

Abstract
The human immune response to an infection by Gram-negative bacteria involves detection of lipopolysaccharides (LPS), also known as endotoxins, which comprise the bacterial outer cell wall. Distinct from mammalian glycolipid structures, LPS have a conserved chemical pattern that is recognized by the pattern recognition receptor complex formed by myeloid differentiation protein 2 (MD-2) and toll-like receptor 4 (TLR4). A remarkable immune-mediated structure-toxicity relationship has been defined that relates to the number of acyl chains in the endotoxin. While there is a clear correlation between endotoxin acylation and elicited agonist or antagonist responses, the 3D structural basis of this relationship remains unclear. In order to explore, at atomic-resolution, the effects of a range of chemically distinct endotoxins on the structure and dynamics of their MD-2·endotoxin complexes, we chose to examine a series of variably acylated lipid A molecules from E. coli and N. meningitidis in complex with human MD-2. Through the application of molecular dynamics simulations, in concert with experimental data, we have identified specific structural and dynamic features of the MD-2-endotoxin complexes that may control dimerization of TLR4 molecules. As dimerization is central to the release of downstream chemical mediators, the results provide a structural foundation for the ability of endotoxins to act as either agonists or antagonists of the TLR4 pathway.
Keywords: endotoxin, MD-2, molecular dynamics simulations, lipid A, lipopolysaccharide, TLR4
1. Introduction
The ability to recognize and respond to an infection by Gram-negative bacteria involves detection of lipopolysaccharides (LPS) that comprise the bacterial outer cell wall. Distinct from mammalian glycolipid structures, a conserved chemical pattern of the LPS, mainly the lipid A and inner core oligosaccharide, bind specifically to the pattern recognition receptor complex of myeloid differentiation protein 2 (MD-2) and toll-like receptor 4 (TLR4) of the human innate immune pathway. This pathway initiates an acute inflammatory response whose downstream effects include the release of endogenous mediators such as cytokines and products of the complement and coagulation cascades (Stewart et al., 2006). Symptomatic responses to Gram-negative bacterial infections in humans range from mild (immunostimulation, pyrexia) to extreme (Gram-negative septic shock/multi-system failure) (Schletter et al., 1995). Decades of research on the innate immune response to LPS from Gram-negative bacteria has identified the relationship between the chemical structure of the LPS and the potency of the innate immune response (Erwin et al., 1991; Golenbock et al., 1991; Hadina et al., 2008; Kawahara et al., 2002; Mullarkey et al., 2003; Munford and Hall, 1986; Riedo et al., 1990; Teghanemt et al., 2005; Zimmer et al., 2007; Zughaier et al., 2006; Zughaier et al., 2005).
Modification of the chemical structure of an endotoxin (defined here as LPS, or precursors to LPS such as lipid A) can impact toxicity. For instance, there is a strong correlation between the number of acyl chains and the immunological response via the TLR4 pathway. Gram-negative bacteria can produce a range of differentially acylated endotoxins as a result of the LPS biosynthesis pathway (Raetz et al., 2007). In general, hexa-acylated endotoxins are agonists, whereas tetra-acylated species are antagonists with very weak endotoxin potentials. The immune response from penta-acylated forms lies somewhere between the two (Golenbock et al., 1991; Hadina et al., 2008; Kawahara et al., 2002; Mullarkey et al., 2003; Teghanemt et al., 2005; Zimmer et al., 2007). While we will focus here on effects related to the number of acyl chains, the linkage position of the secondary chains in the endotoxin can also modulate activity (Ernst et al., 2003). Despite the observed functional correlation between acylation and activity/toxicity, its origin, in terms of the 3D structure and dynamics of the MD-2·endotoxin complexes remains unclear.
In the case of endotoxins that act as agonists, the TLR4·MD-2·endotoxin complex dimerizes, initiating signaling via the interaction of the intra-cellular TLR4 domains (Hyakushima et al., 2004; Saitoh et al., 2004). For clarity, herein we will refer to the TLR4 molecule in the initial TLR4·MD-2·endotoxin complex as simply TLR4, whereas the second TLR4 molecule to interact with MD-2 (as a consequence of dimerization) will be denoted TLR4* (Fig. 1).
Figure 1.
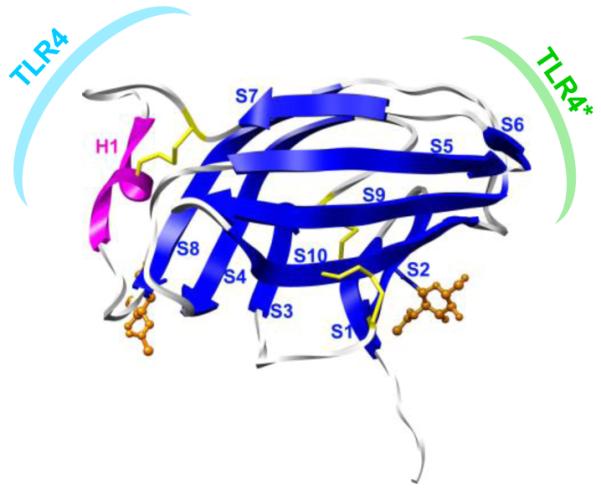
Structure of human MD-2 (2E56): helix H1 (magenta), β-strands S1-S10 (blue), two N-glycans (orange) and three disulfides (yellow). Also noted are the binding surfaces associated with monomeric TLR4·MD-2 (cyan) and with TLR4 dimerization (green).
Antagonists do not induce TLR4 dimerization (Saitoh et al., 2004), despite formation of the TLR4·MD-2·endotoxin complex (Gioannini et al., 2004; Saitoh et al., 2004), and subsequently, they do not lead to an increased release of downstream mediators. In addition, endotoxin antagonists competitively inhibit agonist from signaling via the TLR4 pathway (Saitoh et al., 2004), presumably by preventing TLR4 dimerization. While endotoxin clearly has a role in regulating TLR4 dimerization, variations in the MD-2 sequence can also attenuate dimerization, despite the formation of a MD-2·endotoxin complex. Thus, both the structure of MD-2 and of the endotoxin mediate dimerization (Kobayashi et al., 2006; Saitoh et al., 2004).
Recently, crystal structures of MD-2 in complex with various molecules, including TLR4 fragments, endotoxins, and synthetic endotoxin antagonists have been reported (Kim et al., 2007; Ohto et al., 2007; Park et al., 2009). These structures (determined under cryo-cooled conditions) suggest that the binding mode of the endotoxins in the complexes is remarkably variable, as demonstrated by the opposing orientations of the lipid chains in structures 2E59 and 3FXI, which we denote here as directions “a” and “b”, respectively. Taken together, these complexes provide a basis for relating the 3D structural features of the MD-2·endotoxin complexes to activity. However, there are still missing pieces to the puzzle, including structures of intermediates that would relate a fully agonistic complex like MD-2·LOS to antagonistic complexes such as, MD-2·lipid IVa and MD-2·eritoran, and thus reveal specific 3D structural requirements for activity.
