

Abstract
Apicomplexan parasites encompass several human-pathogenic as well as animal-pathogenic protozoans like Plasmodium falciparum, Toxoplasma gondii, and Eimeria tenella. E. tenella is the causative agent of coccidiosis a disease of chickens, which causes tremendous economic losses to the world poultry industry. Considerable increase of drug resistance makes it necessary to develop and pursue new therapeutic strategies. Cyclin-dependent kinases (CDKs) are key molecules in the regulation of the cell cycle and are therefore prominent target proteins in parasitic diseases. Bioinformatic analysis revealed four potential CDK-like proteins of which one – E. tenella CDK-related kinase 2 (EtCRK2) – is already cloned, expressed and characterized.[1] Using the CDK specific inhibitor Flavopiridol in EtCRK2 enzyme assays and schizont maturation assays we could chemically validate CDK-like proteins as potential drug targets. An X-ray crystal structure of human CDK2 (HsCDK2) served as template to built protein models of EtCRK2 by comparative homology modeling. Structural differences in the ATP-binding site between EtCRK2 and HsCDK2 as well as chicken CDK3 have been addressed for the optimization of selective ATP-competitive inhibitors. Virtual screening and “wet-bench” high throughput screening campaigns on large compound libraries resulted in an initial set of hit compounds. These compounds were further analyzed and characterized leading to a set of four promising lead compounds inhibiting EtCRK2.
Keywords: Coccidiosis, Virtual Screening, Transferases, Drug Discovery, Bioinformatics
Introduction
Infections by parasitic protozoa of the phylum Apicomplexa cause incalculable morbidity and mortality to human and domestic animals. Apicomplexan parasites for example include Plasmodium spp., the causative agent of malaria[2] and Eimeria spp., the causative agent of coccidiosis, a disease mainly of chicken and other poultry.[2] Coccidiosis is a major problem for the global food industry not least in developed countries. Solely, this disease costs the UK poultry industry in excess of 38 million £ per annum as a result of reduced production efficiency (i.e. poor food conversion ratios, retarded growth rates and mortality) and the costs of veterinary and prophylactic intervention.[3,4] The global loss is estimated to range from 500 million £[3] up to the extreme of 3 billion US$[5] per annum and improved strategies for the effective control of eimerian parasites are required. The most significant Eimeria species in poultry include E. tenella, E. acervulina, E. mitis, and E. maxima whereas E. tenella is the most pathogenic one.[2] Although chemo- and immunoprophilactic strategies are available to control these parasites, they are inadequate because in many instances, the Eimeria species have developed resistance against traditional therapeutic agents.[6] Therefore, there is an urgent need to design new affordable and effective pharmaceuticals for control of avian Coccidiosis.[1,5]
Apicomplexan parasites like Plasmodium spp. and Eimeria spp. are characterized by a mostly endogenous developmental life cycle that comprises sequential phases of asexual reproduction (sporogony and schizogony) followed by a terminal phase of sexual reproduction (gamogony).[2] During schizogony the parasites proliferate asexually with a very high cell division rate which in analogy to cell cycles found in other eukaryotes implies a cyclin-dependent kinase (CDK) regulated cell cycle.[7,8] Because of their important role in controlling the progression of the cell cycle, CDKs are prominent drug targets in numerous human diseases, like heart disease[9], cancer[8], neurological disorders[10], and viral infections.[11] Apicomplexan CDKs by virtue of their similarity in sequence and structure to mammalian homologues are also attractive targets of parasitic diseases.[12,13] Therefore, various studies focus on targeting the cell cycle of the malarial parasite P. falciparum by characterizing and inhibiting the related CDKs[14-16], like for example protein kinase 5 (PfPK5), which is one of the best characterized plasmodial CDKs.[17] For Eimeria however, although being of great economic interest[5] and having an almost complete genome sequence available[18], only one CDK-related kinase - EtCRK2 - has been cloned and functionally expressed[1] but no further studies using this protein as a drug target have been published so far.
Here we present a drug discovery workflow which combined in silico and in vitro processes for the discovery of potential anticoccidial lead compounds (Figure 1). After chemical target validation we used in silico screening in combination with in vitro hit and lead confirmation. Our drug discovery efforts led to a set of four confirmed lead structures for EtCRK2, which have great potential for future lead optimization projects.
Figure 1.

Drug discovery workflow for the identification of potential EtCRK2 inhibitors. The workflow consists of 6 steps with different subtasks. Step 4 “Hit Enrichment” and 5 “In vitro Hit Confirmation” was an iterative cycle process and therefore passed multiple times.
Results and Discussion
Target identification and validation
Cyclin-dependent kinases have been identified in almost every eukaryotic organism studied so far.[12] Therefore, it was assumed that CDKs might also be key elements in E. tenella’s cell cycle control and that their knockdown could interfere with the parasite’s proliferation. While several CDKs from the apicomplexan parasite P. falciparum are well characterized, we identified four E. tenella CDK-like proteins (EtCRK2, EtMRK, EtCRK1 and EtCRK3) out of the public available sequencing data (Figure 2). Characteristic CDK-like sequence motifs were found, including the Glycine-rich ATP binding domain, the PSTAIRE box, the Hinge-region, the DFG motif, the T-loop, the conserved residues belonging to the active center K32, D144, and E50 as well as the phosphorylation sites T14, Y15, and T159 (EtCRK2 numbering), provide compelling evidence that deduced proteins are CDKs (Figure 2 and 3).[19-21] These bioinformatics analyzes also revealed EtCRK2, still the only biochemically characterized and physically available EtCRK.[1] Therefore, in this study, we focused on EtCRK2 as a promising CRK target model.
Figure 2.

Sequence alignment and comparison of E. tenella CRKs. (A) Multiple sequence alignment of identified CDK-like homologs of E. tenella (EtCRK2, EtMRK, EtCRK1 and EtCRK3). For EtCRK1 only an incomplete sequence could be determined. Amino acids shaded in gray represent the highest degree of conservation. The major structural features of CDK-like homologs of E. tenella are conserved. The T-Loop (residues 153-162), C-Helix (residues 44-55), G-Loop (residues 10-19), Hinge-Region (residues 78-84) and DFG-Motif (residues 144-146) are colored red, magenta, yellow, green and light-blue, respectively. The residue numberings correspond to the EtCRK2 sequence of E. tenella. The relevant sequence motifs are highly conserved among all CDK homologs. Secondary structure elements are indicated in cartoon representation beneath the alignment. (B) Comparison of known CDK-like sequences from P. falciparum and E. tenella. Besides EtCRK2 blast-searches revealed three new potential CDK-homologous protein sequences of E. tenella. For EtCRK1 and EtCRK3 only partial sequences were identified. Three new potential CDK-like proteins (EtMRK, EtCRK1 and EtCRK3) are homologous to the known CDK-like proteins of P. falciparum. The characteristic vertebrate sequence motif “PSTAIRE” within the C-Helix (vertebrate nomenclature) which is involved in cyclin-binding, shows variations in apicomplexan CDK protein sequences and is colored magenta. All other sequence motives are colored using the same color-code as in Figure 2A.
Figure 3.

