

Abstract
BACKGROUND
The functional effects of N-acetyltransferase 1 (NAT1) polymorphisms and haplotypes are poorly understood, compromising the validity of associations reported with diseases including birth defects and numerous cancers.
METHODS
We investigated the effects of genetic polymorphisms within the NAT1 coding region and the 3′-untranslated region (3′-UTR) and their associated haplotypes on N- and O-acetyltransferase catalytic activities, and NAT1 mRNA and protein levels following recombinant expression in COS-1 cells.
RESULTS
1088T>A (rs1057126; 3′-UTR) and 1095C>A (rs15561; 3′-UTR) each slightly reduced NAT1 catalytic activity and NAT1 mRNA and protein levels. A 9-base pair (TAATAATAA) deletion between nucleotides 1065-1090 (3′-UTR) reduced NAT1 catalytic activity and NAT1 mRNA and protein levels. In contrast, a 445G>A (rs4987076; V149I), 459G>A (rs4986990; T153T), 640T>G (rs4986783; S214A) coding region haplotype present in NAT1*11 increased NAT1 catalytic activity and NAT1 protein, but not NAT1 mRNA levels. A combination of the 9-base pair (TAATAATAA) deletion and the 445G>A, 459G>A, 640T>G coding region haplotypes, both present in NAT1*11, appeared to neutralize the opposing effects on NAT1 protein and catalytic activity, resulting in levels of NAT1 protein and catalytic activity that did not differ significantly from the NAT1*4 reference.
CONCLUSIONS
Since 1095C>A (3′-UTR) is the sole polymorphism present in NAT1*3, our data suggests that NAT1*3 is not functionally equivalent to the NAT1*4 reference. Furthermore, our findings provide biological support for reported associations of 1088T>A and 1095C>A polymorphisms with birth defects.
Keywords: N-acetyltransferase 1 polymorphisms, haplotypes, acetylator genotype, acetylator phenotype, arylamines
INTRODUCTION
The human genome contains two functional arylamine N-acetyltransferase genes, which code for N-acetyltransferase 1 (NAT1) and 2 (NAT2) enzymes (EC 2.3.1.5). NAT1 and NAT2 both possess a single intronless exon with an open reading frame of 870 bp, encoding a 290 amino acid protein. Whereas NAT2 expression is limited primarily to the liver and gut, NAT1 is expressed widely across human tissues (Husain et al., 2007) including placenta (Sim et al., 2000; Smelt et al., 2000).
Human N-acetyltransferase catalyzes conjugation of an acetyl motif, usually from acetyl coenzyme A (AcCoA), to the exocyclic amine (N-acetylation) or hydroxyl (O-acetylation) of substrates. N-acetylation of the exocyclic amine usually results in detoxification (Hein, 2009). However, following N-oxidation, the N-hydroxyl metabolite undergoes O-acetylation (usually activation). The resulting N-acetoxy arylamines are highly unstable, spontaneously forming electrophilic intermediates that readily bind to protein and DNA (Hein, 1988).
Genetic polymorphisms have been observed in both NAT1 and NAT2 (Hein, 2009). NAT2 polymorphisms are well established as the basis of rapid, intermediate and slow acetylation phenotypes. Excellent NAT2 genotype/phenotype correlations have been reported (Chen et al., 2006; Ma et al., 2009; Doll et al., 2010). Although studies have investigated the functional effects of NAT1 single nucleotide polymorphisms (SNPs) and alleles (Walraven et al., 2008), there are remaining gaps regarding effects of NAT1 genetic variation, particularly polymorphisms in the NAT1 3′-untranslated region (3′-UTR).
The functional effects of individual SNPs in the NAT1 coding region recently were reviewed (Hein, 2009). NAT1*11 alleles have SNPs in both the NAT1 coding region and the 3′-UTR. Individuals possessing NAT1*11 alleles have been reported to have significantly lower N-acetyltransferase activities in human red blood cells (Bruhn et al., 1999) but not human leukocytes (Zhangwei et al., 2006). The latter finding is consistent with previous studies following recombinant expression of NAT1*11 in bacteria (Hughes et al., 1998) and COS-1 cells (de Leon et al., 2000). However, recombinant expression in COS-1 cells that focused entirely on the coding region SNPs in NAT1*11 showed increased protein and catalytic activity (Zhu and Hein, 2008).
NAT1 plays an important role in the bioactivation of arylamine carcinogens such as 4-aminobiphenyl in cigarette smoke. NAT1 also has been shown to play an important role in folate metabolism (Wakefield et al., 2007). NAT1 polymorphisms have been investigated for associations with birth defects related to maternal smoking and with periconceptional supplementation of multivitamins that include folic acid. Two of the most common NAT1 polymorphisms are present in the NAT1 putative polyadenylation signal sequence: 1088T>A (rs1057126; 3′-UTR) and 1095C>A (rs15561; 3′-UTR). Oral clefts (lip with or without the palate) were strongly associated with maternal cigarette smoking in infants homozygous for 1088A or 1095A in the NAT1 3′-UTR (Lammer et al., 2004b). Some evidence for an interaction between maternal smoking and the homozygous 1095A genotype also has been shown towards risk of spina bifida (Jensen et al., 2005). Infants homozygous for 1088A or 1095A had a slight increase in oral clefts (lip with or without the palate) that was more robust in those mothers who did not take multivitamins (including folate) during pregnancy (Lammer et al., 2004a). Similarly, infants homozygous for 1088A or 1095A also are associated with increased limb deficiency defects, particularly in mothers with both active and passive smoke exposures, who did not take multivitamin supplements containing folate (Carmichael et al., 2006). As recently reviewed (Shi et al., 2008), although interactions between genes, environment and behavior on birth defects have been reported, further work towards understanding these interactions is needed.
Molecular epidemiological studies have reported associations between NAT1 genetic polymorphisms and disease risk (Hein, 2009) including urinary bladder (Taylor et al., 1998; Katoh et al., 1999; Gago-Dominguez et al., 2003; Sanderson et al., 2007), colorectal (Bell et al., 1995; Chen et al., 1998; Ishibe et al., 2002; Lilla et al., 2006; Shin et al., 2008), breast (Millikan et al., 1998; Zheng et al., 1999; Lee et al., 2003; Ambrosone et al., 2007), lung (Wikman et al., 2001; Gemignani et al., 2007), prostate (Hein, 2002; Rovito, Jr. et al., 2005), and pancreatic cancers (Li et al., 2006; Jiao et al., 2007; Suzuki et al., 2008), and non-Hodgkin lymphoma (Morton et al., 2006; 2007; Kilfoy et al., 2010).
