

Abstract
Synthetic polymer nanoparticles (NPs) that bind venomous molecules and neutralize their function in vivo are of significant interest as “plastic antidotes.” Recently, procedures to synthesize polymer NPs with affinity for target peptides have been reported. However, the performance of synthetic materials in vivo is a far greater challenge. Particle size, surface charge, and hydrophobicity affect not only the binding affinity and capacity to the target toxin but also the toxicity of NPs and the creation of a “corona” of proteins around NPs that can alter and or suppress the intended performance. Here, we report the design rationale of a plastic antidote for in vivo applications. Optimizing the choice and ratio of functional monomers incorporated in the NP maximized the binding affinity and capacity toward a target peptide. Biocompatibility tests of the NPs in vitro and in vivo revealed the importance of tuning surface charge and hydrophobicity to minimize NP toxicity and prevent aggregation induced by nonspecific interactions with plasma proteins. The toxin neutralization capacity of NPs in vivo showed a strong correlation with binding affinity and capacity in vitro. Furthermore, in vivo imaging experiments established the NPs accelerate clearance of the toxic peptide and eventually accumulate in macrophages in the liver. These results provide a platform to design plastic antidotes and reveal the potential and possible limitations of using synthetic polymer nanoparticles as plastic antidotes.
Keywords: antidote, molecular recognition, nanomedicine, plastic antibody, toxicity
Synthetic nanoparticles (NPs) that are capable of recognizing and capturing venomous biomacromolecules and neutralizing their toxicity in vivo are of significant interest as a new class of antidote (1–4). Compared to small molecular antidotes, they cover a much wider area of the surface of target macromolecules through multipoint interactions, which enables efficient neutralization of the targets’ toxicity. In the meantime, the NPs are small enough to diffuse into almost all areas of the body, including blood capillaries, organs, and even inside cells. Furthermore, their adsorbing capacity is substantial since nano-size materials have high surface areas compared to bulk materials.
The design of synthetic NPs capable of capturing target biomacromolecules is a formidable challenge. In nature, strong interactions between biomacromolecules arise from multiple weak interactions comprised of electrostatic, hydrophobic, hydrogen bonding, and van der Waals interactions on complementary three-dimensional binding surfaces. In an effort to mimic these interactions, bulk polymer materials that capture target molecules by multipoint interactions have been synthesized by incorporating moderate amounts of functional monomers that interact with target molecules primarily by electrostatic interactions (5). Nano-size materials with affinity for a target peptide or protein have also been synthesized by optimizing the composition and/or ratio of functional groups on the surface of the synthetic NPs (6). Some success was achieved by a judicious choice of ligands used to coat gold NPs or by stabilizing charged groups on dendritic polymers (7, 8). Linear polyacrylamides that are functionalized with arginine receptors have also been shown to interact with arginine-rich proteins (9). It has also been demonstrated that polymer NPs synthesized with an optimized combination of functional monomers can capture target molecules (10) and neutralize its function (6). However, little has been reported about a general design rationale for achieving NPs with molecular recognition for in vivo applications (3, 8).
For NPs to neutralize the function of target molecules in vivo, they must be stable, biologically inert, and nontoxic. They also have to remain in the bloodstream for a sufficient time to enable capture of target molecules. It has been reported that the NPs smaller than approximately 8 nm will be cleared rapidly from the blood stream by the renal system and NPs larger than 200 nm will be sequestered by the mononuclear phagocytic system (MRS) in the liver and spleen (11–14). Hydrophobicity, charge, flexibility, and shape of NPs are also important; for example hydrophobic particles induce formation of a corona of serum proteins around the surface and strongly charged NPs will be phagocytosed by MRS faster than neutral particles (11–18). Although the size of NPs can be adjusted, surface charges and hydrophobicity of plastic antidotes cannot always be optimized to increase circulation time since surface functionality must be designed to maximize affinity and capacity to target molecules. This limitation requires a fundamentally different rationale for designing nanoparticles for toxin neutralization or applications such as drug carriers or in vivo imaging.