In order to explore the effects of a range of chemically distinct endotoxins on the structure and dynamics of the MD-2-endotoxin complex, we chose to examine a series of variably acylated E. coli and N. meningitidis lipid A molecules in complex with MD-2 (Fig. 2). The internal plasticity of the glycolipid ligands, even when bound to MD-2, poses challenges for atomic-resolution characterization of both the structural and dynamic properties of these complexes under physiological conditions. In order to characterize such variable and dynamic systems under physiologically relevant conditions, we have employed all-atom, explicit solvent molecular dynamics (MD) simulations. A total of 13 simulations were performed: six complexes of MD-2 with N. meningitidis-type endotoxins, six complexes of MD-2 with E. coli-type endotoxins, and one simulation of MD-2 without a ligand (Table 1). The crystallographic data served as critical reference structures for preliminary validation of the computational simulations. The simulations enabled the identification and characterization of the 3D structural and dynamic features of MD-2 and associated endotoxins, which control TLR4 dimerization. Furthermore, differences in the surface structure of the MD-2·endotoxin complex were quantified and correlated with the ability of variably acylated endotoxins to activate the innate immune system via the TLR4 pathway.
Figure 2.
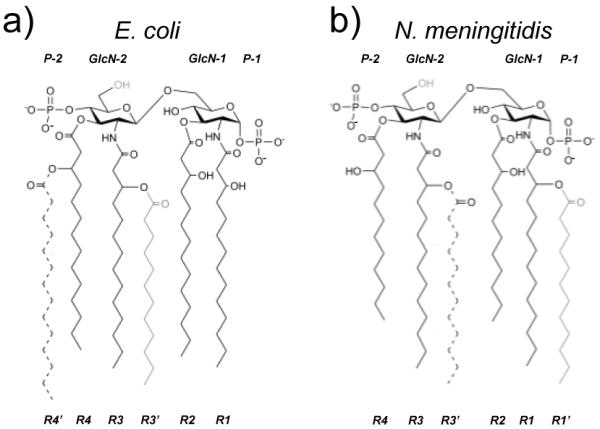
Endotoxin structures from (a) E. coli and (b) N. meningitidis: lipid IVA (solid black chains), penta-acylated (addition of gray chain), and hexa-acylated (addition of dashed chain).
Table 1.
Simulation legend of MD-2-endotoxin complexes where letters a and b denote the two opposing orientations of the ligand in the MD-2 binding site.
| Ligand | tetra-acylated | penta-acylated | hexa-acylated |
|---|---|---|---|
| E. coli | Ec4a, Ec4b | Ec5a, Ec5b | Ec6a, Ec6b |
| N. meningitidis | Nm4a, Nm4b | Nm5a, Nm5b | Nm6a, Nm6b |
2. Materials & Methods
2.1 Modeling
In total 13 MD simulations of the glycoprotein MD-2 were performed, 12 of which were bound to lipid A variants (Table 1), totaling over 0.650 μs of simulation time (50 ns for each production run). Two of these complexes were treated as controls, using crystallographic MD-2·endotoxin structures as reference states; namely MD-2 bound to E. coli lipid IVa (PDB ID: 2E59) (Ohto et al., 2007) and lipid A (PDB ID: 3FXI) (Kim et al., 2007). The lipid A structure was modeled from the LOS structure in 3FXI by removing the core oligosaccharide domain from the LOS, leaving the binding interface between the lipid A portion of the LOS and MD-2 unchanged. Human MD-2 is N-glycosylated at Asn26 and Asn114; however, prior to crystallization trials these glycans were either removed or truncated by treatment with PNGase F (3FXI) or endoglycosidase H (2E59), respectively. Crystal structure 2E59 includes coordinates for residues 17-160 and the core GlcNAc for glycans at Asn26 and Asn114; whereas 3FXI includes coordinates for residues 19-158 and the core GlcNAc at Asn114. Both structures span the structured region of MD-2 (residues 22-155) and differ only in the length of the unstructured N- and C-termini. To assess the ability of MD to realistically model MD-2·endotoxin complexes, simulations of MD-2·lipid A and MD-2·lipid IVa, were run using their respective crystal structures as starting structures, including the core GlcNAc residues. These control simulations of E. coli lipid IVa and lipid A in complex with MD-2 are referred to as “Ec4a” and “Ec6b”, respectively. In this nomenclature system, the first two letters denotes the bacterial origin of the endotoxin, the number is the number of acyl chains, and “a” or “b” denotes the two possible orientations of the lipid in the MD-2 complex, as observed experimentally. In the “a” direction the P-1 phosphate group of the endotoxin is oriented towards the TLR4 interaction site, whereas in “b”, it is oriented towards the face of MD-2 involved in dimerization (Fig. 1).
The six simulations of MD-2 bound to N. meningitidis lipid A-variants included: tetraacylated (Nm4a and Nm4b), penta-acylated (Nm5a and Nm5b) and hexa-acylated (Nm6a and Nm6b) lipid A. The six simulations of MD-2 bound to E. coli lipid A-variants included: tetraacylated (Ec4a and Ec4b), penta-acylated (Ec5a and Ec5b) and hexa-acylated (Ec6a and Ec6b) lipid A. The Ec4a complex (from 2E59) was used to generate models of Nm4a, Nm5a, Nm6a, Ec5a, and Ec6a complexes, whereas the Ec6b complex (from 3FXI) was used to model the Ec4b, Ec5b, Nm4b Nm5b and Nm6b complexes. As the directionality of binding of penta-acylated endotoxins to MD-2 is unknown, and that of hexa-acylated endotoxins is unclear, all pentaacylated and hexa-acylated endotoxin MD-2 complexes were modeled in both “a” (lipid IVa) and “b” (LOS) directions. Although the crystal structures of MD-2 bound to tetra-acylated species (lipid IVa and eritoran) indicate the directionality of binding is in the a-direction, for comparative purposes, and to assess the dependence of the position of the ligand throughout the simulation on its initial orientation in the binding pocket of MD-2, the b-direction complex was also simulated.
Lipid A from N. meningitidis has six symmetric acyl chains that are 12 carbons in length, whereas the acyl chains of E. coli lipid A are asymmetric with four 12 carbon and two 14 carbons chains (Fig. 2). Therefore, initial models of lipids Nm4a, Nm6b were generated by truncation of the extra carbons from the crystal structures of Ec4a and Ec6b, respectively. To model Ec4b, chains R3′ and R4′ were removed from the Ec6b structure; for Ec5b, chain R4′ was removed. For Nm5b, chain R3′ was removed from the Ec6b structure, along with the extra carbons on acyl chains R2 and R4. For Nm4b, chains R1′ and R3′ were removed from the Ec6b structure, along with the extra carbons on acyl chains R2 and R4. For Ec5a, Ec6a, Nm5a and Nm6a, the Ec4a structure was used as a template, the appropriate additional chains were threaded into available space within the binding pocket and for Nm-models, extra carbons were removed from R2 and R4 (Fig. 2). To identify and remove any potential high-energy interactions caused by threading of the additional acyl chains, energy minimization in vacuo was performed. First, minimization of only the acyl chains of the endotoxin in the MD-2 complex was performed (250 steps of steepest decent (SD), 250 steps of conjugate gradient (CG)). This was followed by minimization (250 steps of SD, 250 steps of CG) that permitted flexibility in the positioning of the protein side-chains, by holding the positions of the Cα atoms from the protein and the carbohydrate domain of the endotoxin fixed.