Structural analysis of EtCRK2 and its ATP-binding site. (A) Sequence alignment between EtCRK2, HsCDK2 and GgCDK3. Amino acids shaded in gray represent the highest degree of conservation. Red (conserved) and yellow (amino acid exchange) shades mark residues belonging to the ATP-binding site. A = adenine-binding region, R = ribose-binding region and P = phosphate-binding region. Residues belonging to the two selectivity pockets are marked by roman numbers I and II. (B) Homology model of EtCRK2 (blue) in ribbon-type rendering. For comparison the HsCDK2 template structure (PDB-ID: 1OIR; gray) was superimposed on the model structure. The superimposition exhibits almost identical folding patterns between model and template structure. (C) ATP-binding site of the HsCDK2 template structure with co-crystallized ATP (atom color with surface in gray). ATP binds to the Hinge-region via two hydrogen bonds (dotted yellow lines). Residues belonging to the adenine-binding region, the ribose-binding region, the phosphate-binding region, the selectivity pockets I and II are colored red, magenta, yellow, green and blue, respectively. (D) ATP-binding site of EtCRK2 homology model with docked Indirubine-5-sulfonate.[33] K89 from the HsCDK2 template structure (PDB-ID: 1OIR) was superimposed on E88 of the model structure. The oppositely charged residues E88 and K89 might be addressed in lead optimization approaches to increase the inhibitor selectivity towards EtCRK2.
EtCRK2 shows 68% sequence similarity to human CDK1 (HsCDK1) and human CDK2 (HsCDK2), 77% to Gallus gallus CDK3 (GgCDK3), and between 75% and 80% to several other apicomplexan CRKs, e.g. PfPK5 from P. falciparum. Sequence alignments of EtCRK2 to HsCDK2 and GgCDK3 (Figure 3A) showed that this protein sequence contains all characteristic CDK-like domains. Especially, the ATP-binding domain is highly conserved between EtCRK2 and vertebrate homologs (88% identity to HsCDK2 and to GgCDK3). However, the sequence alignment revealed one significant residue exchange - E88K - (EtCRK2 numbering) between EtCRK2, HsCDK2 and GgCDK3 sequences (Figure 3A). The amino acid replacement is connected to a charge exchange from a negatively charged glutamic acid in the parasite’s protein versus a positively charged lysine in the two vertebrate proteins. This striking active site difference between the parasite and the host protein might become vital in lead optimization approaches towards the development of selective inhibitors targeting the parasite’s protein.
Because no structural information for any of the known eimerian CDK-like proteins was available in the Protein Databank (PDB)[22], we generated a structural model of EtCRK2 using comparative modeling.[23] Many crystal structures of human CDK2 (approx. 160) as well as structures of CDKs from apicomplexan parasites like P. falciparum (4) and Cryptosporidium parvum (2) are available in the PDB. Some of them were co-crystallized with potential inhibitors that are of particular interest for rational structure-based drug design approaches. Due to its excellent structure resolution and low Debye-Waller-Temperature factors[24], we chose an inactive structure of HsCDK2 (PDB-ID: 1OIR) co-crystallized with an Imidazo[1,2-α]pyridine inhibitor as template. Initial docking experiments using 1OIR as the receptor achieved high enrichment rates for known CDK inhibitors (data not shown). Because the ionical interaction of K33 (HsCDK2 numbering) is crucial to the function of the enzyme[25] we paid special attention to the orientation of this residue. In 1OIR K33 can be found in its optimal position and orientation. These findings backed our decision to use 1OIR as the template structure for comparative homology modeling.
The full-length sequence similarity of 68% and the sequence similarity of 81% in structural conserved regions between EtCRK2 and HsCDK2 are very reasonable for the generation of a well qualified homology model.[26] The final homology model structure of EtCRK2 is depicted in Figure 3B. It shows the typical bi-lobal structure.[27] Superimposition of the EtCRK2 model and the HsCDK2 structure showed very high similarity between the two proteins at the structural level. Comparison of the ATP-binding pockets (Figure 3C) indicates high similarity but also two significant residue exchanges between EtCRK2 and HsCDK2 (Y81F and E88K, EtCRK2 numbering). The Y→F amino acid exchange does also occur in GgCDK3. The Tyrosine in EtCRK2 is located in the hinge region and would allow for an additional hydrogen bond. However, this hydrogen bond probably does not determine selectivity and therefore this amino acid exchange was not addressed. The variation of amino acid E in EtCRK2 to K in GgCDK3 is of a higher importance regarding the selective inhibition of EtCRK2. This lysine belongs to one of the selectivity pockets of CDKs, which has been shown previously.[25] The principle of single amino acid mediated selectivity in this CDK pocket was already proven by Pratt et al. who found that the selectivity of the CINK4 inhibitor for human CDK4 (HsCDK4) over HsCDK2 is mediated by the single amino acid substitution K89T (HsCDK2 numbering)[28], which is the positional equivalent to the E88K substitution in EtCRK2. K89 is also critical to the binding of guanine derivatives, that form hydrogen bonds to this lysine in HsCDK2 but not to the threonine in HsCDK4.[29-31] Because the amino acid exchange E88K in EtCRK2 involves a charge exchange we believe that it should be possible to achieve selectivity by placing a hydrogen bond donor or even a positive charge in reasonable distance to E88 (Figures 3D and 6). Therefore, following lead optimization rounds will be directed towards a suitable interaction of the inhibitor with E88 of EtCRK2.
Figure 6.

Hypothetical binding modes of Benzimidazole-carbonitrile derivatives found by molecular docking. (A) BES254242 (3-(chloromethyl)-2-formyl-1-oxo-5H-pyrido[1,2-a]benzimidazole-4-carbonitrile) forms one hydrogen-bond to the hinge region but does not address the selectivity pocket. (B) 9-amino-3-ethyl-2-hydroxy-1-oxo-7-(1H-pyrrol-3-yl)-5H-pyrido[1,2-a]benzimidazole-4-carbonitrile an in silico generated derivative of BES254242 forms two hydrogen-bonds to the hinge region and in particular one hydrogen-bond to E88 via its pyrrole nitrogen. The protein backbone of the EtCRK2 homology model is depicted in ribbon representation. Relevant amino acids and the inhibitor molecule are drawn in stick representation and in ball-and-stick representation, respectively. Hydrogen-bonds are visualized by orange dotted lines.
Although bioinformatics analysis and comparative homology modeling indicate the suitability of EtCRK2 as target protein to fight Coccidiosis, experimental evidence has not been given so far for any of the eimerian kinases. A straight forward approach to prove druggability[32] and validate the target class is the application of known specific chemical inhibitiors in vitro and in cell culture assays. Flavopiridol, a synthetic flavone, derived from a natural source alkaloid[33,34] is known to specifically inhibit CDKs.[35] For chemical in vitro validation, we used Flavopiridol in enzyme assays of EtCRK2 (IC50 33±10 nm, Ki 11±3 nm, n=4) in comparison to HsCDK2 (IC50 36±10 nm, Ki 19±3 nm, n=4). Moreover, we also used Flavopiridol in schizont maturation cell culture assays. Flavopiridol fully inhibited E. tenella schizont development at concentrations of 150 nm and 300 nm. Concentrations below 80 nm showed no inhibitory effects and host cell toxicity was observed at concentrations above 600 nm (n=2). We therefore consider E. tenella CDKs and the eimerian cell cycle to be chemically validated drug targets.
Genetic validation, using gene knock-out or RNA interference would enhance the validation of individual CDKs as drug targets in Eimeria. Genetic validation of CDKs has been used successfully in other Apicomplexa and Kinetoplastida such as Plasmodium[12] and Leishmania[36,37], respectively. In Eimeria however, to our best knowledge, gene knock-out or RNA interference have not proven successful so far.