Previously, we have shown that copolymer NPs consisting of random distributions of both hydrophobic and negatively charged monomers captures the peptide toxin melittin by both hydrophobic and electrostatic interactions (6). However, the combined affinity and specificity of the NPs were not sufficient to detoxify melittin in animal models. Recently, we demonstrated that by applying a molecular imprinting (19) or an affinity purification process (20) together with the combination of functional monomers, NPs with greater affinity to the target peptide was achieved. Furthermore, the NPs with enhanced binding affinity showed neutralization of the toxin in vivo (3). However, other than toxin capture, little has been reported about the important requirements of plastic antidotes for in vivo applications such as biostability, biotoxicity, and biodistribution. In this study, we focus on a design rational that includes optimizing monomer composition for a plastic antidote for in vivo applications. The synthesis of NPs with intrinsically high affinity and selectivity to a target toxin without molecular imprinting or affinity purification would streamline the process of antidote development. Our approach begins with preparation of a library of tailored multifunctional copolymer nanoparticles that allows us to systematically investigate how the composition (hydrophobicity and charge) and size of the particles affects their binding affinity and capacity. The composition of functional groups in the NP is quantified by 1H-NMR and inverse-gated 13C-NMR spectroscopy utilizing a 13C-enriched monomer. This information is used to evaluate the stoichiometry of binding of each NP. The relationship between NP composition and cytotoxicity and biocompatibility are also systematically examined in vitro and in vivo. The detoxification efficiency of optimized NPs is further analyzed in vivo. Finally, the in vivo neutralization mechanism is discussed using biodistribution data of 14C-labeled NPs and fluorescent-labeled melittin together with a histological analysis of organs. From these results, the scope and limitations of using synthetic polymer nanoparticles as plastic antibodies are discussed.
In this report, melittin was selected as the target peptide to establish the design rational of plastic antidotes for in vivo applications. Melittin, a twenty-six amino acid cytolytic peptide isolated from bee venom, is a representative of membrane damaging toxins (21). Although, toxins in this family function as key virulence factors of pathologies resulting from infectious diseases (22, 23) and animal bites/stings (24, 25), little has been reported about designing antidotes since it is hard to capture a wide area of their molecular surface that contributes their toxicity. Thus, an effective strategy to neutralize the activity of such toxins is to capture them on the surface or interior of nanomaterials.
Results
Preparation of NPs.
We first optimized polymerization conditions to produce multifunctional polymer nanoparticles with a monomodal size distribution of approximately 50 nm. As a particle core, we chose N-isopropylacrylamide (NIPAm) cross-linked with 2 mol% N, N′-methylenebisacrylamide (Bis). N-t-butylacrylamide (TBAm), acrylic acid (AAc), and N-3-aminopropyl methacrylamide (APM) were used as hydrophobic, negatively charged and positively charged functional monomers. All NPs except those containing APM were synthesized in the presence of an anionic surfactant (sodium dodecyl sulfate; SDS) by free radical polymerization initiated with ammonium persulfate. NPs with APM were synthesized in the presence of a cationic surfactant (cetyltrimethylammonium bromide; CTAB), initiated with nonionic initiator (azobisisobutyronitrile AIBN) to prevent aggregation during polymerization. By optimizing concentration of surfactant and monomers, and reaction temperature, a library of multifunctional polymer NPs with a monomodal size distribution, consisting of varied combinations of functional monomers, were prepared (Table 1, Fig. 1A). Optimized conditions for the preparation of each NP are described in the SI Text. Diameters of NPs were controlled to approximately 50 nm to minimize renal and MRS clearance.
Table 1.