Lastly, a simulation of apo MD-2, that is MD-2 with no ligand bound was performed. The starting structure for this simulation was a crystal structure of purified MD-2 from P. pastoris that contains no endotoxin ligand in the binding site; however the crystal structure (PDB ID: 2E56) contains three myristic acid molecules in the binding pocket. For the simulation, the myristic acid molecules were removed from the structure. The apo MD-2 structure contains two N-linked N-acetylglucosamines (Asn26 and Asn114) and is composed of 144 amino acids (res. 17-160).
2.2 MD Protocol
Beginning with the 13 starting structures described above (see Table 1), the PTRAJ program in AMBER was used to add TIP3P water molecules to a solvent depth of at least 10 Å from any protein, carbohydrate or lipid atom, and counter ions were added to neutralize the system (28). The apo MD-2 and the 12 MD-2 complexes were simulated with the three native disulfide bonds (Cys25-Cys51, Cys37-Cys148, Cys95-Cys105) intact (Fig. 1). The subsequent minimization and MD steps were performed using AMBER 11 (28) with the AMBER 99sb parameters (Hornak et al., 2006) for the protein and the GLYCAM06 (version f) parameters for carbohydrates (Kirschner et al., 2008) and lipids (Tessier et al., 2008). In order to maintain compatibility with the protein and carbohydrate parameter sets, the partial charges on the acyl chains were derived in a protocol consistent with that employed previously for the simulation of ceramides with GLYCAM (DeMarco and Woods, 2009). Consistent with the GLYCAM development philosophy (DeMarco and Woods, 2009), partial atomic charges were derived for the free acyl chains, and these substituents were then combined with the core oligosaccharide to generate the intact endotoxins. In the charge calculations the carbohydrate attachment points in the acyl chains were truncated with –OCH3 or –NHCH3 groups, where the carbon represents C3 or C2 of the glucosamine ring in the intact endotoxin, respectively. For charge calculations involving the secondary acyl chains, partial charges were fit to the chain up to the branching oxygen, which was capped with a methyl group. For all acyl chains, an ensemble of ten staggered conformations was generated and each structure was geometry optimized using the HF/6-31G(d) basis set using the Gaussian 03 package (Frisch et al., 2004). Charge fitting was performed at the HF/6-31G* level using the RESP program (Bayly et al., 1993) and a restraint weight of 0.010 (Kirschner et al., 2008). Charges for each conformation were then averaged to derive the ensemble-averaged charge sets that attempt to account for the effect of chain flexibility on charge distribution. When combining chains, overall charge neutrality was maintained by adjusting the partial charges on the connecting atoms, as required. GLYCAM partial charges were employed for the monosaccharide residues. Structure files with all partial atomic charges are available at www.glycam.org (Fig. S1).
System equilibration was performed using the AMBER 11 version of SANDER in the following stepwise protocol. 1) Water molecules were energy minimized (500 steps of SD, 500 steps of CG) and then subjected to 10 ps of MD in the isothermal-isobaric (NPT) ensemble at 1 atm with anisotropic pressure scaling. The water molecules were again energy minimized (500 steps of steepest decent, 500 steps of conjugate gradient). 2) For complexes Ec4a, Nm4a, Ec6b, and Nm6b, the glycoprotein and ligand were then minimized (5000 steps of SD, 5000 steps of CG) holding waters fixed, and then the whole system was minimized (500 steps of SD, 500 steps of CG). 3) For complexes Ec4b, Nm4b, Ec5a, Ec5b, Nm5a, Nm5b, Ec6a, and Nm6a, which required addition or deletion of acyl chains to create the starting structures, minimization of the glycoprotein was performed as described above, in addition to holding waters fixed, with Cα, endotoxin ring-atoms restrained (5000 steps of SD, 5000 steps of CG), followed by an energy minimization releasing the waters (500 steps of SD, 500 steps of CG). 4) For Ec4a, Nm4a, Ec6b, and Nm6b, the system was heated to 37°C over 50 ps in the NPT ensemble at 1 atm with anisotropic pressure scaling, as the last step prior to the production run. 5) For complexes Ec4b, Nm4b, Ec5a, Ec5b, Nm5a, Nm5b, Ec6a, and Nm6a, the system was subjected to 50 ps of MD as described above, but holding Cα atoms and the endotoxin carbohydrate-domain fixed. 6) All-atoms in the system (Ec4b, Nm4b, Ec5a, Ec5b, Nm5a, Nm5b, Ec6a, and Nm6a) were then heated in the NPT ensemble at 1 atm with anisotropic pressure scaling, to 37°C over 80 ps.
The production MD simulations of all systems were performed for 50 ns at 37°C in the NPT ensemble at 1 atm. Version 11 of AMBER permits the use of mixed scaling for non-bonded 1-4 van der Waals and electrostatic interactions, as appropriate for the molecule of interest. For the endotoxin and the glycans of the protein, no 1-4 scaling was used (SCEE = SCNB = 1), as is standard when using the GLYCAM parameters. For the protein, 1-4 van der Waals and electrostatic interactions were scaled (SCEE = 1.2 and SCNB = 2.0), as is appropriate with the use of AMBER 99sb force field parameters. A 2 fs time-step was employed throughout for integrating the equations of motion. Hydrogen-containing bonds were constrained with the SHAKE algorithm (Ryckaert et al., 1977) and long range electrostatics were treated using the particle mesh Ewald method (Darden et al., 1993).
2.3 Analysis
Cα root mean square deviation (RMSD), B-factor, root mean square fluctuations (RMSF), and secondary structure data were calculated at 10 ps intervals, and solvent accessible surface area (SASA) at 100 ps intervals. Secondary structure was computed using the DSSP algorithm (Kabsch and Sander, 1983). SASA was computed using NACCESS (Hubbard and Thornton, 1993). Apolar SASA of the endotoxin in the MD-2 complex was computed considering only the SASA of the apolar atoms from the acyl chains. In order to compare the simulations, data requiring ensemble averaging were reported over a timeframe (5-50 ns) where all simulations had equilibrated according to the Cα RMSD data. Where appropriate, analyses were reported over the structured region of the glycoprotein (res. 22-155). Principal component analysis (PCA) was calculated using Dynatraj (Barrett et al., 2004) at 90 ps intervals, and required the use of GROMACS tools (Spoel et al., 2005) for conversion from an AMBER trajectory and VMD (Humphrey et al., 1996) to display the porcupine plots. All other molecular graphics images were produced using UCSF Chimera package (Pettersen et al., 2004).
3. Results
3.1 Structure
3.1.1 Secondary and Tertiary Structure
Apo MD-2 and two MD-2 complexes with E. coli-type ligands: Ec4a (lipid IVA) and Ec6b (lipid A), were simulated as controls, initiated with the experimental crystal structures. These studies were extended to include both orientations of tetra-, penta-, and hexa-acylated E. coli and N. meningitidis lipid A variants bound to MD-2 (Table 1). Global stability of MD-2 over the course of the 13 simulations was monitored via Cα RMSD’s (Fig. S2), secondary structure analysis (Fig. S3) and by visual inspection (Fig. 3). According to the Cα RMSD plots, the simulations required on the order of 1 to 5 ns of equilibration time. To facilitate comparison of all the simulations, we assigned a 5 ns equilibration period to all trajectories. In addition to global structure, local structure was well maintained. Secondary structure analysis showed that native α-strands were preserved (Fig. S3). The lone helical structure, a native 4-residue long helical turn (residues 103-106 in the crystal structures 2E59 and 3FXI, as measured by the DSSP algorithm) was maintained and flickered between 310-helical and turn conformation, with the exception of Ec6b. In Ec6b, a helix-like turn is still visibly present but the structure has relaxed over the course of the simulation and the DSSP algorithm no longer considered it either a 310-helix or a turn. By overlaying snapshots from the simulation at 10 ns intervals secondary and tertiary structure appeared well maintained, with most structural fluctuations occurring in the loop regions, the helical turn and the unstructured N- and C-termini (Fig. 3). Overall good agreement was noted between the crystallographic control structures and the data from the MD simulations.
