

Abstract
Hair fibers are formed by keratinocytes of the hair follicle in a process that involves the breakdown of the nucleus including DNA. Accordingly, DNA can be isolated with high yield from the hair bulb which contains living keratinocytes, whereas it is difficult to prepare from the distal portions of hair fibers and from shed hair. Nevertheless, forensic investigations are successful in a fraction of shed hair samples found at crime scenes. Here, we report that interindividual differences in the completeness of DNA removal from hair corneocytes are major determinants of DNA content and success rates of forensic investigations of hair. Distal hair samples were permeabilized with ammonia and incubated with the DNA-specific dye Hoechst 33258 to label DNA in situ. Residual nuclear DNA was visualized under the fluorescence microscope. Hair from some donors did not contain any stainable nuclei, whereas hair of other donors contained a variable number of DNA-positive nuclear remnants. The number of DNA-containing nuclear remnants per millimeter of hair correlated with the amount of DNA that could be extracted and amplified by quantitative PCR. When individual hairs were investigated, only hairs in which DNA could be labeled in situ gave positive results in short tandem repeat typing. This study reveals that the completeness of DNA degradation during cornification of the hair is a polymorphic trait. Furthermore, our results suggest that in situ labeling of DNA in hair may be useful for predicting the probability of success of forensic analysis of nuclear DNA in shed hair.
Electronic supplementary material
The online version of this article (doi:10.1007/s00414-011-0566-5) contains supplementary material, which is available to authorized users.
Keywords: Hair, DNA, Labeling, Microscopy, STR typing
Introduction
Hair fibers consist of cornified dead keratinocytes that originate from the matrix, i.e., the proliferative compartment, of the hair follicle. Upon moving out of the proliferative zone of the hair follicle, hair keratinocytes, also termed trichocytes, undergo a terminal differentiation program which involves the accumulation and cross-linking of keratins and keratin-associated proteins in the cytoplasm and the breakdown of organelles and the nucleus [1–3]. Fully cornified keratinocytes contain nuclear remnants that can be detected by electron microscopy. However, the molecular composition of these remnants has remained essentially unknown. Recently, we have shown that nuclear DNA of murine hair is degraded by the keratinocyte-specific endonuclease DNase1L2 [4, 5].
Hairs found at crime scenes are an important piece of evidence in forensics [6–8]. Both microscopic analysis of hair structure and molecular analysis of hair-associated DNA are performed to identify the donor. While the discriminative power of microscopic assays is small, genetic data can be a key to the success of forensic investigations. Efficient methods have been established for the extraction of DNA from hair and for genotyping [6]. The latter utilizes both mitochondrial and nuclear DNA, whereby data from nuclear DNA are clearly superior due to increased discrimination power and compatibility with national DNA databases. Short tandem repeat (STR) typing of nuclear DNA allows the unambiguous identification of individuals, provided that sufficient DNA amounts are available [6, 9, 10]. However, in forensic practice, it was found that many hairs contain little, if any, nuclear DNA, whereas others are good sources of DNA. A high yield of DNA has been attributed to the presence of living cells at the base of the hair fibers or to tissue adhering laterally to hair [11, 12].
Here, we have established a novel method for visualizing nuclear DNA in hair shafts. We show that hairs of some individuals contain high numbers of DNA-rich nuclear remnants, whereas hairs from other individuals contain little DNA amenable for in situ labeling or even completely lack DNA in nuclear remnants. Furthermore, our study suggests that the novel protocol, upon further optimization, may be used to predict the success rate of STR typing in shed hair.
Materials and methods
Human hair samples
Hairs from 40 volunteers were obtained by cutting 2–3 cm from the tip of a tuft of undyed hair, thereby excluding the hair bulb. All samples were coded and processed in an anonymous manner. In some experiments, white and pigmented hairs were separated based on visual inspection of the scalp hair of the donor and on inspection of individual hairs against a white background. According to the judgment of trained dermatologists, no pathological abnormalities were detected in any hair specimen. Hairs were stored at −20°C prior to analysis. On average, 1 mg of hair corresponded to a hair length of 200 mm.