Although human epidemiological studies suggested a role for NAT1 genetic polymorphism in susceptibility to birth defects and numerous cancers, the present poor understanding of the relationship between NAT1 genotype and phenotype compromises the accuracy and the validity of the association observed. Understanding the molecular mechanisms of NAT1 polymorphisms is needed to improve understanding of human epidemiological studies. Therefore, the purpose of this study was to investigate functional effects of some of the more common genetic polymorphisms in the NAT1 open reading frame and the 3′-UTR.
MATERIALS AND METHODS
Materials
Cloning vector pCDNA3.1, transfection control plasmid pCMV SPORT-βgal, O-nitrophenyl beta-D-galactopyranoside and lipofectamine plus reagent were obtained from Invitrogen Life Technologies (Carlsbad, CA USA). Restriction endonucleases were obtained from New England Biolabs (Beverly, MA USA). The protein assay kit was purchased from Bio-Rad Laboratories (Hercules CA, USA). P-Amino-benzoic acid (PABA), 4-aminobiphenyl (ABP), acetyl coenzyme A (AcCoA) and polyclonal rabbit anti-β-galactosidase antibody were purchased from Sigma Chemical Company (St. Louis, MO USA). COS-1 cells (SV40-transformed African green monkey kidney cells) were obtained from American Type Culture Collection (Manassas, VA USA). N-hydroxy-2-amino-1-methyl-6-phenylimidazo [4, 5-b] pyridine (N-OH-PhIP) was obtained from Toronto Research Chemicals (North York, ON, Canada). Polyclonal antibody against human NAT1 was kindly provided by Dr. Edith Sim, University of Oxford, Oxford UK.
Vector Construction
Human lung surgical samples (de-identified) were obtained from the National Cancer Institute Cooperative Human Tissue Network. To generate mammalian expression constructs that possess genetic variations in NAT1 3′-UTR (Supplemental Figure), genomic DNA from individuals possessing NAT1*4, NAT1*3, NAT1*10 or NAT1*11 alleles was used as template for polymerase chain reaction (PCR). The PCR amplification was performed with 10 μg genomic DNA, 200 nM each primer (forward primer: 5′-AGTATGGACATTGAAGCATAT-3′, underlined letters indicate the first Metcodon; reverse primer: 5′-GAATTCAACAATAAACCAACATTA-3′, the 5′-end of reverse primer localized at nucleotide 1175 of NAT1), 200 μM each dNTP in 1× reaction buffer (10 mM Tris-HCl (pH 7.3), 50 mM KCl, 1.5 mM MgCl2) with final volume of 50 μl. The PCR cycling parameters were: 94 °C for 5 min, 30 cycles of 94 °C for 30 sec, 55°C for 30 sec, 72°C for 1.5 min, followed by additional extension for 8 min at 72 °C. By this application, an 1175 bp NAT1 fragment containing the 873 bp open reading frame plus 302 bp of the 3′-UTR region was obtained for NAT1*4 reference allele as well as NAT1 variant alleles possessing 1088T>A, 1095C>A or the 9 bp deletion between nucleotides 1065-1090 in the 3′-UTR (Fig. 1). Following PCR amplification of each allele, 120 ng PCR product and 60 ng pCDNA3.1 (Invitrogen Life Technologies, CA USA) were ligated overnight at 15°C with T4 DNA Ligase. Transformants were grown and positive colonies were confirmed by automated DNA sequencing. For mock transfections, 800 ng pCMV sport-βgal plasmid (without NAT1) was delivered into COS-1 cells with identical conditions to the transfection of NAT1 alleles described above.
Figure 1.
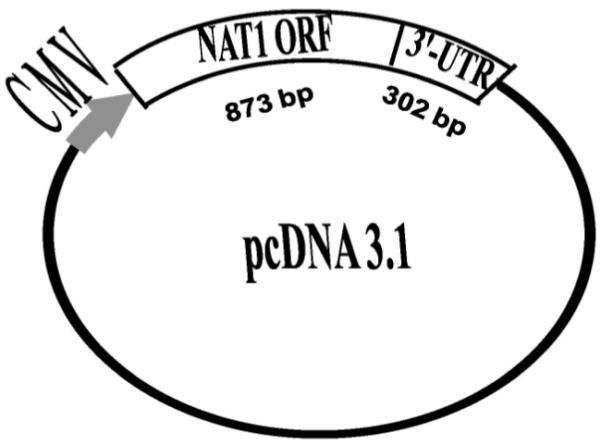
Vector structure of the cloned NAT1 fragments containing the 873-bp NAT1 open reading frame (ORF) and 302 bp of the NAT1 3′-UTR. NAT1 mRNA transcription was driven by cytomegalovirus (CMV) promoter, whereas RNA processing at the 3-end was facilitated by the NAT1 putative polyA signals since the BGH polyA signal in the vector was deleted.
Since the cloned NAT1 fragment contained a putative polyadenylation signal, the original bovine growth hormone (BGH) polyA signal (nucleotides 1110-1324) was deleted with Ava I digestion. Because two Ava I sites have different recognition sequences (c↓tcgac at 1030, and c↓ccggg at 2186, arrow indicates cut position), the linear vector would not be re-ligated directly. It was treated with T4 DNA polymerase to produce blunt ends, followed by re-ligation with T4 DNA Ligase. The deletion of BGH polyA signal for each construct was verified by automated DNA sequencing.
Two additional vectors were constructed in order in order to investigate coding and non-coding genetic polymorphisms separately. One contained 445G>A (rs4987076; V149I), 459G>A (rs4986990; T153T), and 640T>G (rs4986783; S214A) in the open reading frame; the other contained the 9-bp deletion between nucleotides 1065-1090 in the 3′-UTR. The NAT1*4 vector was modified to eliminate one Ssp I site in the vector portion of the plasmid to facilitate the usage of Ssp I site in NAT1 open reading frame. The plasmid was digested with Ssp I, which produced blunt ends. The small fragment that contained part of NAT1 coding region was treated with BspH I, followed by treatment with T4 DNA polymerase to produce a blunt end. This blunt end-produced small fragment and the larger fragment generated in digestion of Ssp I were ligated with T4 DNA Ligase. This modified plasmid (identified as NAT1*11) was confirmed with automated DNA sequencing. The modified NAT1*11 and NAT1*4 vectors were digested with BamH I and Ssp I (New England Biolabs), which cut at a restriction site in the polylinker of the vector and at nucleotide 814 in the NAT1 open reading frame, respectively. NAT1-vector possessing 445G>A (rs4987076; V149I), 459G>A (rs4986990; T153T), and 640T>G (rs4986783; S214A) in the open reading frame was generated by ligation of the small fragment from the NAT1*11 vector and the linear modified NAT1*4 vector. The NAT1-vector possessing the 9-bp deletion between nucleotides 1065-1090 (3′-UTR) was obtained by digestion of the same vectors with Ssp I and EcoR I (in polylinker of vector), and re-ligation of the small fragment from the NAT1*11 vector and the linear modified NAT1*4 vector. The positive colonies were confirmed by automated DNA sequencing.