Figure 3.

Overlay of snapshots from the simulations of MD-2 at 10 ns granularity, relative to the starting structure. Snapshot in ns: 0-yellow, 10-orange, 20-red, 30-magenta, 40-purple, 50-blue. Ligand and glycans removed for clarity.
3.1.2 Quaternary Structure
While the secondary and tertiary structure of MD-2 remained largely independent of the acylation of the bound endotoxin, significant changes in quaternary structure of the complexes was observed. Changing the number of acyl chains, particularly for endotoxins that bound in the “b” orientation, affected the composition of the surface where the complex interacts with dimerizing TLR4*. In all simulations, the endotoxin remained in the binding cavity and fluctuated about, or slightly drifted from, its initial position (Fig. 4). For hexa-acylated species, regardless of the starting orientation, ligands remained (Nm6b, Ec6b) or moved (Nm6a, Ec6a) towards the TLR4* interaction face. As such, the TLR4* interface consisted of both MD-2 and endotoxin surfaces; this in comparison to tetra-acylated species where MD-2 largely occluded the endotoxin from the TLR4* interaction face (Fig. S4). For Ec4b and Nm4b complexes, where the simulations were initiated with the ligands at the TLR4* site, they migrated towards the center of the binding pocket resulting in a TLR4* interaction sites similar to Ec4a and Nm4a. For penta-acylated ligands, we observed both occluded and partially exposed acyl chain conformations at the TLR4* interaction face (Figs 5 and S3).
Figure 4.

Motion of the endotoxin within the binding pocket of MD-2. Snapshots of endotoxins at 10 ns granularity, complexes were aligned by Cα atoms to the starting structure (colored as in Fig. 3).
Figure 5.

Relationship between the volume of the endotoxin and its ensemble average apolar SASA when bound to MD-2. Boxed areas represent antagonist (lower left) and agonist (top right) regions.
To quantify differences in surface composition of MD-2αendotoxin complexes, exposed apolar surface areas of the acyl chains of the bound endotoxin were computed and compared against the molecular volume of the endotoxin (Fig. 5). Ensemble average apolar SASA (αapolar SASAα) for the tetra-acylated species clustered together because all structures converged on a solvent occluded conformation (similar to the crystal structure of Ec4a) (Fig. S4). By contrast, apolar surfaces of the hexa-acylated species were partially accessible to solvent in the complexes (as seen in the crystal structure of MD-2·LOS). Penta-acylated species populated both solvent exposed and occluded conformations; those complexes where the ligand was positioned initially to be solvent accessible (b-direction) remained solvent accessible, and those started as solvent occluded (a-direction) remain occluded.
The correspondence between the final positions of the penta-acylated endotoxins in complex with MD-2, and on their respective starting conformations (a or b-directions), were noted. To exclude the possibility that the Ec5b and Nm5b structures observed during the MD simulations where the endotoxin is solvent accessible at the TLR4* interface were artifacts of their starting conformations, the Ec4b, Nm4b complexes were simulated. These represented endotoxins positioned near the TLR4* interface and not in experimentally defined starting structures. Over the course of the simulation, the endotoxin moved away from the surface, into the interior of the binding pocket (Fig. S5). Conversely, the penta- and hexa-acyled ligands docked in the b-direction maintained greater apolar SASA, with one acyl chain lying at the protein surface, demonstrating volume-dependent (and not starting structure-dependent) positioning of the ligand within the binding pocket (Fig. 5). To further support this size selection, the large ligands Ec6a and Nm6a that were initially positioned within the MD-2 binding pocket moved towards the TLR4* interface during the MD simulations, resulting in increased apolar SASA (Fig. S5).
3.2 Dynamics
The degree of structurally pliability of the MD-2 receptor, while in complex with various endotoxins (Fig. 3) was quantified by examining calculated and observed B-factors (Fig. S6). As seen in the DSSP analysis, the loops and unstructured segments between secondary structure elements correlated with regions with highest B-factors in the crystal structures. Regions, that across all simulations, had consistently relatively high structural plasticity included residues 38-43, 81-88, 96-101, 122-128, and 140-144. Dynamic regions in the bound ligand were examined by calculating fluctuations of individual components of the endotoxin relative to the average structure of the endotoxin (by first aligning the system by Cα RMSD of the protein backbone) (Table 2). Comparisons in Table 2 are best made within the same species as their acyl chains have the same number of atoms. In addition to the carbohydrate domain displaying some flexibility, the position of the acyl chains fluctuated within the binding pocket (Table 2).
Table 2.
Fluctuations of the carbohydrate and lipid domains of the endotoxin in the MD-2 complex.
| RMSF values (Ǎ) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Carbohydrate domaina |
Acyl chains |
||||||||||
| P-1 | GlcN-1 | GlcN-2 | P-2 | R1 | R2 | R3 | R4 | R1′ | R3′ | R4′ | |
| Ec4a | 1.56 | 1.19 | 1.24 | 1.62 | 1.88 | 1.87 | 1.61 | 1.70 | |||
| Ec4b | 4.47 | 3.05 | 2.65 | 2.86 | 2.36 | 2.45 | 2.52 | 2.00 | |||
| Ec5a | 2.01 | 1.54 | 1.51 | 1.36 | 1.48 | 1.35 | 1.46 | 1.43 | 1.52 | ||
| Ec5b | 3.80 | 3.21 | 3.45 | 3.94 | 2.86 | 2.09 | 2.10 | 2.62 | 2.47 | ||
| Ec6a | 2.47 | 1.64 | 2.03 | 3.88 | 3.04 | 2.64 | 1.61 | 2.27 | 4.16 | 1.46 | |
| Ec6b | 3.44 | 2.17 | 2.52 | 3.18 | 2.91 | 1.50 | 1.57 | 1.95 | 2.74 | 1.57 | |
| Nm4a | 1.39 | 1.08 | 1.07 | 1.32 | 1.42 | 1.40 | 1.70 | 1.99 | |||
| Nm4b | 1.56 | 1.26 | 1.37 | 1.75 | 1.68 | 1.41 | 1.61 | 1.64 | |||
| Nm5a | 2.00 | 1.49 | 1.45 | 1.59 | 1.65 | 1.69 | 1.57 | 1.71 | 1.73 | ||
| Nm5b | 2.30 | 1.50 | 1.29 | 1.50 | 1.81 | 2.32 | 2.26 | 1.81 | 2.53 | ||
| Nm6a | 1.47 | 1.20 | 1.49 | 1.87 | 1.10 | 1.73 | 1.77 | 1.97 | 1.82 | 1.76 | |
| Nm6b | 1.92 | 1.39 | 1.46 | 2.66 | 1.88 | 1.44 | 1.69 | 1.73 | 2.58 | 2.06 | |
RMSF’s were calculated after aligning the system by the Cα RMSD of the protein backbone
PCA was used capture and visualize large-magnitude low-frequency motions for each trajectory. This type of PCA, also known as essential dynamics, can identify correlated motions along the protein backbone and rank these sets of correlated motions according to their magnitude. For apo MD-2 and the MD-2·endotoxin complexes, the five largest magnitude eigenvectors were evaluated and found to display a common pattern of motion in all trajectories, and indicated correlated motions from opposite ends of the protein. In the region of the TLR4 interaction site (Fig. 1), H1 as well as the loop preceding H1 and in some cases the loop between S9 and S10, acted as a spring, slightly coiling and uncoiling. This plasticity can also be seen in Fig. S3, as H1 frequently fluctuated between 310-helix and turn conformations. On the other end of MD-2, the loops between S5-S6 and S8-S9, and part of the adjacent α-strands (see the TLR4* interaction site as denoted in Fig. 1) moved in concert with the H1 region, to open and close about the ligand (Fig. 6, S7). Note that strands S5 and S9 connect from either end of H1. While this pattern was observed in each trajectory, the magnitude of this motion varied from trajectory to trajectory (Fig. S7). There were no discernable trends in the magnitude of the proteins motions based on the size, or directionality of the bound ligand.