Virtual Screening and In Vitro Hit Confirmation
Besides limited availability of HTS-facilities in academic environments, virtual screening approaches have two striking advantages over HTS approaches which are lower costs compared to HTS screening campaigns and the possibility to screen arbitrarily sized chemical libraries. The lower costs mainly stem from saved expenses for assay miniaturization and automation as well as from saved costs for compound consumption.[38] Very large compound data sets can be tested without purchasing or synthesis, enabling a fast way to explore unknown parts of the chemical space.[39]
The virtual screening used in this study was set up in a similar way to former studies.[40-42] We employed a customized common multi-step filtering protocol[40-43] as depicted in Figure 4. It consisted of a 3D pharmacophore filtering step, the docking and scoring procedure, and a post-processing for docked ligands. Starting from a screening library of approx. 4,000,000 commercially available compounds we used a receptor-based pharmacophore filter to select only such compounds whose chemical features are principally suited to bind to the active site of CDKs. Key for binding to the CDK active site are two H-bond interactions of the potential inhibitor to the Hinge region of the CDK.[27] This requirement together with a set of common molecular features deduced from an analysis of known CDK inhibitors were encoded in a set of pharmacophore queries, which reduced the number of compounds in the screening library to approx. 800,000 and led to a CDK focused screening library. The pharmacophore filter was favored over any 2D similarity filtering, because no significant reduction in computing time for the overall in silico process was expected. Figure 5 depicts two of the deduced receptor-based pharmacophore queries mapped onto the human CDK inhibitor Indirubin-5-sulfonate and the potential inhibitor BES241415 (see below). Compounds which were unlikely to be lead-like were removed from the screening library by a property filter (for details see Experimental Section) which reduced the number of compounds in the screening library to 529,842.
Figure 4.

Filtering Steps during “Virtual Screening” and “Hit Enrichment”. Four confirmed leads have been identified in the drug discovery workflow. Three resulting from “Virtual Screening” and one from “Hit Enrichment”.
Figure 5.

Examples of pharmacophore queries used for database filtering. Protein-based pharmacophore models were manually derived from the ATP-binding site. The features are colored green for H-bond donors, magenta for H-bond acceptors, light-blue for lipophilic features, dark-blue for negative ionizable features and red for positive ionizable features. (A) Best fit of standard CDK-inhibitor Indirubin-5-sulfonate in one of the derived pharmacophore models. (B) Best fit of one identified lead (Benzimidazole, BES252034) in one of the derived pharmacophore models.
This CDK focused screening library of 529,842 compounds was submitted to a two-step docking protocol using the Jain scoring function (Accelrys Inc., San Diego, USA) for pose generation and the GScore scoring function (Tripos Inc., St. Louis, USA) for the final ranking. Selection of scoring functions for pose generation and ranking was based on the results of a number of re-docking experiments using human CDK crystal structures. In a panel of 15 crystal structures of human CDK/ligand complexes, the ligands were removed from the active site, the ligands’ starting conformers were randomized, and the ligands were docked into their respective crystal structure using various combinations of scoring functions for pose generation and ranking. The optimal combination of scoring functions was selected such that the crystal structure of the ligands was reproduced with minimal RMSD and the in silico ranking correlated with the in vitro ranking deduced from experimental activity data (data not shown).
Docking solutions not fulfilling the requirement of two H-bond interactions between the respective ligand and the CDK hinge region (E80 and L82 for EtCRK2) were removed during the post-processing step using an interaction filter in order to retain only valid ligand poses. As mentioned above, the binding site of EtCRK2 differs from the binding sites of vertebrate CDKs in a prominent amino acid exchange (E88K), which probably could be exploited to achieve selectivity for EtCRK2 over vertebrate CDKs although the overall sequence similarity of CDK active sites is quite high. We therefore employed a second interaction filter which rejected all docking solutions not having placed a suitable donor or polar feature in reasonable distance of 3 Å to the negatively charged E88 residue. Docking, post-processing and interaction filtering led to a result set of approx. 83,500 compounds. The 1000 top-ranked compounds were visually inspected and 195 compounds were manually selected for further testing in the in vitro assay system. The manual selection of compounds was aimed to achieve a manageable diverse set of compounds reflecting the most frequently observed scaffolds of barbiturates, adenines, benzoimides, rhodanines, naphtolactames, and oxindoles (scaffolds are named after the building block interacting with the CDK hinge region). Some of the scaffolds identified in the docking are already reported to be known as CDK inhibitor scaffolds.[13,44,45] From the 195 ordered compounds only 139 were available. The in vitro assay resulted in eleven compounds active with an IC50 below 100 μm. The hit rate (8%) of this virtual screen is 50-fold higher in comparison to high throughput screenings (HTS) where hundreds of thousand compounds have to be tested in an enzyme assay.[38]
Hit Enrichment and In Vitro Hit Confirmation
The stringent pharmacophore and interaction filters used in the in silico screening unintentionally might have dismissed interesting compounds from the screening library. In order to recover such compounds we used the eleven confirmed in silico hit compounds as queries in similarity and substructure searches on the current vendor library (Figure 4). For recovering as many compounds of interest as possible we ran a fingerprint based similarity search using a Tanimoto similarity metric[46] as well as a substructure search on the relevant scaffolds. These two search runs resulted in a combined focused screening library of approx. 46,000 compounds (Figure 4). This focused library was screened using molecular docking as done in the Virtual Screening and In Vitro Hit Confirmation (see above). The 500 top-ranked compounds were inspected visually and 263 compounds were manually selected as virtual hits. Due to insufficient capabilities to test all virtual hits, the number of compounds was again reduced for in vitro testing. Therefore, we focused only on structures from the structural classes “Benzimidazoles”, “Naphtolactames” and “Rhodanine-Oxindoles” which are the most active ones and were identified as hit clusters in the first In Vitro Hit Confirmation step. From the 173 compounds only 98 were available from commercial vendors. Six of these 98 compounds were indeed active in the enzyme assay system showing IC50 values below 100 μm.
In Vitro Lead confirmation
The seventeen in silico hits (Actives in Figure 4) – eleven in silico hits coming from Virtual Screening and In Vitro Hit Confirmation and six in silico hits coming from Hit Enrichment and In Vitro Hit Confirmation – were subjected to an in vitro lead confirmation procedure. First of all, IC50 determination was replicated using freshly dissolved compound from solid stock in order to eliminate false-positives caused e.g. by degradation products. The lower IC50 limit was chosen to be 100 μm in the enzyme assay. Subsequently, the molecular structure of the relevant hit compounds was confirmed by NMR spectroscopy and LC/MS spectronomy, i.e. spectroscopy and spectronomy methods were used to compare the actual molecular structure of the compound in the test tube to the molecular structure stored in the compound database in order to verify the identity of the compound. This structural confirmation was not a full structure determination, but was sufficient to detect differences in the molecular structure of the compound in the tube and the one stored in the database. Only compounds without contradictory findings passed this confirmation step.
The next step in the in vitro lead confirmation was the determination of the compounds’ kinetic aqueous solubility using nephelometry. The lower solubility limit that must have been fulfilled by a compound in order to be accepted as a confirmed in vitro lead was chosen to be 500 μm. Ki values were determined for the five most active compounds (one for each structure class) passing all of the aforementioned confirmation steps, in order to prove their assumed ATP competitive binding mode. All of the compounds tested displayed the same mode of inhibition and were ATP competitive. The four Compounds BES062021, BES143551, BES241415, and BES252034 having the lowest Ki values were selected as confirmed in vitro leads. The activity data of these four compounds are listed in Table 1 together with the data of the standard CDK inhibitor Indirubin-5-sulfonate. The compounds belong to four different compound-classes, namely Rhodanine-oxindols (BES062021), Naphtolactames (BES143551), Benzimidazole-carbonitriles (BES241415), and Benzimidazoles (BES252034). The Naphtolactames, Rhodanines and Oxindoles belong to already known compound classes of human CDK inhibitors [13,44,45]. The cluster of scaffolds with known kinase activity indicates that during virtual screening an enrichment of CDK-inhibitors occured. To our best knowledge, the Benzimidazole-carbonitrile scaffold is a novel CDK inhibitor scaffold that was not reported before. Hypothetical binding modes of derivatives of the Benzimidazole-carbonitrile scaffold are depicted in Figure 6. The Ki-values of the lead compounds are in the single- to double-digit micromolar range, which is a suitable starting point for further lead optimization.