In situ fluorescence labeling of DNA in hair
Human hair was washed under vigorous vortexing three times each in 500 μl of 0.04% sodium dodecyl sulfate (SDS) in water and 70% ethanol. Hairs were then dried overnight at room temperature and subsequently permeabilized with 500 μl 10% ammonia solution for 5 h at room temperature. After washing three times with 500 μl double-distilled water, hairs were incubated with DNA-specific Hoechst 33258 (Invitrogen) at a dilution of 1:5,000 overnight at room temperature. Hair was washed again with water and mounted on glass slides using Fluoprep (Biomerieux). The slides were stored for 1 day to allow excess Hoechst to diffuse out of the hair prior to photodocumentation. The labeled hairs were viewed under an Olympus AX70 microscope and analyzed with Meta Imaging Software (Metamorph). The number of labeled nuclei was determined by counting all signals that were round or oval in shape, exceeded the brightness of the background, and had a diameter in the range of 1–10 μm. Dots of extreme brightness, which were likely to be derived from contaminations with dye, were occasionally observed but excluded from counting.
Preparation of DNA from hair
To remove stratum corneum squames or other cellular components adhering to the surface of hair, it was washed with 1 ml of phosphate-buffered saline (PBS) containing 0.1% Tween, three times for 10 min, each with shaking at 750 rpm. Hair was lysed in buffer containing 50 mM Tris–HCl pH 8.0, 100 mM NaCl, 2.5 mM EDTA pH 8.0, 2% SDS, 40 mM dithiothreitol (DTT), and 0.27 mg/ml proteinase K. Seven hundred fifty microliters of lysis buffer was used for 5 mg of hair. The samples were incubated at 56°C with shaking at 750 rpm for at least 10 h to ensure complete lysis. To achieve complete disintegration of some hair samples, the concentration of proteinase K was increased up to 0.65 mg/ml, and the concentration of DTT was increased up to 70 mM.
DNA was purified with the Qiagen DNeasy Blood and Tissue Kit according to the protocol provided by the manufacturer (Qiagen) with the following modifications. Hairs were completely lysed as described above. The lysate was then combined with a premix of buffer AL (same volume as the lysate) and 96% ethanol (same volume as the lysate). The mixture was vortexed thoroughly and subsequently loaded onto the DNeasy Mini spin column, whereby a maximum of 750 μl was loaded at once, followed by centrifugation and loading of the next aliquot. After extensive washing, the DNA was eluted with two aliquots of 100 μl buffer EB. The final DNA solutions were stored at −20°C until PCR analysis.
Analysis of single hairs
Single-hair samples were obtained from 40 human donors by cutting a 2-cm fragment from the distal end of a scalp hair fiber. The hair samples were washed and stained with Hoechst dye as described above for 5-mg hair samples. Hoechst-labeled nuclei were counted under the microscope, then the hair was removed from the slide, transferred into a 0.5-ml tube, and washed with 500 μl PBS 0.1% Tween three times for 10 min. After washing, DNA was extracted from the hair according to the standard protocol, whereby 300 μl of lysis buffer was used for a single hair.
