterozygotes are shown below the brackets. Heavy dots indicate that the individual is a carrier for the stated mutation.
A. Pedigree Hus29; index case H29 is Y402H heterozygote expressing very low levels of the H402 allele, and a carrier of the R1210C mutation inherited from her father (F). Mother (M), a H402 homozygote, does not carry the R1210C mutation but had low plasma fH levels. The healthy sister, also Y402H heterozygote and R1210C carrier, expresses normal levels of the H402 allele.
B. Pedigree Hus90; index case H90 is Y402H heterozygote expressing very low levels of the H402 allele, and a carrier of the C853R mutation, shared with one daughter and a niece, both healthy. The same niece and his other daughter are Y402H heterozygotes expressing very low levels of the H402 allele.
C. Pedigree Hus169; index case H169, is Y402H heterozygote expressing very low levels of the H allele, and a carrier of the C1218R mutation. Levels of the Y402 allele were also low in this individual.
FIGURE 4. Western blotting of plasma samples from HUS patients and their healthy relatives.
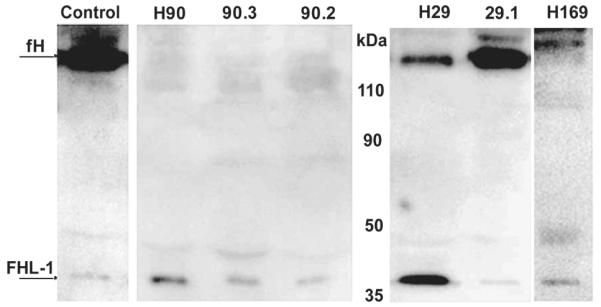
Diluted plasma samples were separated on 10% SDS-PAGE gels under non-reducing conditions, blotted to nitrocellulose and probed with mAb MBI-7 to detect fH-H402. In the Hus90 pedigree, no fH-H402 was detectable in index case (H90), daughter (90.3) or niece (90.2); FHL-1-H402 was present in each case. In the Hus29 pedigree, fH-H402 was detectable in the index case (H29), but at only 12% (by densitometry) that in the control sample or in her sister (H29.1); in contrast, FHL-1-H402 levels were increased five-fold (by densitometry) compared to control or sister samples. Index case H169 had no detectable fH-H402 but FHL-1-H402 was present at levels similar to controls.
Index case Hus90 also had very low levels of fH-H402 in plasma (Fig. 3B). Western blot with fH-H402-specific antibodies showed complete absence of fH-H402 but normal amounts of FHL-1-H402 (Fig. 4), suggestive of a null mutation in the H402 allele downstream of SCR7. Subsequent CFH sequencing revealed the presence in heterozygosity of a novel mutation (C853R; SCR14). Hus90 had three deceased affected siblings; two died during infancy and their mutation status is unknown, while the third though deceased, is an obligate carrier of the C853R mutation because one of his two children is a carrier. This niece of the index case, and one of his two daughters, were the only carriers of the C853R mutation in the next generation; both were Y402H heterozygous and, like the index case, these individuals had very low plasma levels of the H402 allele by ELISA (Fig. 3B). Western blotting confirmed complete absence of fH-H402 and normal expression of FHL-1-H402, as in the index case (Fig. 4). The other daughter and two nephews of the index case were also tested and all were Y402 homozygous, non-carriers with normal plasma fH levels. Segregation analysis in this pedigree confirmed that the H402 allele carried the C853R mutation; likely a null mutation because the cysteine residue is essential for correct folding of SCR 14.
In index case Hus169 was also selected for low plasma fH-H402 level. Subsequent CFH sequencing analysis revealed the presence in heterozygosity of a novel mutation (C1218R) in SCR20 (Fig. 3C). Western blotting with the H402-specific mAb revealed the absence of fH-H402 but normal levels of FHL-1-H402, as in the Hus90 pedigree (Fig. 4). The plasma concentration of fH-Y402 was low at 60μg/ml; however, this concentration, from a single allele, is well within the broad nr for fH noted above (78-437μg/ml from two alleles); these data further emphasise the importance of allele-specific quantification to detect occult null/low expression alleles of fH.