Cell Culture, Transfection and Cell Lysate Preparation
The cells were cultured in Dulbecco’s modified eagle’s medium (DMEM), supplemented with 4.5 g/L glucose, 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 4mM L-glutamine. The cells were maintained at 37 °C with 10% CO2, and sub-cultured every two or three days.
Transfections were repeated three times in duplicate. The three transfections started with cell recovery from cell stock of the same cell passage. After two passages, COS-1 cells were seeded 2.0×106 cells/10 cm dish in DMEM medium with 4 mM L-glutamine, incubated at 37 °C to allow 90-95 % confluence, and transfected using Lipofectamine plus reagent (Invitrogen Life Technology, Carlsbad, CA USA). To facilitate the binding of Lipofectamine to DNA, 8.8 μg plasmid containing NAT1 fragment and 800 ng pCMV SPORT-βgal plasmid were incubated with 40 μl Plus reagent at room temperature for 15 min. After formation of DNA-Plus complex, 60 μl Lipofectamine was added and incubated at room temperature for a second 15 min, allowing the plasmid to be wrapped with Lipofectamine. The cells were pre-washed with basic medium (DMEM with 4 mM L-glutamine) and incubated with 5 ml basic medium for 1 hr. Transfection was initiated by the addition of DNA-Lipofectamine complex into the cells. Three hours after transfection, the medium was brought back to normal (10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 4 mM L-glutamine) with another normal medium change after 24 hr.
Forty-eight hours after transfection, cells were washed with ice cold PBS and scraped in 10 ml PBS. A portion (1.5 ml of 10 ml cell suspension) was collected and pelleted by centrifugation (15,000 rpm 3 min) then stored at −70 °C for future mRNA quantification. The remaining 8.5 ml of cell suspension were pelleted by centrifugation then disrupted by sonication (three pulses for 15 sec at 10 sec intervals) in 1.2 ml lysis buffer (20 mM sodium phosphate pH 7.4, 1 mM EDTA, 1 mM dithiothreitol, 10 μM phenylmethanesulfonyl fluoride, 10 μM leupetin, 1 μM pepstatin, 4 μM aprotinin, 2 mg/ml bovine serum albumin). Precipitation of cell debris was done by centrifugation at 15,000 rpm for 15 min at 4°C. The cell lysate in 50% glycerol was stored at −20 °C for enzyme assays. Total protein concentrations were determined using the Bio-Rad protein assay kit (Bio-Rad, Hercules, CA).
RNA Isolation and Quantitative RT-PCR
RNA was isolated from cell pellets stored at −70 °C using RNeasy® Mini kit and QIAshedder (RNeasy® Mini kit, Qiagen) according to manufacturer’s instructions. Crude RNA products were treated with Turbo-DNase (Ambion) at 37 °C for 30 min to remove any contaminating plasmid DNA, followed by incubation at 70 °C in presence of 5 mM EDTA to inactivate Turbo-DNase. The inactivated DNase and EDTA were removed using RNeasy Mini cleanup protocol. The procedure was repeated once. The third time RNA samples were treated with Turbo-DNase followed by heat-inactivation (70°C for 10 min) of the Turbo-DNase.
First strand of NAT1 cDNA was synthesized and used as template in real-time PCR quantitation of NAT1 mRNA using SuperScript™ First-Strand Synthesis system for RT-PCR (Invitrogen Life Technology, Carlsbad, CA USA). With 50 ng random hexamer and 10 nmoles dNTPs, 500-800 ng template (total RNA) was denatured at 65°C for 5 min in 10 μl volume, and then chilled on ice. The volume of the system was adjusted to 19 μl with final concentrations of 100 nM MgCl2, 200 nM DTT, 40 U RNaseOUT™ (recombinant Ribonuclease inhibitor) in 1 ×RT buffer (120 mM Tris-HCl (pH 8.4) 50 mM KCl). Following pre-warming at 25 °C for 2 min, the reaction was initiated with the addition of 50 units SuperScript™ II RNase H-Reverse transcriptase. The amplification was initiated with a hold at 25 °C for 10 min, followed by incubation at 42° C for 50 min, and then the transcriptase was inactivated at 70°C for 10 min.
The quantitative real time PCR was performed as described previously (Zhu and Hein, 2008). The cycling parameters were 50 °C for 2 min, 95°C for 10 min, followed by 40 cycles of two-step amplification (95 °C for 15 sec, 60 °C for 1 min). Reactions were carried out in 20 μl volume with 1×master mix, 2 μl total RNA from transfected COS-1 cells as templates (200-800 ng), 300 nM forward and reverse primers (forward primer: 5′-AGACATCTCCATCATCTGTGTGTTTACTAGT-3′); (reverse primer: 5′-TTCCTCACTCAGAGTCTTGAACTCTATT-3′); and 100 nM probe (5′-TTCACTGTTTGGTGGGCTTCACCC-3′). The endogenous control COS-1 β-actin was amplified with the same conditions (reaction components, cycling parameters) in separate tubes as previously described (Zhu and Hein, 2008). The primers and probe were (forward primer: 5′-CGCCCAGCACGATGAAA-3′; reverse primer: 5′-CCGCCGATCCACACAGA-3′; probe: 5′-AGATCATTGCTCCTCCTGAGCGCAAGT-3′).