Quantitative real-time PCR
Quantitative real-time PCR (qPCR) was performed using the LightCycler® Carousel-Based System 1.2 and the Light Cycler® Fast Start DNA Master SYBR Green I according to the manufacturer’s protocol (Roche Applied Science). Nuclear DNA was amplified with the primers ALU-forward 5′-GTCAGGAGATCGAGACCATCCC-3′ and 124-bp-reverse 5′-TCCTGCCTCAGCCTCCCAAG-3′ [8]. Mitochondrial DNA was amplified with the primers mito-for 5′-CTCAGATAGGGGTCCCTTGA-3′ and mito-rev 5′-GCACTCTTGTGCGGGATATT-3′ which align to nucleotides 16380–16399 and 16420–16439, respectively of the human mitochondrial genome (GenBank accession number NC_012920.1). Samples with a low ratio of mitochondrial DNA versus nuclear DNA were subjected to sequence analysis of the annealing sites of the qPCR primers in mitochondrial DNA. To this end, a region of the mitochondrial DNA, including the annealing sites of the primers mito-for and mito-rev, was amplified with the primers 5′-CACCCTTAACAGTACATAGTAC-3′ and 5′-CAGATGTCGGATACAGTTCAC-3′. Sequencing of the amplification product confirmed that the annealing sites of the qPCR primers were not mutated in these samples. All qPCRs consisted of an initial denaturation step at 95°C for 10 min and 55 cycles of incubations at 95°C for 5 s, 65°C for 5 s, and 72°C for 15 s. Standard curves for the quantification of nuclear and mitochondrial DNA were generated using dilution series of purified human genomic DNA and of a fragment of human mitochondrial DNA cloned into the pCR2.1-Topo plasmid (Invitrogen), respectively (Supplementary Tables S1 through S7).
STR typing
The co-amplification of five “mini-amplicon” STRs and the gender-specific amelogenin was performed according to a published protocol [9]. A total of 5 μl of DNA (up to 500 pg) was used for amplification using 34 PCR cycles. Preparation of the amplicons and electrophoresis was carried out as described previously [9]. Two independent PCRs were performed, and only STR loci, that were confirmed by both PCRs, were counted as positive.
Statistical analysis
Correlation analyses were performed using the SPSS software (SPSS Inc., Chicago, IL, USA). The bivariate Spearman’s rank correlation coefficient, rho, was determined and the statistical significance of the correlation was evaluated with a two-tailed test. DNA contents of pigmented and white hairs of the same donor were compared with a paired Student's t test. P < 0.05 was considered significant.
Results
Nuclear DNA can be visualized in situ in human hair
A method that we had previously developed to label DNA in murine hair [5] was adapted to human hair. Hairs were obtained from volunteers by cutting the distal portions of hair from the scalp and from other body regions. Since the hair bulb was not included, all samples investigated here may be regarded as models for telogenic hair which lacks living keratinocytes. The hairs were permeabilized with 10% ammonia and subsequently incubated with the DNA-specific fluorescent dye Hoechst 33258 as described in detail in the “Materials and methods” section. Inspection of processed hair specimens revealed that the labeling of DNA was not uniform in all hair corneocytes. Rather, intensive fluorescent labeling of distinct nuclear remnants was observed in some corneocytes, whereas most corneocytes were not labeled (Fig. 1b). The frequency of labeled nuclei did not appear to be associated with the distance from the hair root, i.e., the age of the hair fragment. Importantly, the frequency of nuclei labeled with Hoechst 33258 varied considerably between individual donors. Whereas some individuals lacked DNA-positive nuclei in all scalp hair (Fig. 1a), others contained a low number of labeled nuclei (Fig. 1b) and some contained a high number of nuclei in hair (Fig. 1c). Nuclei were also detected in hair from other body regions (Supplementary Fig. S1).
Fig. 1.