Table 1. Confirmed lead structures inhibiting EtCRK2.
| Structure | Name | Structure class | IC50 [μM] | Ki [μM] | ||
|---|---|---|---|---|---|---|
| HsCDK2 | EtCRK2 | HsCDK2 | EtCRK2 | |||
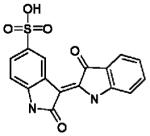
|
Indirubin-5-sulfonate | Oxindole | 0.23±0.11 | 0.67±0.19 | 0.08±0.01 | 0.17±0.03 |

|
BES062021 | Rhodanine-oxindole | 26±19 | 23±14 | 17±8 | 16±9 |

|
BES143551 | Naphtolactame | 12±7 | 36±8 | 2±1 | 6±2 |

|
BES241415 | Benzimidazole-carbonitrile | 41±13 | 61±27 | 41±13 | 83±29 |

|
BES252034 | Benzimidazole | 35±3 | 15±3 | 8±2 | 8±2 |
In silico hits were verified against EtCRK2 and HsCDK2. Due to the high sequence similarity between HsCDK2 and GgCDK3, HsCDK2 served as model for GgCDK3, which was not available. Indirubin-5-sulfonate[33] was used as standard HsCDK2 inhibitor. Four confirmed leads have been identified belonging to the structural class rhodanine-oxindoles, naphtolactames, benzimidazole-carbonitriles and benzimidazoles. Results are expressed as mean IC50 and mean Ki values ± SD in three to four different experiments (n=3-4).
Conclusion
Drug development in parasitology can benefit from cancer drug development research because many similarities exist between cancer cells and parasites. Both share an important feature of living and multiplying in a host organism.[47] During schizogony E. tenella proliferates asexually with a very high cell division rate being faintly reminiscent to fast growing cancer cells. This reminiscence inspired our target selection. CDKs are established as valuable anti-cancer targets due to being a key element in cell cycle control. Cell cycle progression is absolutely essential for the survival of any organism[14] and it is well established that some core mechanisms of cell cycle control in eukaryotes are conserved from yeast to man.[16]
In this study, we provide evidence that CDK-like proteins from E. tenella – like EtCRK2 – are essential for cell cycle progression of the parasite and therefore, could be used as target proteins for the discovery and development of anticoccidial drugs. In order to chemically validate this target class we tested the known human CDK-specific inhibitor Flavopiridol for its effect on EtCRK2 and schizont maturation. Flavopiridol was able to inhibit the enzyme and schizont maturation. We therefore consider CRKs and the cell cycle from E. tenella to be validated as a suitable target complex for chemotherapy.
By using a homology model structure of EtCRK2, which we derived on the basis of a crystal structure of human CDK2, and an in silico screening workflow we were able to select 293 compounds out of a commercial library of approx. 6 million compounds as potential inhibitors of E. tenella CRK2. Seventeen of these 293 compounds prove to be active on EtCRK2 in an in vitro enzyme assay, resulting in an excellent hit enrichment factor of approx. 6%. Among these 17 actives four compounds were classified as confirmed leads of which one belongs to the chemical class of benzimidazole-carbonitriles. To our best knowledge this chemical class has so far not been described to be active on CDKs. A rational in silico derivatization approach lead to a compound (Figure 6B) which was able to form two hydrogen-bonds to the hinge region and to address the important amino acid E88 in the selectivity pocket of EtCRK2. Further lead optimization will follow this approach to extent the different scaffolds towards the selectivity pocket with suitable functional groups.
Taking all together, by combining a wide range of bioinformatics and chemoinformatics in silico technology with “wet bench” laboratory technology, we were able to initiate a target based drug discovery process starting with target identification and resulting in very promising lead compounds. These compounds are ready to be introduced into the next steps of the drug discovery workflow – the lead optimization phase – with the potential outcome of a drug candidate.[48,49]
Experimental Section
Bioinformatic analysis
All sequence data were obtained from the National Center for Biotechnology Information[50] and from the Sanger Institute.[18] Multiple sequence alignments for CDK2 like proteins in E. tenella were conducted using ClustalW[51] with default parameters. The following sequences were used for the multiple alignment: EtCRK2 – GI:40804978; GgCDK3 – GI:126165309; HsCDK2 – GI:29849.
A) Identification of putative genes
Recursive BLAST (BLASTN, TBLASTX or TBLASTN) searches on the E. tenella OmniBlast Server (http://www.sanger.ac.uk/cgi-bin/blast/submitblast/e_tenella/omni) on a set of nucleotide databases available at the Sanger Institute were performed. This BLAST database contains unfinished sequence data. Recursive BLAST searches were performed using previously characterized CDK-like amino acid sequences from Plasmodium falciparum 3d7, Plasmodium yoeli yoeli, Cryptosporidium parvum, Cryptosporidium hominis, Theileria parva, Theileria annulata and Toxoplasma gondii. Sequence alignments with an E-value less than 10−3 and with a bit score greater than 100 were considered significant.
B) Structural identification of putative genes
Exon-Intron boundaries were determined by examining the coding regions with significant matches and adjusted according to the most common consensus sequence of exon-intron junctions (“gt-ag” rule of intronic sequence). Furthermore, gene structures were deduced on the basis of sequence data of the aforementioned organisms. Gene finding was done using fgeneSH.[52] The resulting nucleotide sequences were translated in the correct reading frames using the transeq algorithm implemented in the software Metalife Trinity 1.4 (Metalife AG, Winden, Germany). All multiple and pairwise sequence alignments for CDK-like and cyclin-like proteins in other apicomplexan parasites were conducted using the program ClustalW1.83[51,53] with default parameters. For graphical representation of the alignments the program GeneDoc[54] was used (http://www.psc.edu/biomed/genedoc).
Chemicals
Purity of all screening compounds used was ≥ 90%, if not stated otherwise. Flavopiridol was ordered as Flavopiridol hydrochloride hydrate from Sigma-Aldrich Inc., St. Louis, USA. IUPAC-Name: 2-(2-Chlorophenyl)-5,7-dihydroxy-8-[(3S,4R)-3-hydroxy-1-methyl-4-piperidinyl]-4H-1-benzopyran-4-one. Purity: ≥ 98%. Indirubin-5-sulfonate was ordered from Biomol International LP., Exeter, UK. IUPAC-Name: (3Z)-2-oxo-3-(3-oxo-1H-indol-2-ylidene)-1H-indole-5-sulfonic acid. Purity: ≥ 98%. BES062021 was ordered from Synex Pharma Technologies Co., Ltd., Shanghai, China. IUPAC-Name: 2-[(3Z)-2-oxo-3-(4-oxo-2-thioxo-thiazolidin-5-ylidene)indolin-1-yl]acetic acid. Purity: ≥ 95%. BES143551 was ordered form SPECS GmbH, Berlin, Germany. IUPAC-Name: N,N-diethyl-2-oxo-1,2-dihydrobenzo[cd]indole-6-sulfonamide. Purity: ≥ 90%. BES241415 was ordered from InterBioScreen Ltd., Chernogolovka, Russia. IUPAC-Name: 3-(4-cyano-3-methyl-1-oxo-5H-pyrido[1,2-a]benzimidazol-2-yl)propanoic acid. Purity: ≥ 90%. BES252034 was ordered from Sigma-Aldrich Co., St. Louis, USA. IUPAC-Name: 2-[(5-nitro-1H-benzimidazol-2-yl)imino]imidazolidine-4,5-dione. Purity: ≥ 90%.