Discussion
CFH mutations in aHUS are usually near the C-terminus (50% in SCRs 19/20), present in heterozygosity and cause impaired surface protection from complement activation (16, 27). In these cases, plasma fH levels are unaffected, and plasma fH is a mixture of mutant and normal protein. In a minority of reported aHUS cases, the CFH mutation causes very low or absent expression from that allele (20), usually identified because of reduced fH levels in plasma. However, identification based on fH levels is unreliable not only because of the noted large variability in the normal population, but also because factors such as age and smoking behaviour influence fH levels in plasma (28).
We generated mAb specific for the Y402H polymorphic variants of fH and FHL-1 in order to identify and quantify expression in macular degeneration, where the H402 allele is a major risk factor (21, 22). This polymorphism has not been linked to aHUS; nevertheless, we reasoned that the mAb and assays would enable us to independently quantify expression from normal and mutant alleles in aHUS, facilitating rapid discovery of null mutations. We here describe the use of these mAb to identify in families with a HUS the presence of null or low expression alleles for full-length fH that are associated with disease. In each of the families described, disease was clearly associated with the inheritance of a previously unidentified CFH low/no expression allele revealed by the new assays.
In Hus90 and Hus169 the mutations responsible for low expression of fH-H402 were subsequently identified as novel loss of cysteine mutations in SCR14 (C853R) and SCR20 (C1218R) respectively. Accumulated data on fH mutations suggest that loss of cysteine mutations in fH are null because they disrupt SCR structure (www.fh-hus.org). Indeed, western blotting of plasma from Hus90 and Hus169 with fH-H402-specific mAb confirmed the complete absence of fH-H402, but normal expression of FHL-1 from this allele. Unhindered expression of FHL-1-H402 from the mutated gene explains the traces of fH-H402 reactivity detected in the H402-specific ELISA; all three ELISA used in the study utilise detection mAb that bind in SCR1-7, shared by fH and FHL-1, and thus detect both. Others have reported that null mutations distal to SCR7 permit normal expression of FHL-1 (21, 22), while null mutations in the shared SCRs cause loss of both proteins (29). Indeed, the differential effects on expression of fH and FHL-1 are useful predictors of the location of the mutation, proximal when both are absent and distal to SCR7 when only fH is affected. In this respect, the fact that the described assays detect both full-length fH and the alternative splice product FHL-1 is a considerable advantage for the detection of occult mutations in fH.
In Hus29, the known point mutation R1210C was shown by western blot and segregation in the pedigree to be on the Y402 allele, and hence not responsible for the observed low expression of fH-H402 allele. The R1210C mutation is common, results in the formation of disulphide-bonded albumin-fH complexes that inhibit fluid phase complement activation (19), and in a large series has been shown to be associated with aHUS only in the presence of additional genetic predisposing factors (26). In Hus29 we contend that the additional predisposing factor is the inheritance from the mother of a low expression H402 allele. The sister of the index case, also a Y402H heterozygote and carrier of the R1210C mutation, has not inherited the low expression allele and is therefore likely not at high risk of aHUS, a prediction only made possible by these novel assays. Critically, expression of FHL-1-H402 in the index case was increased five-fold compared to controls, suggesting that a mutation affecting the differential splicing of the CFH gene was responsible for the deficit. Although we have undertaken a thorough search for mutations in the large intronic regions flanking exons 9, 10 and 11 of the CFH gene, this putative mutation remains unidentified. Despite the fact that fH and FHL-1 are products of the same gene, independent regulation of their expression has been described previously (30), and is confirmed in this study.
The families described above provide case histories that prove the value of the variant-specific mAbs and assays we describe here. Because we have targeted a common polymorphism, over 40% of Caucasians are Y402H heterozygous, these reagents will be useful in the large majority of families. Here we showed their value in tracking the disease-associated allele through a family and, critically, in identifying previously unsuspected null or low expression alleles, themselves strong risk factors for the development of aHUS. The specificity of the mAbs permits unambiguous and rapid identification of occult low/no expression alleles, independent risk factors for aHUS that should now be sought in other cohorts.