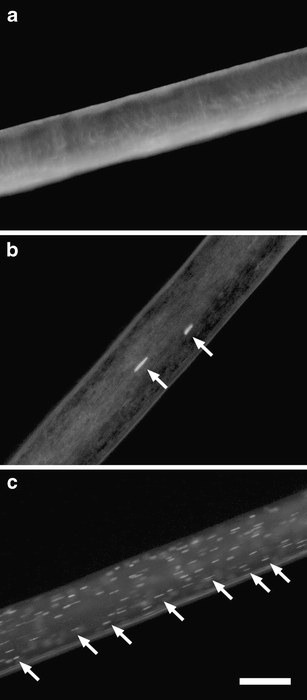
In situ labeling of nuclear DNA in human hair. Human hairs were labeled with Hoechst 33258 and viewed under a fluorescent microscope. Arrows point to labeled nuclei. Note that the hair shown in a lacks Hoechst-positive nuclei, whereas the hair shown in c contains an extremely high number of Hoechst-positive nuclei
Screening of hair from different donors showed that the presence of DNA-positive nuclear remnants was not associated with a specific hair color as hair with and without nuclear remnants were found for each color (not shown). Moreover, the frequency of labeled nuclear remnants in hair did not correlate with the age of the donor (Supplementary Fig. S2). To assess whether the efficiency of in situ labeling of DNA in hair was influenced by the pigmentation status of hair, hair was collected from individuals that had both pigmented (brown or black) and white hairs. Pigmented and white hairs of each donor were separated and labeled with Hoechst dye. Indeed, the frequency of Hoechst-positive nuclei was significantly lower in pigmented hair than in white hair of the same individuals (Fig. 2a). When DNA was extracted from hair and amplified by real-time PCR, the yield was lower in pigmented hair than in white hair, but the difference was not statistically significant (Fig. 2b). These data indicate that the fluorescence signal is partially blocked by the pigment. However, it remains also possible that there is a genuine difference in the frequency of nuclear remnants in pigmented and white hairs.
Fig. 2.
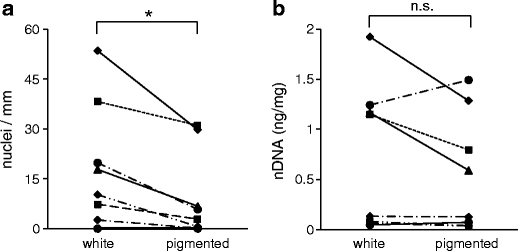
The influence of pigmentation on the detection of nuclear DNA remnants in hair. Hairs of eight individuals were separated in pigmented (brown or black) and non-pigmented (white) hairs. In a fraction of hairs from each group, DNA was labeled in situ, and nuclei were counted under the microscope (a). In another batch of hairs from each group, DNA was extracted and amplified by qPCR with primers specific for nuclear DNA (b). Lines connect results of samples from black and white hair of the same individual. The significance of differences was determined with a paired t test. Asterisk, p < 0.05; n.s., not significant
The abundance of DNA-positive nuclei in hair correlates with the PCR yield of nuclear DNA
Next, we evaluated the relationship of DNA detection in situ and the amount of DNA that could be extracted from hair. Hair samples from 40 donors were analyzed by in situ labeling and by extraction of DNA and subsequent PCR amplification of nuclear and mitochondrial DNA. Initially, hairs of each individual were pooled before labeling and the number of nuclei per millimeter was determined for multiple hairs of each individual. A separate batch of pooled hair (5 mg) of each individual was used for DNA extraction and quantification by PCR. Experiments with single hairs are described below.
The frequency of Hoechst-positive nuclei varied from 0 to more than 60 per millimeter hair. The amount of extractable nuclear DNA was quantified by PCR with primers specific for Alu repeat elements as described in the “Materials and methods” section. For comparison, each DNA preparation was also amplified with PCR primers specific for mitochondrial DNA. The yields of nuclear DNA varied from 0 to 2.5 ng per milligram hair, while the yields of mitochondrial DNA varied from 0 to 370 pg per milligram hair. The frequency of labeled nuclei and the yield of nuclear DNA correlated (Spearman’s rank correlation coefficient rho = 0.65) in a statistically significant manner (Fig. 3a). By contrast, there was no correlation between the number of nuclei and the amount of mitochondrial DNA (Fig. 3b) nor between nuclear and mitochondrial DNA yields from hair (Fig. 3c) which corroborates earlier observations when comparing nuclear and mitochondrial DNA yields in telogenic hair specimens from crime scenes (data not shown). These results confirmed that the in situ labeling method was specific for DNA and indicated that the degree of degradation of nuclear and mitochondrial DNA was determined by different factors.