Yield and diameter of standard NPs
| NP# | Feed ratio of functional monomers [%] * | Yield [%] | Diameter † [nm] | ||
| TBAm | AAc | APM | |||
| 1 | 0 | 0 | 0 | 75 | 56 ± 3 |
| 2 | 40 | 0 | 0 | 90 | 59 ± 11 |
| 3 | 0 | 5 | 0 | 100 | 18 ± 9 |
| 4 | 40 | 5 | 0 | 51 | 67 ± 2 |
| 5 | 0 | 0 | 5 | 70 | 63 ± 9 |
| 6 | 40 | 0 | 5 | 38 | 79 ± 1 |
| 7 | 40 | 1 | 0 | 58 | 56 ± 2 |
| 8 | 40 | 10 | 0 | 85 | 58 ± 1 |
| 9 | 40 | 40 | 0 | 58 | 52 ± 1 |
*NIPAm is present according to the following relationship (NIPAm = 98 - TBAm + AAc + APM). In all cases Bis is present as 2% of the total monomer composition.
†Average—standard deviation of three sequential measurements.
Fig. 1.

Interaction between melittin and NPs synthesized with various feed ratio of TBAm and AAc. (A) AFM image of NPs 9. (B) Neutralization constants of NPs obtained from hemolytic toxicity neutralization assay. C. Apparent binding constant between melittin and NPs obtained from 27-MHz QCM experiments. Red spots on graphs (B) and (C) indicate NPs that did not show neutralization or melittin binding, respectively. Blue spots indicate polymers that precipitated during polymerization or purification. (D) The effect of AAc incorporation on the binding capacities (blue, left axis) and apparent binding constants (green, right axis) of NPs that were polymerized with 40 mol% TBAm. Note that left axis is linear and the right is logarithmic. (E) Effect of AAc incorporation on the stoichiometry between melittin and monomer unit (black, left axis) and melittin and AAc (red, right axis).
NP Binding Capacity of Melittin.
The neutralization efficiency of melittin by NPs 1–6 was tested by the red blood cell lyses test. As we reported, NPs with both carboxylic acid (AAc) and t-butyl groups (TBAm) (NPs 4) significantly neutralized melittin (6). Melittin is comprised primarily of positively charged and hydrophobic amino acids. We attribute the neutralization to a combination of electrostatic and hydrophobic interactions between negatively charged and hydrophobic polymer chains and melittin. In contrast, NPs with both primary amine (APM) and t-butyl groups (TBAm) (NPs 6) accelerated red blood cell lysis. This may due to electrostatic interactions between the cationic polymer and anionic cell surface as well as an interaction between the hydrophobic polymer domain and the cell membrane. Other NPs showed neither neutralization nor acceleration of hemolysis.
To further optimize monomer ratio, a library of NPs with different feed ratios of AAc and TBAm were prepared. The neutralization constants, which reflect the melittin-binding capacity per gram of NP, were calculated from the red blood cell lysis test (6) and plotted against feed ratio of AAc and TBAm (Fig. 1B). The neutralization constants increased with more AAc or TBAm in the monomer feed, although NPs with more than 40 mol% of AAc or TBAm could not be tested since they precipitated during the polymerization process. NPs with the highest AAc (40 mol%) and TBAm (40 mol%) (NPs 9) showed the highest melittin-binding capacity, 180 μmol g-1 (0.5 g melittin per a gram of NP). This capacity is 105 or 102 times larger than that of previously reported protein adsorbing films (5, 26) or nano-fibers (27) and more than 10 times greater than that of immunoglobulins (IgGs; 2 binding sites per 150 kDa = 13 μmol g-1). The binding capacity indicates that on average a 50 mer polymer fragment captures a molecule of melittin (26 amino acid) (Table 2), suggesting that most of the polymer chains in NP 9 are in contact with melittin.
Table 2.