Screening Library
The in silico screening was run on a combined library from commercial vendors (Asinex Ltd., Moscow, Russia; Akos Consulting & Solutions GmbH, Basel, Schweiz; ChemBridge Corporation, San Diego, USA; Chemical Diversity Labs, Inc., San Diego, USA; Enamine Ltd, Kiev, Ukraine; InterBioScreen, Moscow, Russia; LifeChemicals Inc., Burlington, Canada; Maybridge, Cambridge, UK; Otava, Kiev, Ukraine; Specs, Delft, Netherlands; TimTec Corp., Newark; USA; Vitas-M Laboratory Ltd., Moscow, Russia) which was stored in an ISIS database (Symyx Technologies, Inc., Sunnyvale, USA) and which contained about 4 million virtual compounds or subsets of this library resulting from previous filtering steps.
Homology protein modeling
Homology models of EtCRK2 were calculated using the program Modeller implemented in the Insight II software package (Accelrys Inc., San Diego, USA). All calculations were carried out under default conditions unless otherwise stated. To build the homology models, a crystal structure of human CDK2 (pdb ID = 1OIR) was used as template protein, due to its good structure resolution and low Debye-Waller-Temperature factors.[24] The Align123 algorithm of the Insight II multisequence alignment tool was used for the sequence alignment. Four homology models of the EtCRK2 sequence were generated using the highest optimization levels. The final models were refined using standard protocols as implemented in the software. Evaluation of the generated structures was carried out using Ramachandran φ-ψ plot calculations[55] computed with PROCHECK (Accelrys Inc., San Diego, USA). The best quality model was chosen depending on the PROCHECK results.
Generation of protein-based pharmacophore models
Protein-based (receptor-based) pharmacophore models of EtCRK2 were generated using the Cerius2 software package (Accelrys Inc., San Diego, USA). Although receptor-based pharmacophore models were used, special emphasis was placed on the known characteristic interaction patterns of ATP competitive CDK2 inhibitors. In a first step a protein-based interaction map was generated using the “structure based focusing” module. This interaction map contained all H-bond and lipophilic interactions and was manually clustered and refined which resulted in two H-bond donors (L82; K32), four H-bond acceptors (E80; L82; D85; E88) and two lipophilic interactions remained. In order to allow for a more flexible screening the H-bond acceptor and donor features at K32 and E88 were modified using the defaults “positive” and “negative ionisable” respectively. In order to avoid making the pharmacophore queries overly strict no excluded volumes, which describe the topology of the binding site were used. Compounds topologically not fitting the binding site were removed by the following docking procedure. The resulting features were then merged to five point pharmacophore models under the basic condition to contain at least the L82 H-bond acceptor as well as one H-bond donor for hinge interaction and one of two possible hydrophobic features.
Pharmacophore based virtual screening
The pharmacophore based virtual screening campaign was performed on the Screening Library described above. For all compounds in this library, relevant conformations were generated using CATALYST (FAST, max. 150 conformers per compound) and the database was stored in CATALYST’s internal multiconformational data format. The compound’s stereochemistry as stored in the supplier catalogue remained untouched. For virtual screening the single hypotheses were combined to one query using an OR-operator in order to make database searching more efficient. The “BEST Flexible Search” command used for the database search allows for conformational optimization during computation. Hit molecules fulfilling all five features of one of the queries were stored in a 2D SD-file.
Property filtering and focused virtual library preparation
All molecules of the kinase focused library which contained other atoms than C, O, H, N, S, P, F Cl, Br or I, which had a molecular weight >450 Da or which possessed more than eight rotatable bonds were removed from the dataset. Filtering was done using the CACTVS software package.[56] The kinase focused library was then prepared for the virtual screening campaign using molecular ligand docking. For molecular ligand docking relevant ionization states were generated for each compound in the library according to Oellien et al.[57] The 3D conformations were generated using CORINA 3.0[58] with options “ori,rs,r2d,wh”. The final kinase focused library contained approx. 530,000 compounds.
Substructure and similarity searches
All similarity searches were carried out with the Pipline Pilot software package (Accelrys Inc., San Diego, USA) using the Tanimoto coefficient.[46] For each ligand one structural (ECFP_4) and one functional fingerprint (FCFP_4) were calculated and used for similarity searching. Each ligand was used as an individual template for similarity search calculations. A minimal Tanimoto similarity index of 0.6 was set as cut-off value. All substructure searches were carried out using the ISISbase (Symyx Technologies, Inc., Sunnyvale, USA) substructure search. Again, different ionization states were considered for each compound.
Docking
A) Virtual screening
Compounds of the kinase-focused library were docked into the best homology model of EtCRK2, using the software AutoDock 3.05.[59] Docking of the ligands was restricted to a 25 × 25 × 25 Å cubic grid with 0.5 Å grid spacing. The center of the grid was placed at the geometric centre of the ATP-binding site in the EtCRK2 homology model. Dockings were performed using the Lamarckian genetic algorithm[60] in conjunction with a pseudo-Solis-and-Wets local search and in order to ensure an exhaustive search, the genetic algorithm’s population size was set to 50, the maximum number of energy evaluations per run was set to 300.000 and the maximum number of generations that the genetic algorithm run should last was set to 100.000. All other parameters were set to the default values described in the AutoDock manual.[61] The number of poses of each ligand was set to 10. The ligand binding site was defined by a cavity file which contained the residues of the ATP binding site.
B) Hit Enrichment
Compounds which were selected from substructure and similarity searches were docked in the EtCRK2 homology model using the docking software GOLD 3.1.1.[62] Docking studies were performed using default genetic algorithm parameters and automatic settings with “200% search efficiency”. Ten poses per ligand were stored and scored using the scoring function GOLDScore as implemented in the software. An active site of 14 Å diameter was created around the geometric center of the ATP-binding site. The parameters “Early_termination” and “Force_constraints” were set to zero.
Postprocessing of docking results
Docking poses which did not address the hinge-region[63] were discarded using a CACTVS/tcl-script. The script selected only those ligand poses, which may obtain a hydrogen bond to the backbone NH of L82. This interaction is mandatory to almost all CDK inhibitors.[27] Additionally, a second hydrogen bond to a backbone carbonyl of either E80 or L82 must be obtained by a potential ligand. A hydrogen bond was defined as a distance constraint between a donor and an acceptor atom of less than or equal to 3 Å. A bond angle constraint was not implemented. Docking poses of potential ligands fulfilling these criteria were retained and assessed using the Jain scoring function (Accelrys Inc., San Diego, USA) and the overall ranking was done using GScore (Tripos Inc., St. Louis, USA).
Schizont maturation Assay (SMA)
Madin-Darby Bovine Kidney (MDBK) cells[64] (2.5 · 104 cells/well) were seeded in 96-well-microtiterplates (Costar Corning Corp, New York, USA) and incubated in Dulbecco’s Modified Eagle’s Medium (DMEM, PAN Biotech GmbH, Aidenbach, Germany) containing 10% fetal calf serum for approx. 24 h at 37°C, and 5% CO2. E. tenella sporocysts were excysted as described[65] and MDBK cells were infected with 7 · 104 sporozoites/well and incubated at 41°C, 10% CO2, and 95% relative humidity. After incubation for approx. 3 h, non-invaded sporozoites were removed by washing with Earle’s medium 199 (Biochrom AG, Berlin, Germany) followed by the addition of medicated 199 medium containing the CDK inhibitor Flavopiridol[33] to reach final inhibitor concentrations per well of 50 μm, 10 μm, 2 μm, 0,4 μm, and 0.08 μm. Cytotoxicity was determined by exposition of the compounds to non-infected cells. Non-medicated samples served as control.