Figure 6.
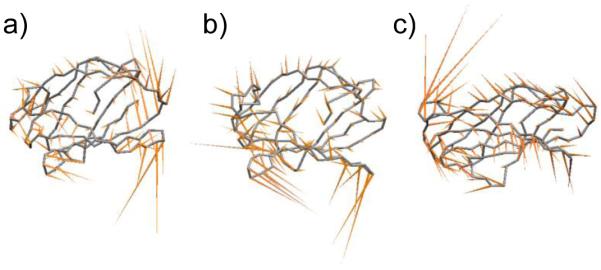
Correlated and endotoxin-independent motions of MD-2. Lowest frequency mode of MD-2 for (a) Ec6b, and (b) Nm6b, and (c) apo, mapped onto the average backbone structure from simulation.
In addition to exploring the effects of differential endotoxin acylation on MD-2 structure and dynamics, the behavior of the bound endotoxins was also analyzed. Both the phospho-glycan and acyl chains showed mobility within the binding pocket. When bound to MD-2, the endotoxin is flanked on one edge by H1 and on the other by the gap between the α-sheets (Fig. 4). The acyl chain nearest the TLR4* interaction site (R4 for the a-direction and, R1′ or R1 for the b-direction) was consistently more mobile than the other acyl chains (see Table 2 and Fig. 4). While the majority of interactions between the endotoxin and protein comprised non-specific hydrophobic contacts between the lipid chains and the hydrophobic binding cavity formed by the α-sandwich, the phospho-glycan and the hydrophilic groups (carbonyl and hydroxyl) of the lipid chains did form hydrogen bonds with the hydrophilic lip of the binding pocket.
4. Discussion
4.1 Modeling MD-2·Endotoxin Complexes
To first assess the ability of MD simulations to model biologically relevant MD-2·endotoxin complexes, we performed three control simulations (apo, Ec4a, Ec6b) initiated with crystallographic structures of MD-2. In all three simulations, the secondary and tertiary structures of MD-2 were preserved over the course of the simulations (Fig. 3, S2, S3). For control simulations of MD-2 bound to E. coli-type endotoxins (Ec4a and Ec6b), the ligands, while dynamic, remained localized in the binding pocket (Fig 5a and j). Based on the ability of these controls to realistically model the structure of MD-2 and MD-2·endotoxin complexes, 10 other models of the E. coli and N. meningitidis lipid A-variants in complex with MD-2 were assembled and simulated (Table 1). In general, the N. meningitidis and E. coli complexes shared similar behaviors including: maintenance of secondary and tertiary structure of MD-2, and the dynamic behavior of the ligand, while remaining in the binding pocket, as compared to the control simulations (Fig. 3, 4, S2, S3).
4.2 How MD-2 Acts as a Pattern-Recognition Receptor
MD-2 is known as pattern-recognition receptor (Poltorak et al., 1998; Shimazu et al., 1999); it can recognize a host of chemically related LPS molecules by their common phosphoglucosamine core and acylation pattern (Fig. 2). The ability to accommodate such a broad range of acylation patterns is achieved by the apolar interior surface of the MD-2 binding cavity, allowing non-specific MD-2·endotoxin contacts. There are several modes in which the acyl chains of the endotoxins are able to occupy the large hydrophobic binding pocket (Kim et al., 2007; Ohto et al., 2007; Park et al., 2009). The simulations indicated that once bound, the lipid tails nevertheless remained dynamic (Fig. 4). This lack of specificity reflects part of the pattern recognition (a hydrophobic component) by MD-2 and allows the recognition of endotoxins with various numbers, distributions and lengths of acyl chains. Relaxed binding specificity has been observed for other glycolipid-binding proteins including GM2-activator protein, which can recognize a host of chemically distinct glycolipids. Its relaxed binding specificity arises from the non-specific apolar interactions of the lipid tails within the binding pocket. This tolerance for variations in ligand composition is augmented by the flexibility of the protein itself (Wright et al., 2005). The same can be said of MD-2, whose crystal structures bound to myristic acid, lipid IVa, LOS (Ohto et al., 2007; Park et al., 2009) and the antagonist eritoran (Kim et al., 2007) demonstrate a high degree of heterogeneity in placement of the lipid tails within the binding pocket. Moreover, the MD simulations provided additional insight into the role played by glycolipid acylation in modulating the dynamics of MD-2 (Fig. 3, S7), as will be discussed in greater detail below.
Despite accommodating a series of E. coli and N. meningitidis ligands, no gross changes in secondary and tertiary structure of MD-2 were observed (Fig. 3, S3). This result is consistent with experiments using radiolabeled endotoxins showing tetra-, penta- and hexa-acylated N. meningitidis endotoxins formed monomeric complexes with soluble MD-2 (Teghanemt et al., 2005). The simulations demonstrated that MD-2 was able to accommodate the various endotoxins without significant structural changes, particularly on the edge of the protein that interacts with the initial TLR4 molecule (Fig. 1). This finding is consistent with the formation of the TLR4·MD-2 complex in the presence or absence of endotoxin, and the formation of the TLR4·MD-2·endotoxin complex, independent of the acylation pattern of the endotoxin (Coats et al., 2005; Gioannini et al., 2005); indicating at least a portion of the 3D structure of the MD-2 is preserved upon binding a variety of endotoxins.