Stoichiometry between melittin and NP monomer composition
| NP# | AAc Feed | AAc Incorp. | Unit per Mel | TBAm per Mel | AAc per Mel | Ka(app) [105 M-1] |
| 7 | 1 | 0.5 | 500 | 200 | 2 | N.D. |
| 4 | 5 | 2 | 100 | 41 | 2 | 4.6 |
| 8 | 10 | 5 | 76 | 31 | 4 | 16 |
| 9 | 40 | 19 | 50 | 25 | 10 | 660 |
To investigate the importance of particle size on the binding capacity, polymer NPs were reconstructed as a bulk film by casting from ethanol solution. In this study, the films showed little neutralization of melittin, indicating the small size of NPs (approximately 50 nm) enabled the particles to capture vast amounts of target peptide by exposing a large area of the affinity surface to the solution.
NP Binding Affinity of Melittin.
The apparent binding constant of NPs was calculated from the Langmuir isotherm obtained from a quartz crystal microbalance analysis (6, 19). Only NPs with a high feed ratio of both AAc and TBAm showed measurable apparent binding constants for melittin, NPs with highest AAc and TBAm (NPs 9) showed the highest apparent binding constant of 660 × 105 (M-1) (Fig. 1C, Table 2).
Stoichiometry between Melittin and NP Functional Groups.
From solution 1H-NMR and inverse-gated 13C-NMR spectroscopy we confirmed that TBAm was almost quantitatively incorporated in the copolymers. From inverse-gated 13C-NMR spectroscopy of NPs copolymerized with 13C-enriched acrylic acid, we quantified the incorporation of AAc to be 46% of the feed ratio. The binding capacity and stoichiometry between each functional monomer and melittin are calculated from the neutralization constants (Table 2). Binding capacity and affinity were also plotted against the feed ratio of AAc (Fig. 1D). Interestingly, the binding capacity increased linearly with increasing AAc at low AAc levels (0.1% ∼ 10%) then reached saturation at higher levels of AAc; in contrast, the apparent binding constant to melittin increased exponentially over the entire range of AAc. Analysis of the binding stoichiometry between AAc and melittin (Table 2, Fig. 1E) reveals that each melittin is captured by approximately 2 carboxylic acids that are distributed on a 500 ∼ 100 mer segment of polymer in the low AAc feed range (1% ∼ 10%), increasing after 10% up to 10 AAc per melittin distributed over 50 mer polymer segments. This analysis suggests that multipoint electrostatic interactions between the positive charges on melittin and the carboxylate anions on the polymer enable the strong interaction between melittin and NP 9 (40 mol% AAc feed), whereas weaker melittin-NP interactions are observed for the lower AAc containing NPs such as NP 4 (5 mol% AAc feed).
Cytotoxicity of NPs In Vitro.
The cytotoxicity of each NP in the library was tested in vitro using HT-1080 cells. In the assay, only the particles with both hydrophobic and positively charged functional groups (NPs 6) showed cytotoxicity at concentrations of 0.3 μg mL-1 (Fig. 2A). At 3 μg mL-1, particles with only hydrophobic groups (NP 2) also showed some cytotoxicity. However, other particles consisting of AAc and TBAm (NPs 4 and NP 9) showed no cytotoxicity (up to 3000 μg mL-1, Fig. S1). Those results indicate that particle hydrophobicity induces toxicity and the addition of positive charge enhances the toxicity. Negative charges on the hydrophobic particle create electrostatic repulsion to the negatively charged phosphate groups on the cell surfaces minimizing interaction with the hydrophobic cell membrane.
Fig. 2.

Biocompatibility of NPs in vitro and in vivo. (A) Cytotoxicity of NPs towards HT-1080 cells, determined by the Alamar Blue® assay. NPs at the indicated concentrations (0.3 μg mL-1 and 3 μg mL-1) were incubated with cells for 24 h. The error bars indicate s.d. (B) Amount of NP aggregation that formed by incubation with mouse plasma (37 °C, 1 hour) followed with centrifugation (13,000 rpm, 5 min). (C) Change in body mass of mice injected with NP4 (blue) and NP9 (red)(n = 3, dose = 10 mg kg-1) compared with control (isotonic glucose solution, n = 3). There is no statistically significant difference in the mass change between control and NPs over a period of 2 weeks. The error bars indicate s.d.