Fig. 3.
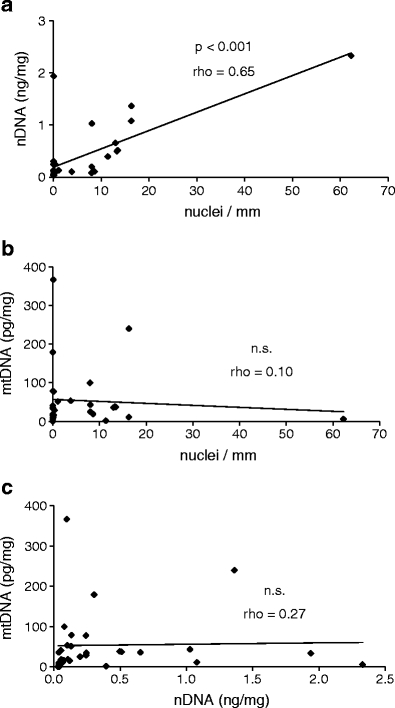
Correlation between the number of nuclei labeled for DNA in situ and the amount of extractable nuclear DNA in hair. Hairs of different individuals were stained for DNA, and nuclei were counted under the microscope. A separate batch (5 mg) of hairs of the same individual was then used for DNA extraction and quantification of nuclear and mitochondrial DNA by real-time PCR. Spearman’s rank correlation coefficient (rho) and the significance of correlation were determined for the frequency of DNA-positive nuclei and nuclear DNA (a), the frequency of DNA-positive nuclei and mitochondrial DNA (b), and the amounts of nuclear DNA and mitochondrial DNA (c). rho, Spearman’s rank correlation coefficient; n.s., not significant
The abundance of DNA-positive nuclei in hair correlates with the success rate of STR typing of hair
Next, we combined the assays of in situ labeling of DNA and DNA extraction from individual hairs. Single hairs from 40 donors were first labeled with Hoechst 33258 and inspected under the microscope. Then the same hairs were washed and subjected to DNA extraction and quantification. Like pooled hair (Fig. 3a), single-hair samples showed a positive and highly significant correlation between the number of nuclei and the yield of nuclear DNA (Fig. 4a). By contrast, the frequency of nuclei did not correlate with the amount of mitochondrial DNA extracted from the same hairs (Fig. 4b).
Fig. 4.
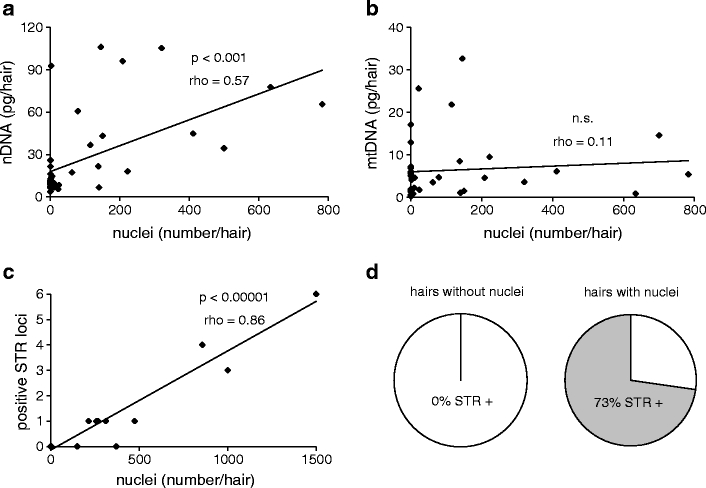
Analysis of single hairs by in situ labeling of DNA, DNA quantification, and STR typing. Samples were prepared by cutting 2 cm from the distal end of single hairs of different individuals. The hairs were stained for DNA, and nuclei were counted under the microscope. Subsequently, the dye was removed by washing and the same hairs were then subjected to DNA extraction and quantification of nuclear and mitochondrial DNA by real-time PCR. Spearman’s rank correlation coefficient (rho) and the significance of correlation were determined for the frequency of DNA-positive nuclei and nuclear DNA (a) and the frequency of DNA-positive nuclei and mitochondrial DNA (b). In a separate experiment, STR typing was performed on nine hairs lacking nuclei and 11 hairs containing more than eight nuclei per millimeter. The correlation between the total number of nuclei labeled in situ and the number of positive STR loci is shown (c). All hairs that lacked Hoechst-positive nuclei did not yield positive STR results, whereas 73% of hairs with Hoechst-positive nuclei yielded at least one positive STR result. The pie charts illustrate the portions of all hairs investigated (d). rho, Spearman’s rank correlation coefficient; n.s., not significant