Methods
Generation and characterisation of fH Y402H variant-specific mAb
Mice were immunised as described before using fusion proteins comprising fH-Y402 SCRs 6-8 linked to human IgG4 Fc (fH-Fc) (24, 31). Hybridoma supernatants were screened for binding to native fH-H402 and fH-Y402 as described (24). Two hybridomas producing mAb specific for fH-Y402 were identified, re-cloned, isotyped, expanded and purified as described (24). Specificity was confirmed by western and dot blotting on pure fH-H402 and fH-Y402 as described (24). Affinities of the mAb for fH-H402 and fH-Y402 variants were determined by surface plasmon resonance on a Biacore T-100 (GE Healthcare) as described (24).
Development of fH variant-specific ELISA
Maxisorp (Nunc) plates were coated with affinity-purified polyclonal rabbit anti-fH (100μl, 5mg/l) overnight at 4°C and blocked with 1% BSA in PBS. Purified protein standards or serum samples (diluted 1:3000 in BSA/PBS, though lower dilutions (1:100) were used to confirm low fH levels) were added in triplicate and incubated. Wells were washed and incubated with HRP-labelled Y402-specific or H402-specific mAb (1mg/l). All incubations were 1 hour, 37°C. Wells were washed and bound mAb detected using OPD substrate. Absorbance (492nm) was measured. Standards were included on each plate and samples from controls and patients were randomly assigned to eliminate assay bias. A nonlinear regression model was used to fit standard curves generated by ELISA. Total fH (mg/l) was calculated by summing results for fH-Y402 and fH-H402. Total fH was also measured in an ELISA where HRP-labelled non-selective anti-fH mAb, OX-24, specific for an epitope in SCR5 of fH (32; Prof. R.B. Sim, personal communication) was used as detection. Detection limits, working ranges, normal ranges and assay performance were determined as described (24), using plasma from 46 local healthy controls (Ethics approval from the Research and Ethics Committee for Wales. Ref 09/MRE09/35). Of note, because the capture antibody is polyclonal and the detecting mAbs identify epitopes in the shared SCRs 1-7, both the variant-specific and total fH assays will measure the combined levels of fH and FHL-1. However, none of the assays detect any of the FH-related proteins because none contain a SCR5-homologous SCR, and the SCR7-homologous SCR present in FHR-3 alone of these proteins is not conserved around the Y402H relevant region.
Western and dot-blot assays for fH variants
Western and dot-blot assays were performed as described (24). To confirm the specificity of the mAbs, plasma from patients of known Y402H variant status was diluted 10-fold, 20-fold or 40-fold and dotted onto nitrocellulose. Separate sets were then probed with either Y402-specific (MBI-6), H402-specific (MBI-7) or non-selective (OX-24) mAb. Positive dots were developed using HRP-labelled anti-mouse IgG and chemiluminescent detection. As further proof of specificity, pure fH protein, Y402 and H402 in separate lanes, was run on 10% SDS-PAGE under non-reducing conditions, and transferred to nitrocellulose. Separate strips were probed either with MBI-6 or MBI-7, and then developed using HRP-labelled anti-mouse IgG and chemiluminescence detection.
To test the presence of fH-H402 in patient samples, plasma (diluted 1:100; 20μl) was separated on 10% SDS-PAGE under non-reducing conditions, transferred to nitrocellulose (30 minute transfer to retain FHL-1 on the membrane) and probed with MBI-7. Blots were developed as above. To quantify the relative amounts of fH and/or FHL-1 in and between samples, the specific bands were analyzed by densitometry.
Genomic analyses
The base change (T1277C) responsible for the Y402H polymorphism was analyzed by PCR amplification and sequencing of CFH exon 9 as described (24). Patients and relatives were screened for other mutations and polymorphisms in CFH by automatic sequencing of each exon as described (26).
Measurement of fH Variants in Plasma Samples from aHUS Patients and relatives
Patient samples were collected with the approval of the Ethics Committee of the Consejo Superior de Investigaciones Científicas in accordance with the Declaration of Helsinki. All participants provided written informed consent. EDTA plasma was stored in aliquots at −80°C. Study cases were selected for genetically determined heterozygosity at the locus encoding the fH-Y402H polymorphism. Concentrations of the Y402H variants were measured; index cases were identified with aberrant expression of either variant. Plasma and DNA were collected from all available family members of these index cases.
Statistical Analysis
Data evaluation was performed using GraphPad Prism software (version 5.0 for Windows; GraphPad Inc., La Jolla, USA). The data were checked for normality using the D’Agostino-Pearson normality test.