4.3 Volume-Dependent Localization of Endotoxins within MD-2
4.3.1 Human MD-2
The secondary and tertiary structure of MD-2 was largely independent of the acylation of the bound endotoxin; however, significant differences in the quaternary structure of the complexes were observed. Most notably, the region of the TLR4* interface varied based on the degree of acylation of the bound endotoxin (Fig. S4). Increasing the volume of the ligand by the addition of lipid chains resulted in increased exposure of apolar surfaces in the complex (Fig. 5, 7, S8). Consistent with the crystal structure of E. coli lipid IVa with MD-2, in the MD-2 simulations the lipid domains of tetra-acylated endotoxins were encapsulated by MD-2 and revealed little apolar SASA (Fig. 5). On the other hand for agonists Nm6b and Ec6b, one acyl chain remained at the MD-2 surface, as with the E. coli LOS·MD-2·TLR4 dimer crystallographic structure, and contributes to the MD-2·TLR4* interface. In the absence of TLR4*, we found that these conformations are not strictly TLR4* dependent and that acyl chain-volume guides placement of the endotoxin in the binding pocket. Furthermore, by modeling the Ec4b, Nm4b, Ec6a and Nm6a complexes, it was shown that the endotoxins were able to translate within the binding pocket over the timescale of the simulations (Fig. S5), as such, the positions of the endotoxins were not fixed by the initial starting conformations chosen for the simulations, but influenced by the size of the lipid domain of the endotoxin.
Figure 7.

A change in the number of acyl chains of an endotoxin modifies the TLR4* interaction face. Complexes taken from the 30 ns snapshots of Nm4a, Nm5b and Nm6b simulations (examples from all simulations can be found in Fig. S4). MD-2 (blue) with residues within 3.5 Å of TLR4* in the crystal structure 3FXI highlighted in green, and the endotoxin (phospho-glycan domain, magenta; acyl chains, grey).
While there exists data for the tetra- and hexa-acylated species, the position of a ligand within the binding cavity of MD-2 with intermediary activity, such as penta-acylated endotoxins is unknown. In the MD simulations, both occluded and partially exposed acyl chain conformations were present, suggesting that these endotoxins form between antagonist (Ec4a/b, Nm4a/b) and more agonistic-type (Ec6a/b, Nm6a/b) complexes with MD-2, consistent with assays that demonstrate their moderate potency (Teghanemt et al., 2005). The simulations revealed that unlike E. coli-type hexa-acyled endotoxins, the presence of TLR4* may be required to stabilize near-surface conformations of penta-acylated endotoxins. In general, in the absence of TLR4*, maintaining an acyl chain near the surface of MD-2 is unfavorable unless the binding site is essentially filled. Transient near-surface conformations of weak agonists may allow TLR4 dimerization, and subsequently initializing signaling; albeit to a lesser extent than hexa-acylated lipid A complexes that consistently present greater apolar SASA.
The observed relationship between acyl chain volume, apolar SASA and the activity of an endotoxin, correlates with mechanism employed by bacteria to evade immune detection. In patients with N. meningitidis infections, a mutation in the endotoxin synthesis pathway produces hypo-acylated species, which correlates with tempered immune responses (Fransen et al., 2009). These in vivo findings have been reproduced in vitro via enzymatic deacylation of LPS from E. coli, Haemophilus influenza, N. meningitidis, and Salmonella typhimurium, which results in attenuated toxicity (Erwin et al., 1991; Munford and Hall, 1986; Riedo et al., 1990; Teghanemt et al., 2005).
With a clearer understanding of the molecular mechanisms that drive TLR4 dimerization we can better predict the toxicity of an endotoxin based on chemical modifications of the lipid domain. In the case of N. meningitidis and E. coli endotoxins (tetra- to hexa-acylated lipid A variants), an antagonist can be defined as having an acyl chain volume of less than 1100 Å3 and an αapolar SASAα of less than 130 Å2, whereas an agonist has an acyl chain volume greater than 1400 Å3 with an αapolar SASAα greater than 130 Å2 (Fig. 5). A weakly agonistic endotoxin, would lie between the two and be capable of forming complexes with apolar SASA similar to both antagonist- and agonist-type structures (Fig. 5, 7). Quantification of this relationship provides a testable hypothesis that may advance the rational development of endotoxin-based therapeutics and vaccine adjuvants.
4.3.2 Murine MD-2
From crystallographic data, it is known that antagonists such as lipid IVa or eritoran do not reside at the surface of MD-2, but bury themselves in the binding pocket (Kim et al., 2007; Ohto et al., 2007). The MD simulations demonstrated that the binding site needed to be mostly filled to sterically force a hydrophobic acyl chain to reside at the surface of the MD-2·endotoxin complex (Fig. 5). Interestingly, tetra-acylated species such lipid IVa (Ec4a/b, Nm4a/b) are known as strong antagonists but can become agonists when murine MD-2 is substituted for human MD-2 in the TLR4 pathway. Residues 57, 61 and 122 are critical for this switch in toxicity (Muroi and Tanamoto, 2006). By mutating several residues in murine MD-2 to the corresponding amino acid in the human sequence, or to alanine, the following mutations were found to critical in controlling toxicity: T57S/A, V61L/A and E122K/A (Muroi and Tanamoto, 2006). The role of residue 122 can be accounted for by its proximity to the TLR4* interaction surface (Park et al., 2009) and the non-conservative E→K mutation. Altering the charge of an amino acid in this region disrupts the surface complementarity with the binding site on the corresponding species’ TLR4* (Walsh, et al, 2008). On the other hand, both residues 57 and 61 are found deep in the MD-2 binding cavity and present no obvious role in assisting TLR4* binding. The results presented here suggest a plausible mechanism for their role in signaling via modulation of shape of the binding pocket. Further simulations of MD-2·endotoxin complexes could be studied to assess the result of mutations of residues 57, 61 and 122 on the shape of the binding pocket. It is conceivable that, given the importance of the volume of the binding pocket on acyl group exposure, mutations in the binding cavity may result in the endotoxin being relocated closer to the TLR4* interaction surface, thus presenting an agonist-like surface to TLR4*. This would be consistent with the view here that the location of the acyl chains within MD-2, and with respect to the TLR4* interaction face, is an important determinant of TLR4 dimerization.
4.4 Role for Dynamics in Function and Activity of MD-2
Interestingly, the regions with the highest degree of plasticity across all trajectories, residues 81-88, 96-101 and 122-128, corresponded to TLR4 interfaces (Fig. 8). Employing PCA, we found that the dynamic motions of these three segments were correlated: loop-H1 (res. 96-101) compresses and decompresses as the loops between S5-S6 (res. 81-88) and S8-S9 (res. 122-128) open or close about the binding pocket (Fig. 6, 8). The presence of these motions in apo MD-2 and all liganded complexes, suggests that this is an inherent motion of the protein that is independent of the chemical structure of the bound ligand. This dynamic behavior accounts for the ability of MD-2 to accommodate endotoxins with a variable number, length and distribution of lipid chains.
Figure 8.
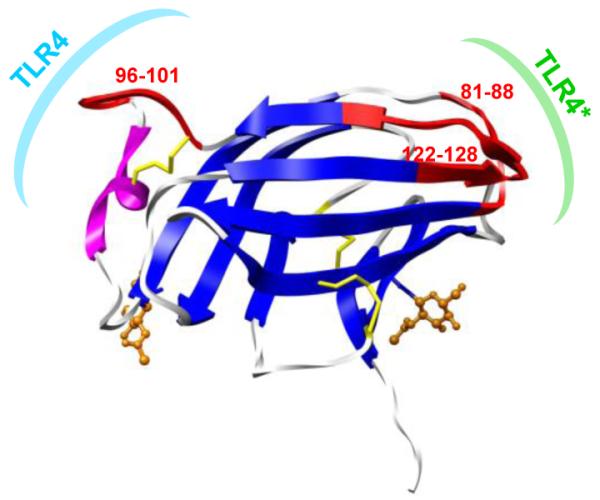
Regions of MD-2 that experience the highest positional fluctuations in all simulations, mapped to the structure of MD-2 (red), correspond to surfaces that interact with TLR4 molecules. These regions also correspond to key dynamic segments identified via PCA.