Western Blot Analysis
NAT1 protein (1.25 μg total protein) was loaded onto 12% PAGEr® Gold Tris-Glycine SDS-PAGE gel (Carmbex Bio Science Rockland, INC., Rockland, ME USA), and subjected to electrophoresis at 120V for 60 min. Protein was transferred to Immuno-blot PVDF membrane at 100V for one hour using Mini Trans-Blot®cell (Bio-Rad) apparatus. The blockage of nonspecific sites was performed at 4°C overnight with 5% nonfat milk in 0.05 % T-TBS (20 mM Tris, 500 mM sodium chloride pH7.5, 0.05% Tween-20). The NAT1 specific bands were detected by hybridization with anti-NAT1 rabbit derived polyclonal antiserum (1:4000) in 2.5% milk in 0.01 % T-TBS for 2 hours. NAT1 bands were detected by peroxidase labeled anti-rabbit secondary antibody (1: 20,000) for one hour, visualized with ECL plus western blotting detection reagents. The membrane then was stripped, and re-probed to visualize β-galactosidase bands as loading control. The membrane was incubated at 50 °C for 30 min with occasional agitation in strip buffer (100 mM Tris-HCl (pH 6.8), 2% SDS, 100 mM β-mercaptoethanol) and washed twice with 0.01 % T-TBS for 10 min. With the exception of the primary antibody (rabbit anti-β-galactosidase in 1:2000 dilution), the conditions for blotting β-galactosidase were the same as that for NAT1. The intensity of the individual protein (NAT1 or β-galactosidase) band was quantified by densitometry analysis and expressed as density units.
Enzyme Assays
Cell lysates were assayed for N-acetyltransferase and O-acetyltransferase catalytic activities as described previously (Zhu and Hein, 2008). Briefly, arylamine substrate PABA (750 μM), ABP (300 μM) or N-OH-PhIP (100 μM) were incubated with AcCoA (1 mM) and diluted cell lysate at 37 °C. The cell lysate dilution and incubation time was varied to ensure less than 5 percent substrate converted to product. Reactants and products were separated and quantitated by high performance liquid chromatography. The activities were normalized to co-transfected β-galactosidase activity as previously described (Zang et al., 2004).
Statistical Analysis
One-way analysis of variance followed by Newman-Keuls Multiple Comparison test was employed to test for differences among NAT1 haplotypes in catalytic activities, and NAT1-specific mRNA and protein. Statistical significance was set at p<0.05.
RESULTS
Vector Construction
Vectors constructed for this study all possessed 1175 bp NAT1 fragments including the 873 bp coding sequence and 302 bp of the 3′-UTR (Fig. 2) and the native NAT1 polyA signal altered by the 1088T>A SNP (supplemental figure). The original BGH polyA signal was deleted for each vector to eliminate the influence of vector pcDNA3.1V5-His-TOPO© BGH polyadenylation signal on NAT1 mRNA processing.
Figure 2.
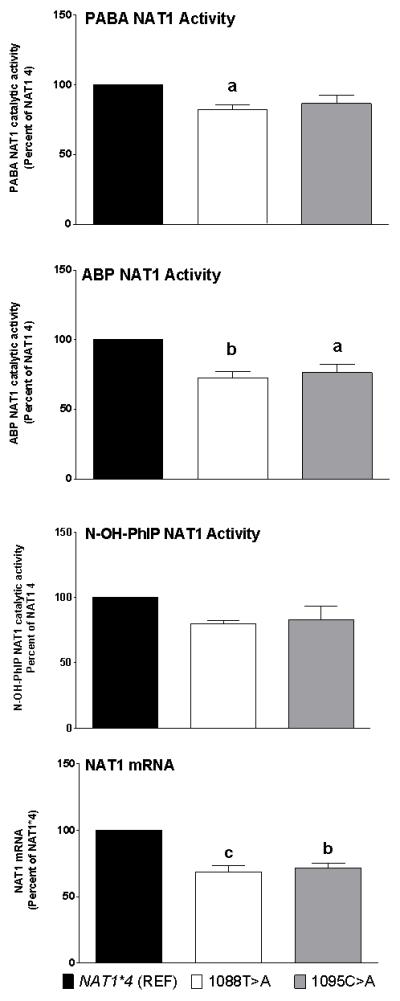
Effects of 1088T>A and 1095C>A on NAT1 activities towards PABA and ABP N-acetyltransferase, and N-OH-PhIP O-acetyltransferase activities and NAT1 mRNA. Each bar represents Mean ± SEM for three transfections. a) p<0.05; b) p<0.01; c) p<0.001 significantly different from the NAT1*4 reference.
Effects of 1088T>A and 1095C>A in NAT1 3′-UTR
As shown in Figure 1, both 1088T>A and 1095C>A resulted in slight reduction in N- and O-acetylation activities compared to the NAT1*4 reference allele. The reduction caused by 1088T>A was significant (p<0.01) for the N-acetylation of PABA and ABP, but not towards the O-acetylation of N-hydroxy-PhIP. The reduction caused by 1095C>A was significant (p<0.05) only for the N-acetylation of ABP. Both 1088T>A and 1095C>A caused significant reduction (p<0.01) in NAT1 mRNA (Fig. 1). NAT1 protein levels also were reduced significantly (p < 0.001) compared to the NAT1*4 reference allele (Fig. 3).
Figure 3.

Effects of NAT1 polymorphisms on protein expression. Ratios of NAT1 protein to beta-galactosidase protein are shown following densitometric scanning of the representative Western blots shown above the graph. Top Western blot: A: Positive control (recombinant NAT1 from yeast); B: Negative control (mock transfection); C: NAT1*4 reference; D: 1095C>A; E: 1088T>A. Bottom Western blot: A: 9-bp deletion haplotype; B: 445G>A, 459G>A, 640T>G haplotype; C: NAT1*4 reference; D: Combination 445G>A, 459G>A, 640T>G, 9-bp deletion haplotype; E: Negative control (mock transfection); F: Positive control (recombinant NAT1 from yeast). Each bar represents Mean ± SEM for three transfections. a) p<0.001 from NAT1*4 reference; b) p<0.01 from 445G>A, 459G>A, 640T>G haplotype; c) p<0.05 from combination 445G>A, 459G>A, 640T>G, 9-bp deletion haplotype.
Effects of 9-bp (TAATAATAA) Deletion Between Nucleotides 1065-1090 in NAT1 3′-UTR
As shown in Figure 4, the 9-bp (TAATAATAA) deletion between nucleotides 1065-1090 in the 3′-UTR resulted in significant reductions in N-acetylation of PABA (p<0.001), ABP (p<0.01) and the O-acetylation of N-OH-PhIP (p<0.05). The 9-bp deletion also caused significant reduction in NAT1 mRNA (Fig. 4). NAT1 protein levels also were reduced significantly (p < 0.05) compared to the 445G>A, 459G>A, and 640T>G and 9-bp deletion haplotype (Fig. 3).