After 60 h of incubation at 41°C, completion of schizont development was visually confirmed by light microscopy. Cells were washed several times and fixed with 4% formaldehyde (50 μl/well) for approx. 45 min. Plates were washed three times with PBS containing 0.5% Tween20 followed by 45 min incubation with PBS containing 0.5% Tween20 and 5% milk powder (washing buffer). After several washing steps an anti-merozoite serum, which was produced in-house using standard procedures[66], was added and incubated for 1 h followed by several washing steps and a 1 h incubation with a FITC-conjugated AffiniPure Fab-Fragment Goat-Anti-Rabbit IgG (H+L) (Dianova GmbH, Hamburg, Germany) cells were washed and the number of mature schizonts was determined in an ELISA reader (Tecan Group Ltd., Männedorf, Switzerland) at an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Expression and purification of recombinant EtCRK2 and XlRINGO
Plasmids (pQE60-EtCRK2[1] and pMALc2X-XlRINGO[67]) were kindly provided by Dr. Jane H. Kinnaird (University of Glasgow, UK) and by Dr. Ario de Marco (European Molecular Biology Laboratory, Heidelberg, Germany), respectively. Both plasmids were transformed in Escherichia coli BL21 (DE3) pLysS (Invitrogen, Carlsbad, USA). These cells were used to express the recombinant EtCRK2-his and MBP-XlRINGO fusion proteins separately overnight at 24°C using 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (Molecula Research Laboratories, Columbia, USA). Cell pellets were harvested by centrifugation at 9,000g for 40 min.
The pellet containing EtCRK2-his was resuspended in 50 mM phosphate buffer, pH 7.4, 300 mM NaCl and 10 mM imidazole (Sigma-Aldrich Corp., St. Louis, USA) and after addition of 25 U/ml benzonase (Merck KGaA, Darmstadt, Germany), the cells were disrupted by a French Press (Thermo Spectronic, Madison, USA). The resulting homogenate was centrifuged at 10,000g and EtCRK2-his was purified from the supernatant by a Ni-NTA affinity chromatography column (Ni-NTA agarose matrix beads, Sigma-Aldrich Corp., St. Louis, USA). The protein was eluted in the same buffer with 250 mM imidazole. A size exclusion chromatography (Hiload 16/60 Superdex 75 column, GE Healthcare Bio-Sciences AB, Uppsala, Sweden) was used for a further purification of the kinase and to remove the imidazole. Buffer A (10 mM TRIS-HCl, pH 7.4, 100 mM NaCl) was used for all size exclusion chromatographies.
A pellet containing MBP-XIRINGO was resuspended in 50 mM phosphate buffer, pH 7.4, 50 mM NaCl, and after addition of 25 U/ml benzonase, the cells were disrupted by a French press. After centrifugation at 10,000g, MBP-XlRINGO was purified from the supernatant by amylose affinity chromatography (amylose resin matrix beads from New England Biolabs, Ipswich, USA) and eluted in the same buffer supplemented with 10 mM maltose (Sigma-Aldrich Corp., St. Louis, USA). A size exclusion chromatography (Hiload 26/60 Superdex 200 column, GE Healthcare Bio-Sciences AB, Uppsala, Sweden) was performed to separate MBP-XlRINGO from the free maltose binding protein (MBP). EtCRK2-his and MBP-XlRINGO were pooled, incubated over night at 4°C to build a complex and loaded onto a Hiload 26/60 Superdex 200 column for a further size exclusion chromatography, in order to separate the excess of EtCRK2-his from the complex. All affinity and size exclusion chromatographies were carried out in an ÄKTA FPLC System (GE Healthcare Bio-Sciences AB, Uppsala, Sweden).
Time-resolved fluorescence energy transfer (TR-FRET) assay
TR-FRET assays were performed in 384-well black plates (Greiner Bio-One GmbH, Kremsmünster, Austria) according to the manufacturer’s instructions (IMAP™ TR-FRET Screening Express Kit, Molecular Devices, MDS Analytical Technologies, Sunnyvale, USA) using the components included in the assay kit. The assay was carried out in a final reaction volume of 20 μl IMAP™ reaction buffer (10 mM TRIS-HCl, pH: 7.2, 10 mM MgCl2, 0.05% NaN3 and 0.1% phosphate free BSA) containing 1 mM dithiothreitol (DTT, Sigma-Aldrich Corp., St. Louis, USA), 100 nm FAM-histone H1-derived peptide (fluorescent substrate, Molecular Devices, MDS Analytical Technologies, Sunnyvale, USA), compound (for IC50 determination in a range from 0.02 to 200 μM), depending on the used kinase complex 0.05 μg/μl of EtCRK2-XlRINGO or 0.1 U/ml hCDK2-CYCA (Upstate Biotechnology, Lake Placid, USA) and started with an addition of 30 μm ATP (Sigma-Aldrich Corp., St. Louis, USA). Reactions were stopped after 45 min with progressive binding buffer (stop/detection solution) containing a 1:900 dilution of IMAP™ TR-FRET donor (Terbium based phosphor conjugate, called “Tb donor”) and IMAP™ progressive binding reagent (containing the IMAP™ binding entity). After phosphorylation by the kinase, the fluorescent phosphorylated substrate and the “Tb donor” (phosphate-containing linker, sensitizer and Tb complex combined in one molecule) are brought in close proximity by binding simultaneously to the same IMAP™ binding entity. This enables the resonance energy transfer. Because of the long fluorescence lifetime of the terbium ion, the produced resonance energy transfer can be detected in time resolved mode (Molecular Devices, MDS Analytical Technologies, Sunnyvale, USA).
The TR-FRET measurement was performed using a Wallac Victor 2 multilabel counter (PerkinElmer, Waltham, USA) with the following parameters: number of repeats 1; measurement height 3.00 mm; emission filter 490 nm; second measuring emission filter 520 nm; excitation filter 340 nm; delay 500 μs; window time 1400 μs; cycle 2,000 μs; light integrator capacitors 1; light integrator ref. level 21; flash energy area high; flash energy level 119; no flash absorbance measurement; beam normal.
The raw data measured at 490nm and at 520 nm were analyzed using the calculation module for IMAP™ TR-FRET data (www.moleculardevices.com; Molecular Devices, MDS Analytical Technologies, Sunnyvale, USA). IC50 values were calculated using the application XLfit version 4.3.2, Build 11 (Microsoft Corporation, Redmond, USA) included in the Activitybase version 7.2.1.5., a data management software application for drug discovery, (ID Business Solutions Limited, Guildford, Surrey, UK).
Ki Determination
TR-FRET assays for Ki determinations of the compounds were performed as described above, using 4 different ATP concentrations between 5-15 μm and 5 different inhibitor concentrations 1-100 μm for rhodanine-oxindole and naphtolactame, 2-200 μm for benzimidazol-carbononitrile and 2.5-250 μm for benzimidazole. The raw data were analyzed using the calculation module for IMAP™ TR-FRET data. Ki values were calculated using the Enyzme Kinetics Module 1.1 of SigmaPlot 2002 for Windows version 8.0 (SPSS Software, Chicago, USA).
Compound solubility test
A two-fold serial dilution of the compounds was performed (0.5-500 μm) in dimethyl sulfoxide (DMSO, Acros Organics, Fischer Scientific, Morris Plains, USA) and added in duplicate to 96-well microtiter plates. 10 mM sodium phosphate buffer, pH 7.4 was added to give a total volume of 200 μl and a final DMSO concentration of 5% in all wells. The plates were incubated at room temperature for 22 min and the relative solubility of the compounds was determined by measuring forward scattered light using a NEPHELOstar laser-based microplate nephelometer (BMG LABTECH GmbH, Offenburg, Germany). Wells containing only buffer and 5% DMSO were used as controls. Data analysis was carried out using the application XLFit in the Activitybase version 7.2.1.5. (ID Business Solutions Limited, Guildford, Surrey, UK).