In addition, the opening of the α-sandwich in MD-2 (at the TLR4* interface) creates a direct channel to the binding pocket (Fig. 9, S7), which could facilitate the transfer of endotoxin into MD-2. Endotoxins are recognized and transferred sequentially by lipid binding protein, to CD14 and then to cell-surface MD-2. This serial transfer of endotoxin and recognition by MD-2 is observed for both antagonist and agonist endotoxins of the TLR4 pathway (Gioannini et al., 2004; Saitoh et al., 2004). Our hypothesis that concerted and endotoxin-independent motions of MD-2 facilitate transfer of endotoxin is supported by mutational analysis of MD-2. Residues Phe121 and Tyr131 have been identified as important in the interaction of CD14 with MD-2 (Teghanemt et al., 2008), and these residues reside on one edge of the identified dynamic channel (Fig. 8).
Figure 9.

Open (magenta) and closed (black) loop conformations of MD-2 may serve key functional roles in endotoxin transfer and TLR4* dimerization. Snapshots from MD simulations focusing on the loops between strands S5-S6 and S8-S9 of (a) Ec6b, (b) Nm4a, and (c) apo.
For the region of MD-2 that interacts with TLR4 (Fig. 8), dampening of the dynamics of this region (via binding to TLR4) may contribute to the 10-20 fold increase in the estimated dissociation constant (Kd) reported for LPS from soluble MD-2 compared to cell surface TLR4·MD-2 (Akashi et al., 2003). Overall, the plasticity of these three regions of MD-2 accommodates the quaternary structure differences necessary for the formation of agonist- versus antagonist-type complexes (Fig. 7).
5. Conclusions
The results of the MD studies help to define the connection between the temporal and spatial properties of MD2·endotoxin complexes, and their ability to activate the innate immune system via the TLR4 pathway. Due to the spectrum of immune responses potentiated by endotoxins, these structures are currently being used as the starting point for rational drug design (Hackett et al., 2006; Kawata et al., 1999; Wang et al., 2010). Moderate agonists are desired for use as efficacious vaccine adjuvants, while antagonists are being sought as inhibitors of TLR4-dependent signal transduction to combat sepsis (Christ et al., 1995; Divanovic et al., 2005; Mullarkey et al., 2003; Tidswell et al., 2010). Volumetric and apolar SASA characteristics of the lipid domain, as well as structural and dynamic information for MD-2·endotoxin complexes, described herein, can be applied to the design of lipid A-based molecules. These studies suggest that selective modulation of the TLR4 response could be accomplished through reductions in the hydrophobic surface presented by an agonist MD-2αendotoxin complex. For instance, truncation of the solvent-exposed acyl chain of a hexa-acylated endotoxin would allow specific modification of hydrophobic-hydrophobic contacts at the TLR4* dimerization interface (Fig. 10). Alternatively, a more dramatic switch in activity could potentially be achieved via the introduction of different functional groups to the edge acyl chain, disrupting the complementarily between the MD-2αendotoxin complex and the dimerizing TLR4* molecule (Fig. 10). Herein are presented methods and results that can be employed to guide this complex design process.
Figure 10.

Changing the length and/or chemical structure of the acyl chain that interacts with TLR4* may be a good strategy for manipulating the TLR4-mediated immune response. Examples of N. meningitidis lipid A modifications of the edge acyl chain, (a) based on the 30 ns snapshot: (b) truncation, to reduce hydrophobic endotoxin·TLR4* contacts, (c) introduction of new functional groups (red) to modify surface complementarity with TLR4*.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. David Stephens and Dr. Russ Carlson for many helpful discussions. Grant support provided by the Georgia Research Alliance (GRA.VAC.09.J), the National Institutes of Health (RR05351), the National Science Foundation (0821263), and the Science Foundation of Ireland (08/IN.1/B2070).
Abbreviations
- GlcN
d-p-glucosamine
- KDO
3-deoxy-d-manno-oct-2-ulosonic acid
- LOS
lipooligosaccharide
- LPS
lipopolysaccharide
- MD
molecular dynamics
- MD-2
myeloid differentiation protein 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix. Supplementary Figures S1-S8.
References
- Akashi S, et al. Lipopolysaccharide Interaction with Cell Surface Toll-like Receptor 4-MD-2. J. Exp. Med. 2003;198:1035–1042. doi: 10.1084/jem.20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CP, et al. Dynamite: a simple way to gain insight into protein motions. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2280–2287. doi: 10.1107/S0907444904019171. [DOI] [PubMed] [Google Scholar]
- Bayly CI, et al. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- Christ W, et al. E5531, a pure endotoxin antagonist of high potency. Science. 1995;268:80–83. doi: 10.1126/science.7701344. [DOI] [PubMed] [Google Scholar]
- Coats SR, et al. MD-2 Mediates the Ability of Tetra-Acylated and Penta-Acylated Lipopolysaccharides to Antagonize Escherichia coli Lipopolysaccharide at the TLR4 Signaling Complex. J Immunol. 2005;175:4490–4498. doi: 10.4049/jimmunol.175.7.4490. [DOI] [PubMed] [Google Scholar]
- Darden T, et al. Particle Mesh Ewald - an Nlog(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- DeMarco ML, Woods RJ. Atomic-resolution conformational analysis of the GM3 ganglioside in a lipid bilayer and its implications for ganglioside-protein recognition at membrane surfaces. Glycobiology. 2009;19:344–355. doi: 10.1093/glycob/cwn137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divanovic S, et al. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst RK, et al. Pseudomonas aeruginosa lipid A diversity and its recognition by Toll-like receptor 4. J. Endotoxin Res. 2003;9:395–400. doi: 10.1179/096805103225002764. [DOI] [PubMed] [Google Scholar]
- Erwin AL, et al. Enzymatically deacylated Neisseria lipopolysaccharide (LPS) inhibits murine splenocyte mitogenesis induced by LPS. Infect. Immun. 1991;59:1881–1887. doi: 10.1128/iai.59.6.1881-1887.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen F, et al. Naturally occurring lipid A mutants in neisseria meningitidis from patients with invasive meningococcal disease are associated with reduced coagulopathy. PLoS Pathog. 2009;5:e1000396. doi: 10.1371/journal.ppat.1000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch MJ, et al. Gaussian 03. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- Gioannini TL, et al. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4186–4191. doi: 10.1073/pnas.0306906101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioannini TL, et al. Monomeric endotoxin:protein complexes are essential for TLR4-dependent cell activation. J. Endo. Res. 2005;11:117–123. doi: 10.1179/096805105X35198. [DOI] [PubMed] [Google Scholar]