Figure 4.

Effects of combination 445G>A, 459G>A, and 640T>G and 9-bp (TAATAATAA) deletion between 1065-1090 in the 3′-UTR on PABA and ABP N-acetyltransferase and N-OH-PhIP O-acetyltransferase activities and NAT1 mRNA. Each bar represents Mean ± SEM for three transfections. a) p<0.001 from 9-bp deletion; b) p<0.05 from NAT1*4 (REF); c) p<0.01 from 9-bp deletion; d) p<0.01 from NAT1*4 (REF); e) p<0.01 from combination 445G>A, 459G>A, and 640T>G and 9-bp deletion haplotype; f) p<0.05 from 9-bp deletion.
Effects of 445G>A, 459G>A, and 640T>G Haplotype
The 445G>A, 459G>A, and 640T>G haplotype resulted in significantly higher PABA (p<0.05) and ABP (p<0.01) NAT1 catalytic activities but not in NAT1 mRNA (p>0.05) compared to the NAT1*4 reference (Fig. 4). This haplotype resulted in significantly (p<0.01) higher levels of NAT1 protein than the NAT1*4 reference allele (Fig. 3).
Effects of Combination 445G>A, 459G>A, and 640T>G and 9-bp (TAATAATAA) Deletion Between Nucleotides 1065-1090 in NAT1 3′-UTR
Although the 445G>A, 459G>A, and 640T>G haplotype enhanced catalytic activities and the 9-bp (TAATAATAA) deletion between nucleotides 1065-1090 in the 3′-UTR reduced catalytic activities, the combination 445G>A, 459G>A, 640T>G, and 9-bp deletion haplotype resulted in N- and O-acetylation catalytic activities that did not differ significantly (p>0.05) from the NAT1*4 reference allele (Fig. 4). The NAT1 protein level for the combination 445G>A, 459G>A, 640T>G, 9-bp deletion haplotype was significantly higher (p<0.05) than the 9-bp deletion haplotype and significantly lower (p<0.05) than the 445G>A, 459G>A, and 640T>G haplotype (Fig. 3).
DISCUSSION
The functional effects of NAT1 polymorphisms and haplotypes are poorly understood, compromising the validity of associations reported with diseases including birth defects and numerous cancers. The NAT1 reference allele is NAT1*4. We investigated the effects of polymorphisms within the NAT1 coding region and the 3′-UTR and their associated haplotypes on N- and O-acetyltransferase catalytic activities, and mRNA and protein levels via recombinant expression of cDNAs possessing the polymorphisms in COS-1 cells.
1088T>A (3′-UTR) and 1095C>A (3′-UTR) reduced NAT1 catalytic activity slightly secondary to a reduction in NAT1 mRNA and protein levels. As noted in the introduction, these polymorphisms have been associated with oral clefts (Lammer et al., 2004b) and evidence for an interaction between maternal smoking and the homozygous 1095A genotype has been shown towards risk of spina bifida (Jensen et al., 2005). Infants homozygous for these polymorphisms show increased oral clefts that was more robust in those mothers who did not take multivitamins such as folate during pregnancy (Lammer et al., 2004a). Similarly, infants homozygous for 1088A or 1095A also are associated with increased limb deficiency defects, particularly in mothers with both active and passive smoke exposures, who did not take multivitamin supplements containing folate (Carmichael et al., 2006). Transgenic A/J mice overexpressing human NAT1 are protected from both teratogen-induced and sporadic cleft palate suggesting a protective role of human NAT1 (Erickson et al., 2008). Whether or not the slight reductions in NAT1 caused by 1088A and 1095A in our study are sufficient to explain the increased risk for oral clefts and limb deficiencies observed previously requires further study.
Since 1095C>A (3′-UTR) is the sole polymorphism present in NAT1*3, our data suggest that NAT1*3 is not functionally equivalent to the NAT1*4 reference. This result has major impact for investigations of associations between NAT1 alleles and disease risk (see introduction), since the NAT1*3 allele is commonly grouped with the NAT1*4 as reference alleles.
Previous studies have investigated the role of individual NAT1 coding region polymorphisms following recombinant expression in bacteria (Grant et al., 1997), yeast (Fretland et al., 2001, 2002) and COS-1 cells (Zhu and Hein, 2008). Our current study focused on functional effects of the 445G>A, 459G>A, and 640T>G coding region haplotype present in NAT1*11. We observed that this haplotype caused an increase in NAT1 catalytic activity in association with an increase in NAT1 protein, but not mRNA levels, a result different from that observed following recombinant expression in yeast (Fretland et al., 2001) but in agreement with a previous study following recombinant expression in COS-1 cells (Zhu and Hein, 2008).
Individuals possessing NAT1*11 alleles have been reported to have significantly lower N-acetyltransferase activities in human red blood cells (Bruhn et al., 1999) but not human leukocytes (Zhangwei et al., 2006). The latter finding is consistent with previous studies following recombinant expression of NAT1*11 in bacterial (Hughes et al., 1998) and COS-1 cells (de Leon et al., 2000). Recombinant expression studies in COS-1 cells that focused entirely on the coding region SNPs in NAT1*11 showed increased protein and catalytic activity (Zhu and Hein, 2008). The present results clarify some of these conflicting findings. Although the 445G>A, 459G>A, and 640T>G coding region haplotype present in NAT1*11 increased NAT1 catalytic activity in association with an increase in NAT1 protein, but not mRNA levels (as noted before in Zhu and Hein, 2008), the 9-bp (TAATAATAA) deletion between 1065-1090 also present in NAT1*11, reduced NAT1 catalytic activity associated with a reduction in NAT1 mRNA and protein levels. Consistent with these results, the combination of the 9-bp (TAATAATAA) deletion between 1065-1090 in the 3′-UTR and the 445G>A, 459G>A, 640T>G coding region haplotypes, both present in NAT1*11, resulted in levels of NAT1-specific protein and catalytic activity that did not differ significantly from the NAT1*4 reference. Thus, our data suggest that NAT1*11 does not encode NAT1 protein with a higher activity than that encoded by the NAT1*4 reference as had been suggested from observations of the functional effects of the NAT1*11 coding region polymorphisms alone.