Liquid chromatography, mass spectroscopy (LC/MS) and nuclear magnetic resonance (NMR) analyses
LC/MS analysis were undertaken in order to check the molecular weight of the compounds using an Agilent Technologies LC7MSD1100 (liquid chromatography, mass selective detector, from Agilent Technologies, Santa Clara, USA). The liquid chromatography conditions were: Zorbax SB (stable bound)-C18, 1.8 μm, 4.6 × 30 mm column; 0.1 μl/min flow; gradient 10-100% eluent B over 3 minutes (eluent A is 95:5 H2O: acetonitrile + 0.1% formic acid and eluent B is acetonitrile) and a column temperature of 40°C. The mass spectroscopy detection was performed using a MSD 1100 ESI&API (mass selective detector; electron spray ionisation & advanced chemical ionisation; Agilent Technologies, Santa Clara, USA) single quadrupole mass spectrometer. NMR tests were carried out with the compounds in order to verify their chemical structure. Data were obtained on a Bruker Avance DRX 400 MHz NMR spectrometer (Bruker AXS, Madison, USA).
Supplementary Material
Acknowledgements
We thank Dipl.-Ing. J. Hofmann, Dr. A. Heckeroth, and Dipl.-Ing. B. von Oepen for performing the chemical in vitro validation using the schizont maturation assay, Dr. Markus Wagener for the preparation of figure 5, and Dr. J. Cramer for his assistance and helpful discussions. We are very grateful to Intervet Innovation GmbH for providing facilities and Intervet colleagues for their support in this project. J.C. Mottram is supported by an SRDG grant (HR04013) from the Scottish Funding Council. We thank Dr. Jane H. Kinnaird (University of Glasgow, UK) and Dr. Ario de Marco (European Molecular Biology Laboratory, Heidelberg, Germany) for the pQE60-EtCRK2 and the pMALc2X-XlRINGO plasmids, respectively.
Reference List
- [1.].Kinnaird JH, Bumstead JM, Mann DJ, Ryan R, Shirley MW, Shiels BR, Tomley FM. Int. J. Parasitol. 2004;34:683–692. doi: 10.1016/j.ijpara.2004.01.003. [DOI] [PubMed] [Google Scholar]
- [2.].Mehlhorn H. In: Encyclopedia of parasitology. 3rd Edition ed. Mehlhorn H, editor. Springer; Berlin: 2008. [Google Scholar]
- [3.].Shirley MW, Smith AL, Blake DP. Vaccine. 2007;25:5540–5547. doi: 10.1016/j.vaccine.2006.12.030. [DOI] [PubMed] [Google Scholar]
- [4.].Williams RB, Carlyle WW, Bond DR, Brown IA. Int. J. Parasitol. 1999;29:341–355. doi: 10.1016/s0020-7519(98)00212-4. [DOI] [PubMed] [Google Scholar]
- [5.].Dalloul RA, Lillehoj HS. Expert Rev. Vaccines. 2006;5:143–163. doi: 10.1586/14760584.5.1.143. [DOI] [PubMed] [Google Scholar]
- [6.].Stephen B, Rommel M, Daugschies A, Haberkorn A. Vet. Parasitol. 1997;69:19–29. doi: 10.1016/s0304-4017(96)01096-5. [DOI] [PubMed] [Google Scholar]
- [7.].Doerig C, Chakrabarti D, Kappes B, Matthews K. Prog. Cell Cycle Res. 2000;4:163–183. doi: 10.1007/978-1-4615-4253-7_15. [DOI] [PubMed] [Google Scholar]
- [8.].Malumbres M, Barbacid M. Nat. Rev. Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- [9.].Tamamori M, Ito H, Hiroe M, Terada Y, Marumo F, Ikeda MA. Am. J. Physiol. 1998;275:H2036–H2040. doi: 10.1152/ajpheart.1998.275.6.H2036. [DOI] [PubMed] [Google Scholar]
- [10.].Monaco EA, Vallano ML. Curr. Med. Chem. 2003;10:367–379. doi: 10.2174/0929867033368277. [DOI] [PubMed] [Google Scholar]
- [11.].Schang LM, St Vincent MR, Lacasse JJ. Antivir. Chem. Chemother. 2006;17:293–320. doi: 10.1177/095632020601700601. [DOI] [PubMed] [Google Scholar]
- [12.].Doerig C. Biochim. Biophys. Acta. 2004;1697:155–168. doi: 10.1016/j.bbapap.2003.11.021. [DOI] [PubMed] [Google Scholar]
- [13.].Gray N, Detivaud L, Doerig C, Meijer L. Curr. Med. Chem. 1999;6:859–875. [PubMed] [Google Scholar]
- [14.].Doerig C, Meijer L. Expert Opin. Ther. Targets. 2007;11:279–290. doi: 10.1517/14728222.11.3.279. [DOI] [PubMed] [Google Scholar]
- [15.].Doerig C, Billker O, Pratt D, Endicott J. Biochim. Biophys. Acta. 2005;1754:132–150. doi: 10.1016/j.bbapap.2005.08.027. [DOI] [PubMed] [Google Scholar]
- [16.].Hammarton TC, Mottram JC, Doerig C. Prog. Cell Cycle Res. 2003;5:91–101. [PubMed] [Google Scholar]
- [17.].Holton S, Merckx A, Burgess D, Doerig C, Noble M, Endicott J. Structure. 2003;11:1329–1337. doi: 10.1016/j.str.2003.09.020. [DOI] [PubMed] [Google Scholar]
- [18.].Wellcome Trust Sanger Institute 2009 http://www.sanger.ac.uk/
- [19.].Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- [20.].Malumbres M, Barbacid M. Trends Biochem. Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- [21.].Morgan DO. Annu. Rev. Cell Dev. Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- [22.].Berman H, Henrick K, Nakamura H. Nat. Struct. Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- [23.].Cavasotto CN, Phatak SS. Drug Discov. Today. 2009;14:676–683. doi: 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- [24.].Gao HX, Peng LM. Acta Crystallogr. A. 1999;55:926–932. doi: 10.1107/s0108767399005176. [DOI] [PubMed] [Google Scholar]
- [25.].Branden C, Tooze J. In: Introduction to Protein Structure - Folding and Flexibility. 2 ed. Brandon C, Tooze J, editors. Garland Publishing Inc.; London: 1999. pp. 89–120. [Google Scholar]
- [26.].Xiang Z. Curr. Protein Pept. Sci. 2006;7:217–227. doi: 10.2174/138920306777452312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27.].Kubinyi H, Müller G. Chemogenomics in Drug Discovery. A Medicinal Chemistry Perspective. 1 ed. Wiley-VCH Verlag; Weinheim: 2004. pp. 191–219. [Google Scholar]
- [28.].Pratt DJ, Bentley J, Jewsbury P, Boyle FT, Endicott JA, Noble ME. J. Med. Chem. 2006;49:5470–5477. doi: 10.1021/jm060216x. [DOI] [PubMed] [Google Scholar]
- [29.].Heady L, Fernandez-Serra M, Mancera RL, Joyce S, Venkitaraman AR, Artacho E, Skylaris CK, Ciacchi LC, Payne MC. J. Med. Chem. 2006;49:5141–5153. doi: 10.1021/jm060190+. [DOI] [PubMed] [Google Scholar]