Acknowledgments
This work was supported by the UK Multiple Sclerosis Society no 884/08 (to BPM), MRC Project Grant no. 84908 (to CLH and BPM), and Ministerio de Ciencia e Innovación SAF 2005-00913, the CIBER de Enfermedades Raras and Fundación Renal Iñigo Alvarez de Toledo (to SRdeC).
Footnotes
Disclosure. As the authors of this article, we certify that we have no conflict of interest with any entity in connection with the submitted article.
References
- 1.Richards A, Goodship J, Goodship T. The genetics and pathogenesis of haemolytic uraemic syndrome and thrombotic thrombocytopenic purpura. Curr Opin Nephrol Hypertens. 2002;11:431–436. doi: 10.1097/00041552-200207000-00010. [DOI] [PubMed] [Google Scholar]
- 2.Noris M, Remuzzi G. Non-shiga toxin–associated hemolytic uremic syndrome. In: Zipfel P, editor. Complement and Kidney Disease. Birkhäuser; Basel: 2005. pp. 65–83. [Google Scholar]
- 3.Becker S, Fusco G, Fusco J, Balu R, et al. Collaborations in HIV Outcomes Research/US Cohort. HIV-associated thrombotic microangiopathy in the era of highly active antiretroviral therapy: an observational study. Clin Infect Dis. 2004;39:S267–S275. doi: 10.1086/422363. [DOI] [PubMed] [Google Scholar]
- 4.Constantinescu AR, Bitzan M, Weiss LS, et al. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis. 2004;43:976–982. doi: 10.1053/j.ajkd.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Mungall S, Mathieson P. Hemolytic uremic syndrome in metastatic adenocarcinoma of the prostate. Am J Kidney Dis. 2002;40:1334–1336. doi: 10.1053/ajkd.2002.36929. [DOI] [PubMed] [Google Scholar]
- 6.McMinn JR, George JN. Evaluation of women with clinically suspected thrombotic thrombocytopenic purpura-hemolytic uremic syndrome during pregnancy. J Clin Apher. 2001;16:202–209. doi: 10.1002/jca.10005. [DOI] [PubMed] [Google Scholar]
- 7.Dlott JS, Danielson CF, Blue-Hnidy DE, et al. Drug-induced thrombotic thrombocytopenic purpura/hemolytic uremic syndrome: a concise review. Ther Apher Dial. 2004;8:102–111. doi: 10.1111/j.1526-0968.2003.00127.x. [DOI] [PubMed] [Google Scholar]
- 8.Kind T, Levy J, Lee M, et al. Cobalamin C disease presenting as hemolytic-uremic syndrome in the neonatal period. J Pediatr Hematol Oncol. 2002;24:327–329. doi: 10.1097/00043426-200205000-00023. [DOI] [PubMed] [Google Scholar]
- 9.Pirson Y, Lefebvre C, Arnout C, et al. Hemolytic uremic syndrome in three adult siblings: a familial study and evolution. Clin Nephrol. 1987;28:250–255. [PubMed] [Google Scholar]
- 10.Kaplan BS, Papadimitriou M, Brezin JH, et al. Renal transplantation in adults with autosomal recessive inheritance of hemolytic uremic syndrome. Am J Kidney Dis. 1997;30:760–765. doi: 10.1016/s0272-6386(97)90079-2. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez de Cordoba S, Esparza-Gordillo J, de Jorge E Goicoechea, et al. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. 2004;41:355–367. doi: 10.1016/j.molimm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Fremeaux-Bacchi V, Moulton EA, Kavanagh D, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17:2017–2025. doi: 10.1681/ASN.2005101051. [DOI] [PubMed] [Google Scholar]
- 13.Kavanagh D, Kemp E, Mayland E, et al. Mutations in complement factor I (IF) predispose to the development of atypical HUS. J Am Soc Nephrol. 2005;16:2150–2155. doi: 10.1681/ASN.2005010103. [DOI] [PubMed] [Google Scholar]
- 14.Frémeaux-Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112:4948–4952. doi: 10.1182/blood-2008-01-133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2007;104:240–245. doi: 10.1073/pnas.0603420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saunders RE, Abarrategui-Garrido C, Frémeaux-Bacchi V, et al. The interactive Factor H-atypical hemolytic uremic syndrome mutation database and website: update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum Mutat. 2007;28:222–234. doi: 10.1002/humu.20435. [DOI] [PubMed] [Google Scholar]