Stability of NPs in Plasma.
To test the stability of NPs in plasma, each NP was labeled with the 5-(dimethylamino)naphthalene-1-sulfonyl (DNS) group (fluorescent dye) by incorporating 1 mol% of N-[2-[[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]amino]ethyl]-2- propenamide (DNS methacrylate) in the polymerization reaction. Then, DNS-labeled NPs (0.1 mg mL-1) were incubated with mouse plasma for 1 h at 37 °C and centrifuged (15,000 × G). NPs in the plasma before and after the incubation-centrifugation process were quantified by the florescent intensity of the solution. The amount of NPs that were aggregated with plasma were quantified from the difference of fluorescent intensity and plotted against functional monomer feed ratio (Fig. 2B). Most of the NPs with high TBAm (hydrophobic) loadings with or without positively charged monomers aggregated in plasma. In contrast most NPs with high TBAm and a critical amount of AAc (negatively charged) remained in the plasma. This same trend is observed in the cytotoxicity studies (Fig. 2A). Poly NIPAm NPs with a high TBAm ratio without AAc are known to interact with abundant plasma proteins such as albumin and fibrinogen (15). In our case, the hydrophobicity of TBAm containing nanoparticles can induce nonspecific interactions with these same abundant plasma proteins resulting in aggregation. However, introduction of a certain amount of negative charge from AAc monomers prevents aggregation perhaps due to electrostatic repulsion since albumin and fibrinogen are negatively charged under physiological conditions (28).
Biocompatibility of NPs 4 and 9 In Vivo.
NPs that were effective in neutralizing melittin (NPs 4 and NPs 9) were injected (10 mg kg-1) intravenously into mice to evaluate in vivo toxicity. Over a period of 2 weeks, there was no significant difference in body weight between groups administered NPs and control mice (Fig. 2C). There was no significant toxicity observed histopathologically in liver, lung, and kidney tissue 2 weeks after injection (Fig. S2).
Detoxification of Melittin by NPs In Vivo.
The ability of NPs to neutralize melittin’s toxicity was tested in vivo by systemic administration. A high dose of melittin is known to induce cell lyses including hemolysis and myolysis, eventually resulting in death due to renal failure and/or cardiac complications. In this study, mice were injected intravascularly with melittin followed by intravascular injection of NPs. The controls did not receive the injection of NPs. A 100 percent mortality rate was observed in mice that were intravenously administered melittin at a dose of 4.5 mg kg-1 (8 animals, Fig. 3A). Upon intravenous infusion of NP 9 (30 mg kg-1) 20 s after 4.5 mg kg-1 of melittin, a significant decrease in mortality was observed (p value is 0.0002; 8 animals). In contrast, NP 4 did not significantly detoxify melittin in vivo (p value 0.05, 9 animals), although NP 4 showed significant detoxification in vitro. This indicates particles with high binding affinity and capacity are necessary for melittin detoxification in vivo. Furthermore, administration of NP 2 (30 mg kg-1) (polymerized without AAc) showed greater mortality than without administration of NPs (p value 0.014,12 animals), suggesting the importance of reducing cytotoxicity and nonspecific interactions with plasma proteins by incorporating negatively charged monomers in the hydrophobic NPs. Surprisingly, NP 9 (30 mg kg-1) significantly decreased mortality of the mice when the particles were injected 5 min after melittin injection (3.4 mg kg-1) as well (5 animals, p value 0.046, Fig. S3). These results indicate the potential use of these particles for therapeutic applications.
Fig. 3.