To evaluate the implications of our previous results in a forensic setting, we performed STR typing. Nine hairs lacking DNA-positive nuclei and 11 hairs containing at least eight Hoechst-labeled nuclei per millimeter were subjected to DNA extraction and miniSTR-multiplex PCR according to standard procedures [9]. The STR results of each hair were then verified by STR typing of buccal scraping samples from the donors of hairs. The total number of nuclei in each hair correlated in a highly significant manner with the number of STR loci that could be typed successfully (Fig. 4c). The correlation was also significant when only hairs containing stainable nuclei were considered (Supplementary Fig. S3). Strikingly, all hairs that lacked stainable nuclei were also negative in STR typing, whereas in approximately 75% of hairs that contained stainable nuclei, at least one STR locus could be typed successfully (Fig. 4d).
Discussion
Our results demonstrate that there is great variance in the degree of completion of DNA degradation in hair and that the incomplete breakdown of nuclear DNA is a prerequisite for successful DNA extraction and STR genotyping. Together, both findings represent a significant advance in our understanding of the molecular and cellular basis for forensic investigations of hair specimens.
The methodology of labeling DNA in human hair was developed by modifying a protocol for labeling murine hair [5]. DNA in hair was first visualized in DNase1L2-deficient mice that had been generated to investigate the process of DNA degradation in keratinocytes [5]. The characterization of these mice and their comparison with wild-type mice revealed that the keratinocyte-specific endonuclease DNase1L2 is essential for the degradation of nuclear DNA during the formation of hair corneocytes. Accordingly, hair from mice that lack DNase1L2 contains aberrantly high amounts of nuclear DNA. Permeabilization with 1% ammonia facilitated the labeling of murine hair with DNA-specific fluorescent dyes such as Hoechst 33258 and DAPI [5]. In the present study, we have adapted this method to human hair by increasing the concentration of ammonia to 10%. This modification sufficed to demonstrate the proof of principle for human hair. However, further improvements of the method are possible and required with regard to forensic applications. That will involve the thorough monitoring of potential sources of contamination and appropriate actions to enable reliable forensic investigations. Also, the performance of DNA forensic analysis systems, such as commercial STR multiplex kits, mitochondrial DNA sequencing, and single base extension assays need to be evaluated in this perspective.
Our data show that the amount of residual DNA varies greatly between corneocytes of the same hair. Only a very small fraction of hair corneocytes was labeled with a DNA-specific dye, indicating that in the great majority of differentiating keratinocytes of the human hair matrix, nuclear DNA is degraded almost to completion. The reason for the incomplete breakdown of DNA in the other cells is not clear at present. Because of the critical role of DNase1L2 in DNA degradation during hair cornification [5], we hypothesize that either low expression or low catalytic activity of DNase1L2 in a fraction of keratinocytes causes incomplete DNA breakdown.