- 17.Perkins SJ, Goodship TH. Molecular modelling of the C-terminal domains of factor H of human complement: a correlation between haemolytic uraemic syndrome and a predicted heparin binding site. J Mol Biol. 2002;316:217–224. doi: 10.1006/jmbi.2001.5337. [DOI] [PubMed] [Google Scholar]
- 18.Esparza-Gordillo J, Jorge EG, Garrido CA, et al. Insights into hemolytic uremic syndrome: segregation of three independent predisposition factors in a large, multiple affected pedigree. Mol Immunol. 2006;43:1769–1775. doi: 10.1016/j.molimm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 19.Sánchez-Corral P, Perez-Cabalerro D, Huarte O, et al. Structural and functional characterisation of factor H mutations associated with atypical Hemolytic Uremic Syndrome. Am J Hum Genet. 2002;71:1285–1295. doi: 10.1086/344515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dragon-Durey MA, Fremeaux-Bacchi V, Loirat C, et al. Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol. 2004;15:787–795. doi: 10.1097/01.asn.0000115702.28859.a7. [DOI] [PubMed] [Google Scholar]
- 21.Ault BH, Schmidt BZ, Fowler NL, et al. Human factor H deficiency. Mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem. 1997;272:25168–25175. doi: 10.1074/jbc.272.40.25168. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt BZ, Fowler NL, Hidvegi T, et al. Disruption of disulfide bonds is responsible for impaired secretion in human complement factor H deficiency. J Biol Chem. 1999;274:11782–11788. doi: 10.1074/jbc.274.17.11782. [DOI] [PubMed] [Google Scholar]
- 23.Edwards AO, Ritter R, 3rd, Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 24.Hakobyan S, Harris CL, Tortajada A, et al. Measurement of factor H variants in plasma using variant-specific monoclonal antibodies: application to assessing risk of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:1983–1990. doi: 10.1167/iovs.07-1523. [DOI] [PubMed] [Google Scholar]
- 25.Zipfel PF, Skerka C. FHL-1/reconectin: a human complement and immune regulator with cell-adhesive function. Immunol Today. 1999;20:135–40. doi: 10.1016/s0167-5699(98)01432-7. [DOI] [PubMed] [Google Scholar]
- 26.Martinez-Barricarte R, Pianetti G, Gautard R, et al. The complement factor H R1210C mutation is associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2008;19:639–646. doi: 10.1681/ASN.2007080923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pérez-Caballero D, González-Rubio C, Gallardo ME, et al. Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet. 2001;68:478–484. doi: 10.1086/318201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esparza-Gordillo J, Soria JM, Bui A, et al. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics. 2004;56:77–82. doi: 10.1007/s00251-004-0660-7. [DOI] [PubMed] [Google Scholar]
- 29.Falcao DA, Reis ES, Paixao-Cavalcante D, et al. Deficiency of the human complement regulatory protein factor H associated with low levels of component C9. Scand J Immunol. 2008;68:445–455. doi: 10.1111/j.1365-3083.2008.02152.x. [DOI] [PubMed] [Google Scholar]
- 30.Friese MA, Hellwage J, Jokiranta TS, et al. FHL-1/reconectin and factor H: two human complement regulators which are encoded by the same gene are differentially expressed and regulated. Mol Immunol. 1999;36:809–818. doi: 10.1016/s0161-5890(99)00101-7. [DOI] [PubMed] [Google Scholar]
- 31.Harris CL, Lublin DM, Morgan BP. Efficient generation of monoclonal antibodies for specific protein domains using recombinant immunoglobulin fusion proteins: pitfalls and solutions. J Immunol Methods. 2002;268:245–58. doi: 10.1016/s0022-1759(02)00207-7. [DOI] [PubMed] [Google Scholar]
- 32.Sim E, Palmer MS, Puklavec M, et al. Monoclonal antibodies against the complement control protein factor H (beta 1 H) Biosci Rep. 1983;3:1119–1131. doi: 10.1007/BF01120205. [DOI] [PubMed] [Google Scholar]