In summary, our data suggest that NAT1*3 should not be included in the reference group and NAT1*11 should not be included in a “rapid” acetylator group in gene-environment association studies. Since transgenic A/J mice overexpressing human NAT1 are protected from both teratogen-induced and sporadic cleft palate (Erickson et al., 2008) the findings of the present study provide biological support for reported associations of NAT1 1088T>A and 1095C>A polymorphisms with birth defects.
Supplementary Material
Supplemental Figure. Cloned 1175 bp NAT1 fragment (Reference sequence published in Genbank Assession Number X17059). The fragment consists of the 873-bp coding region and 302-bp of the 3′-UTR; ATG; start codon; TAG: stop codon; AATAAA: putative polyadenylation signal polyA-1. The locations of the 445G>A, 459G>A, 640T>G, 1088T>A, and 1095C>A SNPs are shown by increased font size. The region of the 9-bp deletion (three TAA triplets) is underlined.
ACKNOWLEDGEMENTS
We thank Professor Edith Sim, University of Oxford, for her kind donation of human NAT1 antiserum.
Grant sponsors: United States Public Health Service grants R01-CA034627 and P30-ES014443 and grants from the Kentucky Lung Cancer Research Program and the University of Louisville Center for Genetics and Molecular Medicine.
Footnotes
Current address: Department of Pediatrics, University of Chicago, Chicago, Illinois
Reprints should be addressed to: David W. Hein, Clinical and Translational Research – Room 303, University of Louisville Health Sciences Center, 505 South Hancock Street, Louisville, KY 40202-1617. d.hein@louisville.edu.
Portions of this work constituted partial fulfillment for the PhD in pharmacology and toxicology at the University of Louisville awarded to Yuanqi Zhu.
REFERENCES
- Ambrosone CB, Abrams SM, Gorlewska-Roberts K, Kadlubar FF. Hair dye use, meat intake, and tobacco exposure and presence of carcinogen-DNA adducts in exfoliated breast ductal epithelial cells. Arch Biochem Biophys. 2007;464:169–175. doi: 10.1016/j.abb.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Bell DA, Stephens EA, Castranio T, Umbach DM, Watson M, Deakin M, Elder J, Hendrickse C, Duncan H, Strange RC. Polyadenylation polymorphism in the acetyltransferase 1 gene (NAT1) increases risk of colorectal cancer. Cancer Res. 1995;55:3537–3542. [PubMed] [Google Scholar]
- Bruhn C, Brockmoller J, Cascorbi I, Roots I, Borchert HH. Correlation between genotype and phenotype of the human arylamine N-acetyltransferase type 1 (NAT1) Biochem Pharmacol. 1999;58:1759–1764. doi: 10.1016/s0006-2952(99)00269-5. [DOI] [PubMed] [Google Scholar]
- Carmichael SL, Shaw GM, Yang W, Iovannisci DM, Lammer E. Risk of limb deficiency defects associated with NAT1, NAT2, GSTT1, GSTM1, and NOS3 genetic variants, maternal smoking, and vitamin supplement intake. Am J Med Genet A. 2006;140:1915–1922. doi: 10.1002/ajmg.a.31402. [DOI] [PubMed] [Google Scholar]
- Chen B, Zhang WX, Cai WM. The influence of various genotypes on the metabolic activity of NAT2 in a Chinese population. Eur J Clin Pharmacol. 2006;62:355–359. doi: 10.1007/s00228-006-0110-6. [DOI] [PubMed] [Google Scholar]
- Chen J, Stampfer MJ, Hough HL, Garcia-Closas M, Willett WC, Hennekens CH, Kelsey KT, Hunter DJ. A prospective study of N-acetyltransferase genotype, red meat intake, and risk of colorectal cancer. Cancer Res. 1998;58:3307–3311. [PubMed] [Google Scholar]
- de Leon JH, Vatsis KP, Weber WW. Characterization of naturally occurring and recombinant human N-acetyltransferase variants encoded by NAT1. Mol Pharmacol. 2000;58:288–299. doi: 10.1124/mol.58.2.288. [DOI] [PubMed] [Google Scholar]
- Doll MA, Zang Y, Moeller T, Hein DW. Codominant expression of N-acetylation and O-acetylation activities catalyzed by N-acetyltransferase 2 in human hepatocytes. J Pharmacol Exp Ther. 2010;334:540–544. doi: 10.1124/jpet.110.168567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RP, Cao W, Acuna DK, Strnatka DW, Hunter RJ, Chau BT, Wakefield LV, Sim E, McQueen CA. Confirmation of the role of N-acetyltransferase 2 in teratogen-induced cleft palate using transgenics and knockouts. Mol Reprod Dev. 2008;75:1071–1076. doi: 10.1002/mrd.20852. [DOI] [PubMed] [Google Scholar]
- Fretland AJ, Doll MA, Leff MA, Hein DW. Functional characterization of nucleotide polymorphisms in the coding region of N-acetyltransferase 1. Pharmacogenetics. 2001;11:511–520. doi: 10.1097/00008571-200108000-00006. [DOI] [PubMed] [Google Scholar]
- Fretland AJ, Doll MA, Zhu Y, Smith L, Leff MA, Hein DW. Effect of nucleotide substitutions in N-acetyltransferase-1 on N-acetylation (deactivation) and O-acetylation (activation) of arylamine carcinogens: implications for cancer predisposition. Cancer Detect Prev. 2002;26:10–14. doi: 10.1016/s0361-090x(02)00005-3. [DOI] [PubMed] [Google Scholar]
- Gago-Dominguez M, Bell DA, Watson MA, Yuan JM, Castelao JE, Hein DW, Chan KK, Coetzee GA, Ross RK, Yu MC. Permanent hair dyes and bladder cancer: risk modification by cytochrome P4501A2 and N-acetyltransferases 1 and 2. Carcinogenesis. 2003;24:483–489. doi: 10.1093/carcin/24.3.483. [DOI] [PubMed] [Google Scholar]
- Gemignani F, Landi S, Szeszenia-Dabrowska N, Zaridze D, Lissowska J, Rudnai P, Fabianova E, Mates D, Foretova L, Janout V, Bencko V, Gaborieau V, Gioia-Patricola L, Bellini I, Barale R, Canzian F, Hall J, Boffetta P, Hung RJ, Brennan P. Development of lung cancer before the age of 50: the role of xenobiotic metabolizing genes. Carcinogenesis. 2007;28:1287–1293. doi: 10.1093/carcin/bgm021. [DOI] [PubMed] [Google Scholar]