Importantly, human individuals differed greatly with regard to the content of DNA-positive nuclei in hair. Several individuals in our population sample completely lacked nuclear remnants in scalp hair. In others, nuclei were either detected in all hairs or, if the frequency of nuclear remnants was low, in a fraction of all hairs. Although polymorphisms in the human gene encoding DNase1L2 have not been confirmed yet [13], genetic factors are likely to determine the propensity to retain nuclear DNA in hair. It remains to be tested whether the presence of DNA in nuclear remnants in hair is stable over time and constant across different sites of the scalp of a given individual. The efficiency of DNA degradation may change during the hair cycle and in response to hormones or other physiological factors. Furthermore, inflammation of the hair follicle may influence DNA breakdown, possibly by affecting the expression of DNase1L2 [4, 14]. A potential influence of aging on DNA retention in hair was not supported by our data. Importantly, our results showed that dark pigmentation is associated with a lower frequency of detectable nuclear remnants in hair. It is likely that the pigment merely decreases the visibility of the fluorescent label in situ because melanin absorbs radiation both at the excitation wavelength of 350 nm and at the emission wavelength which peaks at 461 nm. Thus, future studies must consider both genuine differences in DNA abundance in hair and methodological issues.
The number of DNA-positive nuclear remnants correlated with the amount of extractable nuclear DNA both when pooled hair samples and when individual hairs were investigated. A Spearman’s correlation coefficient of rho = 0.65 indicated that some, but not all, variability in DNA yield could be explained by differences in the numbers of stainable nuclei. It is not surprising that the correlation was not perfect because influencing factors, such as differences in the DNA content of individual residual nuclei, were not considered in the present study. Here, each nucleus was counted if the label could be clearly distinguished from the background signal. It may be possible to better predict DNA content of hair by measuring the intensity of the fluorescent signal from each labeled nucleus and summing up all signals from each hair.
In contrast to the amount of nuclear DNA, the yield of mitochondrial DNA from hair did not correlate with the number of labeled nuclei, nor did the mitochondrial DNA content correlate with the nuclear DNA content of hair. Previous studies in the mouse have revealed a difference in the mechanisms of degradation of nuclear and mitochondrial DNA in hair with only the former being dependent on DNase1L2 [5]. The lack of correlation between the amounts of nuclear and mitochondrial DNA in human hair suggests that, also in human hair, different mechanisms of DNA degradation are active in the nucleus and in mitochondria.
The association of the results of in situ labeling and the amount of extractable DNA has implications for forensics as the amount of DNA extracted from hair is the most important factor for the success rate of forensic genotyping. Indeed, our results establish that the frequency of nuclear remnants correlates with the success rate of STR typing. Most strikingly, we found that the absence of signals in in situ labeling of DNA in hair was associated with the complete failure to determine STR loci, whereas more than 70% of hairs containing in situ-labeled nuclei yielded at least one positive STR result.
In summary, this study provides novel insights into the cellular basis of the variability in the success rate of DNA typing of hair and represents a starting point for the development of a novel method of forensics. Our results suggest that in situ labeling of DNA can be used to evaluate the likelihood of success of STR typing of a given hair sample. Such a pre-evaluation is desirable if the capacity of STR typing is limited. However, since the existing protocol of in situ labeling of DNA in hair is time-consuming, further optimization is needed before this method can be applied in forensic practice.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