- Golenbock DT, et al. Lipid A-like molecules that antagonize the effects of endotoxins on human monocytes. J. Biol. Chem. 1991;266:19490–19498. [PubMed] [Google Scholar]
- Hackett CJ, et al. Monophosphoryl Lipid A and Synthetic Lipid A Mimetics As TLR4-Based Adjuvants and Immunomodulators. In: Georgiev VS, editor. Vaccine Adjuvants, Humana Press; 2006. pp. 235–255. [Google Scholar]
- Hadina S, et al. MD-2-Dependent Pulmonary Immune Responses to Inhaled Lipooligosaccharides: Effect of Acylation State. Am. J. Respir. Cell Mol. Biol. 2008;38:647–654. doi: 10.1165/rcmb.2007-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V, et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Struct., Funct., Bioinf. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SJ, Thornton JM. ‘NACCESS’, Computer Program. Department of Biochemistry and Molecular Biology, University College of London; 1993. [Google Scholar]
- Humphrey W, et al. VMD: Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Hyakushima N, et al. Interaction of Soluble Form of Recombinant Extracellular TLR4 Domain with MD-2 Enables Lipopolysaccharide Binding and Attenuates TLR4-Mediated Signaling. J. Immunol. 2004;173:6949–6954. doi: 10.4049/jimmunol.173.11.6949. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kawahara K, et al. Modification of the Structure and Activity of Lipid A in Yersinia pestis Lipopolysaccharide by Growth Temperature. Infect. Immun. 2002;70:4092–4098. doi: 10.1128/IAI.70.8.4092-4098.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawata T, et al. E5531, a synthetic non-toxic lipid A derivative blocks the immunobiological activities of lipopolysaccharide. British Journal of Pharmacology. 1999;127:853–862. doi: 10.1038/sj.bjp.0702596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, et al. Crystal Structure of the TLR4-MD-2 Complex with Bound Endotoxin Antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Kirschner KN, et al. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, et al. Regulatory Roles for MD-2 and TLR4 in Ligand-Induced Receptor Clustering. J. Immunol. 2006;176:6211–6218. doi: 10.4049/jimmunol.176.10.6211. [DOI] [PubMed] [Google Scholar]
- Mullarkey M, et al. Inhibition of Endotoxin Response by E5564, a Novel Toll-Like Receptor 4-Directed Endotoxin Antagonist. J. Pharmacol. Exp. Ther. 2003;304:1093–1102. doi: 10.1124/jpet.102.044487. [DOI] [PubMed] [Google Scholar]
- Munford RS, Hall CL. Detoxification of Bacterial Lipopolysaccharides (Endotoxins) by a Human Neutrophil Enzyme. Science. 1986;234:203–205. doi: 10.1126/science.3529396. [DOI] [PubMed] [Google Scholar]
- Muroi M, Tanamoto K.-i. Structural Regions of MD-2 That Determine the Agonist-Antagonist Activity of Lipid IVa. J. Biol. Chem. 2006;281:5484–5491. doi: 10.1074/jbc.M509193200. [DOI] [PubMed] [Google Scholar]
- Ohto U, et al. Crystal Structures of Human MD-2 and Its Complex with Antiendotoxic Lipid IVa. Science. 2007;316:1632–1634. doi: 10.1126/science.1139111. [DOI] [PubMed] [Google Scholar]
- Park BS, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, et al. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Poltorak A, et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Raetz CRH, et al. Lipid A Modification Systems in Gram-Negative Bacteria. Annu. Rev. Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedo F, et al. Deacylated lipopolysaccharide inhibits plasminogen activator inhibitor-1, prostacyclin, and prostaglandin E2 induction by lipopolysaccharide but not by tumor necrosis factor-alpha. J. Immunol. 1990;144:3506–3512. [PubMed] [Google Scholar]
- Ryckaert JP, et al. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- Saitoh S.-i., et al. Lipid A antagonist, lipid IVa, is distinct from lipid A in interaction with Toll-like receptor 4 (TLR4)-MD-2 and ligand-induced TLR4 oligomerization. Int. Immunol. 2004;16:961–969. doi: 10.1093/intimm/dxh097. [DOI] [PubMed] [Google Scholar]
- Schletter J, et al. Molecular mechanisms of endotoxin activity. Arch. Microbiol. 1995;164:383–9. doi: 10.1007/BF02529735. [DOI] [PubMed] [Google Scholar]
- Shimazu R, et al. MD-2, a Molecule that Confers Lipopolysaccharide Responsiveness on Toll-like Receptor 4. J. Exp. Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoel DVD, et al. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Stewart I, et al. Cyanobacterial lipopolysaccharides and human health - a review. Environ. Health. 2006;5:7. doi: 10.1186/1476-069X-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teghanemt A, et al. Novel Roles in Human MD-2 of Phenylalanines 121 and 126 and Tyrosine 131 in Activation of Toll-like Receptor 4 by Endotoxin. J. Biol. Chem. 2008;283:1257–1266. doi: 10.1074/jbc.M705994200. [DOI] [PubMed] [Google Scholar]
- Teghanemt A, et al. Molecular Basis of Reduced Potency of Underacylated Endotoxins. J. Immunol. 2005;175:4669–4676. doi: 10.4049/jimmunol.175.7.4669. [DOI] [PubMed] [Google Scholar]
- Tessier MB, et al. Extension of the GLYCAM06 Biomolecular Force Field to Lipids, Lipid Bilayers and Glycolipids. Mol. Simul. 2008;34:349–364. doi: 10.1080/08927020701710890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidswell M, et al. Phase 2 trial of eritoran tetrasodium (E5564), a Toll-like receptor 4 antagonist, in patients with severe sepsis. Crit. Care Med. 2010;38:72–83. doi: 10.1097/CCM.0b013e3181b07b78. [DOI] [PubMed] [Google Scholar]
- Walsh C, et al. Elucidation of the MD-2/TLR4 Interface Required for Signaling by Lipid IVa. J. Immun. 2008;181:1245–1254. doi: 10.4049/jimmunol.181.2.1245. [DOI] [PubMed] [Google Scholar]
- Wang X, et al. Synthetic and Natural TLR4 Agonists as Safe and Effective Vaccine Adjuvants. In: Harris JR, et al., editors. Endotoxins: Structure, Function and Recognition. Springer Netherlands; 2010. pp. 303–321. [DOI] [PubMed] [Google Scholar]
- Wright CS, et al. Crystal Structure Analysis of Phosphatidylcholine-GM2-Activator Product Complexes: Evidence for Hydrolase Activity. Biochemistry. 2005;44:13510–13521. doi: 10.1021/bi050668w. [DOI] [PubMed] [Google Scholar]
- Zimmer SM, et al. Human MD-2 discrimination of meningococcal lipid A structures and activation of TLR4. Glycobiology. 2007;17:847–856. doi: 10.1093/glycob/cwm057. [DOI] [PubMed] [Google Scholar]
- Zughaier S, et al. Hexa-acylation and KDO2-glycosylation determine the specific immunostimulatory activity of Neisseria meningitidis lipid A for human monocyte derived dendritic cells. Vaccine. 2006;24:1291. doi: 10.1016/j.vaccine.2005.09.039. [DOI] [PubMed] [Google Scholar]
- Zughaier SM, et al. Differential Induction of the Toll-Like Receptor 4-MyD88-Dependent and -Independent Signaling Pathways by Endotoxins. Infect. Immun. 2005;73:2940–2950. doi: 10.1128/IAI.73.5.2940-2950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.