- Grant DM, Hughes NC, Janezic SA, Goodfellow GH, Chen HJ, Gaedigk A, Yu VL, Grewal R. Human acetyltransferase polymorphisms. Mutat Res. 1997;376:61–70. doi: 10.1016/s0027-5107(97)00026-2. [DOI] [PubMed] [Google Scholar]
- Hein DW. Acetylator genotype and arylamine-induced carcinogenesis. Biochim Biophys Acta. 1988;948:37–66. doi: 10.1016/0304-419x(88)90004-2. [DOI] [PubMed] [Google Scholar]
- Hein DW. N-acetyltransferase SNPs: emerging concepts serve as a paradigm for understanding complexities of personalized medicine. Expert Opin Drug Metab Toxicol. 2009;5:353–366. doi: 10.1517/17425250902877698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein DW, Leff MA, Ishibe N, Sinha R, Frazier HA, Doll MA, Xiao GH, Weinrich MC, Caporaso NE. Association of prostate cancer with rapid N-acetyltransferase 1 (NAT1*10) in combination with slow N-acetyltransferase 2 acetylator genotypes in a pilot case-control study. Environ Mol Mutagen. 2002;40:161–167. doi: 10.1002/em.10103. [DOI] [PubMed] [Google Scholar]
- Hughes NC, Janezic SA, McQueen KL, Jewett MA, Castranio T, Bell DA, Grant DM. Identification and characterization of variant alleles of human acetyltransferase NAT1 with defective function using p-aminosalicylate as an in-vivo and in-vitro probe. Pharmacogenetics. 1998;8:55–66. doi: 10.1097/00008571-199802000-00008. [DOI] [PubMed] [Google Scholar]
- Husain A, Zhang X, Doll MA, States JC, Barker DF, Hein DW. Functional analysis of the human N-acetyltransferase 1 major promoter: quantitation of tissue expression and identification of critical sequence elements. Drug Metab Dispos. 2007;35:1649–1656. doi: 10.1124/dmd.107.016485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibe N, Sinha R, Hein DW, Kulldorff M, Strickland P, Fretland AJ, Chow WH, Kadlubar FF, Lang NP, Rothman N. Genetic polymorphisms in heterocyclic amine metabolism and risk of colorectal adenomas. Pharmacogenetics. 2002;12:145–150. doi: 10.1097/00008571-200203000-00008. [DOI] [PubMed] [Google Scholar]
- Jensen LE, Hoess K, Whitehead AS, Mitchell LE. The NAT1 C1095A polymorphism, maternal multivitamin use and smoking, and the risk of spina bifida. Birth Defects Res A Clin Mol Teratol. 2005;73:512–516. doi: 10.1002/bdra.20143. [DOI] [PubMed] [Google Scholar]
- Jiao L, Doll MA, Hein DW, Bondy ML, Hassan MM, Hixson JE, Abbruzzese JL, Li D. Haplotype of N-acetyltransferase 1 and 2 and risk of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:2379–2386. doi: 10.1158/1055-9965.EPI-06-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T, Inatomi H, Yang M, Kawamoto T, Matsumoto T, Bell DA. Arylamine N-acetyltransferase 1 (NAT1) and 2 (NAT2) genes and risk of urothelial transitional cell carcinoma among Japanese. Pharmacogenetics. 1999;9:401–404. doi: 10.1097/00008571-199906000-00017. [DOI] [PubMed] [Google Scholar]
- Kilfoy BA, Zheng T, Lan Q, Han X, Holford T, Hein DW, Qin Q, Leaderer B, Morton LM, Yeager M, Boyle P, Zhao P, Chanock S, Rothman N, Zhang Y. Genetic variation in N-acetyltransferases 1 and 2, cigarette smoking, and risk of non-Hodgkin lymphoma. Cancer Causes Control. 2010;21:127–133. doi: 10.1007/s10552-009-9442-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammer EJ, Shaw GM, Iovannisci DM, Finnell RH. Periconceptional multivitamin intake during early pregnancy, genetic variation of acetyl-N-transferase 1 (NAT1), and risk for orofacial clefts. Birth Defects Res A Clin Mol Teratol. 2004;70:846–852. doi: 10.1002/bdra.20081. [DOI] [PubMed] [Google Scholar]
- Lammer EJ, Shaw GM, Iovannisci DM, Van Waes J, Finnell RH. Maternal smoking and the risk of orofacial clefts: Susceptibility with NAT1 and NAT2 polymorphisms. Epidemiology. 2004;15:150–156. doi: 10.1097/01.ede.0000112214.33432.cc. [DOI] [PubMed] [Google Scholar]
- Lee KM, Park SK, Kim SU, Doll MA, Yoo KY, Ahn SH, Noh DY, Hirvonen A, Hein DW, Kang D. N-acetyltransferase (NAT1, NAT2) and glutathione S-transferase (GSTM1, GSTT1) polymorphisms in breast cancer. Cancer Lett. 2003;196:179–186. doi: 10.1016/s0304-3835(03)00311-2. [DOI] [PubMed] [Google Scholar]
- Li D, Jiao L, Li Y, Doll MA, Hein DW, Bondy ML, Evans DB, Wolff RA, Lenzi R, Pisters PW, Abbruzzese JL, Hassan MM. Polymorphisms of cytochrome P4501A2 and N-acetyltransferase genes, smoking, and risk of pancreatic cancer. Carcinogenesis. 2006;27:103–111. doi: 10.1093/carcin/bgi171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilla C, Verla-Tebit E, Risch A, Jager B, Hoffmeister M, Brenner H, Chang-Claude J. Effect of NAT1 and NAT2 genetic polymorphisms on colorectal cancer risk associated with exposure to tobacco smoke and meat consumption. Cancer Epidemiol Biomarkers Prev. 2006;15:99–107. doi: 10.1158/1055-9965.EPI-05-0618. [DOI] [PubMed] [Google Scholar]
- Ma JJ, Liu CG, Li JH, Cao XM, Sun SL, Yao X. Effects of NAT2 polymorphism on SASP pharmacokinetics in Chinese population. Clin Chim Acta. 2009;407:30–35. doi: 10.1016/j.cca.2009.06.025. [DOI] [PubMed] [Google Scholar]
- Millikan RC, Pittman GS, Newman B, Tse CK, Selmin O, Rockhill B, Savitz D, Moorman PG, Bell DA. Cigarette smoking, N-acetyltransferases 1 and 2, and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 1998;7:371–378. [PubMed] [Google Scholar]