- [30.].Alzate-Morales J, Caballero J. J. Chem. Inf. Model. 2010;50:110–122. doi: 10.1021/ci900302z. [DOI] [PubMed] [Google Scholar]
- [31.].Gray NS, Wodicka L, Thunnissen AM, Norman TC, Kwon S, Espinoza FH, Morgan DO, Barnes G, LeClerc S, Meijer L, Kim SH, Lockhart DJ, Schultz PG. Science. 1998;281:533–538. doi: 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]
- [32.].Caffrey CR, Rohwer A, Oellien F, Marhofer RJ, Braschi S, Oliveira G, McKerrow JH, Selzer PM. PLoS One. 2009;4:e4413. doi: 10.1371/journal.pone.0004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33.].Senderowicz AM. Invest. New Drugs. 1999;17:313–320. doi: 10.1023/a:1006353008903. [DOI] [PubMed] [Google Scholar]
- [34.].Shapiro GI. Clin. Cancer Res. 2004;10:4270s–4275s. doi: 10.1158/1078-0432.CCR-040020. [DOI] [PubMed] [Google Scholar]
- [35.].Dai Y, Grant S. Curr. Opin. Pharmacol. 2003;3:362–370. doi: 10.1016/s1471-4892(03)00079-1. [DOI] [PubMed] [Google Scholar]
- [36.].Hassan P, Fergusson D, Grant KM, Mottram JC. Mol. Biochem. Parasitol. 2001;113:189–198. doi: 10.1016/s0166-6851(01)00220-1. [DOI] [PubMed] [Google Scholar]
- [37.].Mottram JC, McCready BP, Brown KG, Grant KM. Mol. Microbiol. 1996;22:573–583. doi: 10.1046/j.1365-2958.1996.00136.x. [DOI] [PubMed] [Google Scholar]
- [38.].Phatak SS, Stephan C, Cavasotto CN. Expert. Opin. Drug Discov. 2009;4:947–959. doi: 10.1517/17460440903190961. [DOI] [PubMed] [Google Scholar]
- [39.].Keil M, Marhofer RJ, Rohwer A, Selzer PM, Brickmann J, Korb O, Exner TE. Front. Biosci. 2009;14:2559–2583. doi: 10.2741/3398. [DOI] [PubMed] [Google Scholar]
- [40.].Perola E. Proteins. 2006;64:422–435. doi: 10.1002/prot.21002. [DOI] [PubMed] [Google Scholar]
- [41.].Muthas D, Sabnis YA, Lundborg M, Karlen A. J. Mol. Graph. Model. 2008;26:1237–1251. doi: 10.1016/j.jmgm.2007.11.005. [DOI] [PubMed] [Google Scholar]
- [42.].Oellien F, Engels K, Cramer J, Marhöfer RJ, Kern C, Selzer PM. In: Drug Discovery in Infectious Diseases - Antiparasitic and Antibacterial Drug Discovery: From Molecular Targets to Drug Candidates. Selzer PM, editor. Vol. 1. WILEY-VCH Verlag GmbH & Co. KGaA; Weinheim: 2009. pp. 323–338. [Google Scholar]
- [43.].Jacobsson M, Garedal M, Schultz J, Karlen A. J. Med. Chem. 2008;51:2777–2786. doi: 10.1021/jm7016144. [DOI] [PubMed] [Google Scholar]
- [44.].Bramson HN, Corona J, Davis ST, Dickerson SH, Edelstein M, Frye SV, Gampe RT, Jr., Harris PA, Hassell A, Holmes WD, Hunter RN, Lackey KE, Lovejoy B, Luzzio MJ, Montana V, Rocque WJ, Rusnak D, Shewchuk L, Veal JM, Walker DH, Kuyper LF. J. Med. Chem. 2001;44:4339–4358. doi: 10.1021/jm010117d. [DOI] [PubMed] [Google Scholar]
- [45.].Liu JJ, Dermatakis A, Lukacs C, Konzelmann F, Chen Y, Kammlott U, Depinto W, Yang H, Yin X, Chen Y, Schutt A, Simcox ME, Luk KC. Bioorg. Med. Chem. Lett. 2003;13:2465–2468. doi: 10.1016/s0960-894x(03)00488-8. [DOI] [PubMed] [Google Scholar]
- [46.].Hert J, Willett P, Wilton DJ, Acklin P, Azzaoui K, Jacoby E, Schuffenhauer A. Org. Biomol. Chem. 2004;2:3256–3266. doi: 10.1039/B409865J. [DOI] [PubMed] [Google Scholar]
- [47.].Klinkert MQ, Heussler V. Mini Rev. Med. Chem. 2006;6:131–143. doi: 10.2174/138955706775475939. [DOI] [PubMed] [Google Scholar]
- [48.].Rohwer A, Marhöfer RJ, Caffrey CR, Selzer PM. In: Drug Discovery in Infectious Diseases - Molecular Approaches towards Targeted Drug Discovery of Apicomplexan Parasites. Selzer PM, Becker K, editors. Vol. 1. WILEY-VCH Verlag GmbH & Co. KGaA; Weinheim: 2010. [Google Scholar]
- [49.].Chassaing C, Sekljic H. In: Drug Discovery in Infectious Diseases - Antiparasitic and Antibacterial Drug Discovery: From Molecular Targets to Drug Candidates. Selzer PM, editor. Vol. 1. WILEY-VCH Verlag GmbH & Co. KGaA; Weinheim: 2009. pp. 117–133. [Google Scholar]
- [50.].NCBI 2009 http://www.ncbi.nlm.nih.gov/
- [51.].Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52.].Salamov AA, Solovyev VV. Genome Res. 2000;10:516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53.].S. D. U. Accelrys Inc. Computer Program. 2003 [Google Scholar]
- [54.].Nicholas KB, Nicholas HB, Deerfield DW. “GeneDoc: Analysis and Visualization of Genetic Variation”. 1997 [Google Scholar]
- [55.].Ramachandran GN, Ramakrishnan C, Sasisekharan V. J.Mol.Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- [56.].Ihlenfeldt WD, Voigt JH, Bienfait B, Oellien F, Nicklaus MC. J. Chem. Inf. Comput. Sci. 2002;42:46–57. doi: 10.1021/ci010056s. [DOI] [PubMed] [Google Scholar]
- [57.].Oellien F, Cramer J, Beyer C, Ihlenfeldt WD, Selzer PM. J. Chem. Inf. Model. 2006;46:2342–2354. doi: 10.1021/ci060109b. [DOI] [PubMed] [Google Scholar]
- [58.].Sadowski J, Gasteiger J. Chem. Reviews. 1993;93:2567–2581. [Google Scholar]
- [59.].Morris GM, Goodsell DS, Huey R, Olson AJ. J. Comput. Aided. Mol. Des. 1996;10:293–304. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- [60.].Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- [61.].The Scripps Research Institute 2009 http://autodock.scripps.edu/faqs-help/manual/autodock-3-user-s-guide.
- [62.].Cambridge Cystallographic Database Center, Cambridge,UK 2008.
- [63.].Traxler P, Green J, Mett H, Sequin U, Furet P. J. Med. Chem. 1999;42:1018–1026. doi: 10.1021/jm980551o. [DOI] [PubMed] [Google Scholar]
- [64.].MADIN SH, Darby NB. Proc. Soc. Exp. Biol. Med. 1958;98:574–576. doi: 10.3181/00379727-98-24111. [DOI] [PubMed] [Google Scholar]
- [65.].Whitmire WM, Kyle JE, Speer CA, Burgess DE. Infect. Immun. 1988;56:2538–2543. doi: 10.1128/iai.56.10.2538-2543.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66.].Olson JA. Antimicrob. Agents Chemother. 1990;34:1435–1439. doi: 10.1128/aac.34.7.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67.].Ferby I, Blazquez M, Palmer A, Eritja R, Nebreda AR. Genes Dev. 1999;13:2177–2189. doi: 10.1101/gad.13.16.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.