(PDF 203 kb)
Acknowledgments
We thank Jennifer Scherz, Ramona Gmeiner, Cordula Berger, Gabriela Huber, and Harald Niederstätter for the excellent technical support. This work was supported by grants P20043 and P21312 of the Austrian Science Fund (FWF) to L.E.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Linch CA, Whiting DA, Holland MM. Human hair histogenesis for the mitochondrial DNA forensic scientist. J Forensic Sci. 2001;46:844–853. [PubMed] [Google Scholar]
- 2.McNevin D, Wilson-Wilde L, Robertson J, Kyd J, Lennard C. Short tandem repeat (STR) genotyping of keratinised hair. Part 1. Review of current status and knowledge gaps. Forensic Sci Int. 2005;153:237–246. doi: 10.1016/j.forsciint.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Linch CA. Degeneration of nuclei and mitochondria in human hairs. J Forensic Sci. 2009;54:346–349. doi: 10.1111/j.1556-4029.2008.00972.x. [DOI] [PubMed] [Google Scholar]
- 4.Fischer H, Eckhart L, Mildner M, Jaeger K, Buchberger M, Ghannadan M, Tschachler E. DNase1L2 degrades nuclear DNA during corneocyte formation. J Invest Dermatol. 2007;127:24–30. doi: 10.1038/sj.jid.5700503. [DOI] [PubMed] [Google Scholar]
- 5.Fischer H, Szabo S, Scherz J, Jaeger K, Rossiter H, Buchberger M, Ghannadan M, Hermann M, Theussl HC, Tobin DJ, Wagner EF, Tschachler E, Eckhart L (2011) Essential role of the keratinocyte-specific endonuclease DNase1L2 in the removal of nuclear DNA from hair and nails. J Invest Dermatol. doi:10.1038/jid.2011.13 [DOI] [PMC free article] [PubMed]
- 6.Hellmann A, Rohleder U, Schmitter H, Wittig M. STR typing of human telogen hairs—a new approach. Int J Leg Med. 2001;114:269–273. doi: 10.1007/s004140000175. [DOI] [PubMed] [Google Scholar]
- 7.Müller K, Klein R, Miltner E, Wiegand P. Improved STR typing of telogen hair root and hair shaft DNA. Electrophoresis. 2007;28:2835–2842. doi: 10.1002/elps.200600669. [DOI] [PubMed] [Google Scholar]
- 8.Opel KL, Fleishaker EL, Nicklas JA, Buel E, McCord BR. Evaluation and quantification of nuclear DNA from human telogen hairs. J Forensic Sci. 2008;53:853–857. doi: 10.1111/j.1556-4029.2008.00777.x. [DOI] [PubMed] [Google Scholar]
- 9.Grubwieser P, Mühlmann R, Berger B, Niederstätter H, Pavlic M, Parson W. A new “miniSTR-multiplex” displaying reduced amplicon lengths for the analysis of degraded DNA. Legal Med. 2006;120:115–120. doi: 10.1007/s00414-005-0013-6. [DOI] [PubMed] [Google Scholar]
- 10.Butler JM. Short tandem repeat typing technologies used in human identity testing. Biotechniques. 2007;43:ii–v. doi: 10.2144/000112582. [DOI] [PubMed] [Google Scholar]
- 11.Linch CA. The ultrastructure of tissue attached to telogen hair roots. J Forensic Sci. 2008;53:1363–1366. doi: 10.1111/j.1556-4029.2008.00868.x. [DOI] [PubMed] [Google Scholar]
- 12.McNevin D, Wilson-Wilde L, Robertson J, Kyd J, Lennard C. Short tandem repeat (STR) genotyping of keratinised hair. Part 2. An optimised genomic DNA extraction procedure reveals donor dependence of STR profiles. Forensic Sci Int. 2005;153:247–259. doi: 10.1016/j.forsciint.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Fujihara J, Yasuda T, Iida R, Kimura-Kataoka K, Soejima M, Koda Y, Kato H, Panduro A, Yuasa I, Takeshita H. Global analysis of single nucleotide polymorphisms in the exons of human deoxyribonuclease I-like 1 and 2 genes. Electrophoresis. 2010;31:3552–3557. doi: 10.1002/elps.201000319. [DOI] [PubMed] [Google Scholar]
- 14.Shiokawa D, Matsushita T, Kobayashi T, Matsumoto Y, Tanuma S. Characterization of the human DNAS1L2 gene and the molecular mechanism for its transcriptional activation induced by inflammatory cytokines. Genomics. 2004;84:95–105. doi: 10.1016/j.ygeno.2004.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 203 kb)