- Morton LM, Bernstein L, Wang SS, Hein DW, Rothman N, Colt JS, Davis S, Cerhan JR, Severson RK, Welch R, Hartge P, Zahm SH. Hair dye use, genetic variation in N-acetyltransferase 1 (NAT1) and 2 (NAT2), and risk of non-Hodgkin lymphoma. Carcinogenesis. 2007;28:1759–1764. doi: 10.1093/carcin/bgm121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton LM, Schenk M, Hein DW, Davis S, Zahm SH, Cozen W, Cerhan JR, Hartge P, Welch R, Chanock SJ, Rothman N, Wang SS. Genetic variation in N-acetyltransferase 1 (NAT1) and 2 (NAT2) and risk of non-Hodgkin lymphoma. Pharmacogenet Genomics. 2006;16:537–545. doi: 10.1097/01.fpc.0000215071.59836.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovito PM, Jr., Morse PD, Spinek K, Newman N, Jones RF, Wang CY, Haas GP. Heterocyclic amines and genotype of N-acetyltransferases as risk factors for prostate cancer. Prostate Cancer Prostatic Dis. 2005;8:69–74. doi: 10.1038/sj.pcan.4500780. [DOI] [PubMed] [Google Scholar]
- Sanderson S, Salanti G, Higgins J. Joint effects of the N-acetyltransferase 1 and 2 (NAT1 and NAT2) genes and smoking on bladder carcinogenesis: a literature-based systematic HuGE review and evidence synthesis. Am J Epidemiol. 2007;166:741–751. doi: 10.1093/aje/kwm167. [DOI] [PubMed] [Google Scholar]
- Shi M, Wehby GL, Murray JC. Review on genetic variants and maternal smoking in the etiology of oral clefts and other birth defects. Birth Defects Res C Embryo Today. 2008;84:16–29. doi: 10.1002/bdrc.20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin A, Shrubsole MJ, Rice JM, Cai Q, Doll MA, Long J, Smalley WE, Shyr Y, Sinha R, Ness RM, Hein DW, Zheng W. Meat intake, heterocyclic amine exposure, and metabolizing enzyme polymorphisms in relation to colorectal polyp risk. Cancer Epidemiol Biomarkers Prev. 2008;17:320–329. doi: 10.1158/1055-9965.EPI-07-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim E, Payton M, Noble M, Minchin R. An update on genetic, structural and functional studies of arylamine N-acetyltransferases in eucaryotes and procaryotes. Hum Mol Genet. 2000;9:2435–2441. doi: 10.1093/hmg/9.16.2435. [DOI] [PubMed] [Google Scholar]
- Smelt VA, Upton A, Adjaye J, Payton MA, Boukouvala S, Johnson N, Mardon HJ, Sim E. Expression of arylamine N-acetyltransferases in pre-term placentas and in human pre-implantation embryos. Hum Mol Genet. 2000;9:1101–1107. doi: 10.1093/hmg/9.7.1101. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Morris JS, Li Y, Doll MA, Hein DW, Liu J, Jiao L, Hassan MM, Day RS, Bondy ML, Abbruzzese JL, Li D. Interaction of the cytochrome P4501A2, SULT1A1 and NAT gene polymorphisms with smoking and dietary mutagen intake in modification of the risk of pancreatic cancer. Carcinogenesis. 2008;29:1184–1191. doi: 10.1093/carcin/bgn085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JA, Umbach DM, Stephens E, Castranio T, Paulson D, Robertson C, Mohler JL, Bell DA. The role of N-acetylation polymorphisms in smoking-associated bladder cancer: evidence of a gene-gene-exposure three-way interaction. Cancer Res. 1998;58:3603–3610. [PubMed] [Google Scholar]
- Wakefield L, Cornish V, Long H, Griffiths WJ, Sim E. Deletion of a xenobiotic metabolizing gene in mice affects folate metabolism. Biochem Biophys Res Commun. 2007;364:556–560. doi: 10.1016/j.bbrc.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walraven JM, Trent JO, Hein DW. Structure-function analyses of single nucleotide polymorphisms in human N-acetyltransferase 1. Drug Metab Rev. 2008;40:169–184. doi: 10.1080/03602530701852917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikman H, Thiel S, Jager B, Schmezer P, Spiegelhalder B, Edler L, Dienemann H, Kayser K, Schulz V, Drings P, Bartsch H, Risch A. Relevance of N-acetyltransferase 1 and 2 (NAT1, NAT2) genetic polymorphisms in non-small cell lung cancer susceptibility. Pharmacogenetics. 2001;11:157–168. doi: 10.1097/00008571-200103000-00006. [DOI] [PubMed] [Google Scholar]
- Zang Y, Zhao S, Doll MA, States JC, Hein DW. The T341C (Ile114Thr) polymorphism of N-acetyltransferase 2 yields slow acetylator phenotype by enhanced protein degradation. Pharmacogenetics. 2004;14:717–723. doi: 10.1097/00008571-200411000-00002. [DOI] [PubMed] [Google Scholar]
- Zhangwei X, Jianming X, Qiao M, Xinhua X. N-Acetyltransferase-1 gene polymorphisms and correlation between genotype and its activity in a central Chinese Han population. Clin Chim Acta. 2006;371:85–91. doi: 10.1016/j.cca.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Zheng W, Deitz AC, Campbell DR, Wen WQ, Cerhan JR, Sellers TA, Folsom AR, Hein DW. N-acetyltransferase 1 genetic polymorphism, cigarette smoking, well-done meat intake, and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 1999;8:233–239. [PubMed] [Google Scholar]
- Zhu Y, Hein DW. Functional effects of single nucleotide polymorphisms in the coding region of human N-acetyltransferase 1. Pharmacogenomics J. 2008;8:339–348. doi: 10.1038/sj.tpj.6500483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure. Cloned 1175 bp NAT1 fragment (Reference sequence published in Genbank Assession Number X17059). The fragment consists of the 873-bp coding region and 302-bp of the 3′-UTR; ATG; start codon; TAG: stop codon; AATAAA: putative polyadenylation signal polyA-1. The locations of the 445G>A, 459G>A, 640T>G, 1088T>A, and 1095C>A SNPs are shown by increased font size. The region of the 9-bp deletion (three TAA triplets) is underlined.