Detoxification of melittin in mice by systemic administration of NPs. (A) Survival rates of mice over a 24 h period after intravenous injection of 4.5 mg kg-1 melittin (green). 30 mg kg-1 of NP2 (blue), NP4 (red), and NP9 (black) was systemically administrated via a tail vein 20 s after melittin injection. P values are calculated by the Willcoxon test. (B) Level of inflammation of mice quantified by gross pathology (96 h after poisoning). Left two columns; without melittin with/without 30 mg kg-1 of NP9, right two columns; with 3.8 mg kg-1 of melittin followed with 30 mg kg-1 of NP9. (C) Weight change of surviving mice 48 h after melittin injection, followed by (red) and without (black) administration of 30 mg kg-1 of NP 9. Horizontal bars indicate the mean weight change percentage. (D). Survival rate of mice 24 h after intravenous injection of melittin, followed with (green) or without (blue) administration of 30 mg kg-1 of NP9. 50% lethal doses (LD50) of each condition are printed on the 50% surviving line.
In addition to mortality, systemic administration of melittin induced significant peritoneal inflammation (Fig. 3B) and weight loss (melittin 4.5 mg kg-1, p value 0.005; Fig. 3C) in surviving mice. These symptoms were significantly alleviated by systemic administration of 30 mg kg-1 NPs 9 (Fig. 3B, p value 0.0037; 3C, p value 0.0007).
To estimate the detoxification capacity of NPs in vivo, the survival rate of mice was plotted against the melittin dose with and without post administration of 30 mg kg-1 (Fig. 3D) of NPs 9. From the shift of 50% lethal dose from 3.2 mg kg-1 to 5.8 mg kg-1, the detoxification capacity of NP 9 in vivo was calculated as 87 mg g-1 (30 μmol g-1). This is about five times smaller than the binding capacity estimated in vitro from the red blood cell test. The difference may due to competition from plasma proteins and biodistribution of melittin and NPs. Although, the detoxification capacity is still twice that of IgG (13 μmol g-1).
Distribution of NPs and melittin In Vivo.
In vivo imaging of fluorescent-labeled melittin (Cy7- or Cy5-melittin) reveals that the biodistribution of melittin is significantly altered by post administration of NP 9. Cy7- melittin is distributed throughout the body immediately after injection and circulates in the body for more than 30 min (Fig. 4A, Left, deep blue regions). The biodistribution did not change significantly by post administration of NP 4 (Fig. 4A, Middle, deep blue regions), however, the fluorescent intensity of Cy7-melittin diminished immediately after administration of NPs 9 (Fig. 4A, Right, white regions). Ex vivo results show the distribution of Cy5- melittin in each organ 10 min after administration of melittin with or without a follow on dose of NP 9. Cy5-melittin does not accumulate in any organ during this time unless administered NP 9 (Fig. 4B, Left). With NP 9, melittin is concentrated in the liver (Fig. 4B, Right, yellow to red on liver).
Fig. 4.

Biodistribution of melittin and NPs. (A) Left; fluorescent images of Cy7-melittin after intravenous injection of Cy7-melittin (0.3 mg kg-1). 10 mg kg-1 of NP4 (Center) or NP9 (Right) were injected 20 s after the injection of melittin. (B) Fluorescent ex vivo images of Cy5-melittin (0.3 mg kg-1, 10 min after injection) of mice followed with (Left) and without (Right) 10 mg kg-1 NP9. Li, Sp, SI, K, H, and Lu indicate liver, spleen, small intestine, kidney, heart, and lung, respectively. (C) Biodistribution of 14C-labeled NP9 (30 mg kg-1) in mice (n = 5) 30 min after administration. (D) Fluorescence histology images of a liver 70 min after injection of Cy5-melittin 0.3 mg kg-1 and 10 mg kg-1 of NP9. Green; fluoroscein-NP9, red; Cy5-melittin, yellow; merged. The scale bars; 25 μm.
To study the biodistribution of NPs, NPs 9 was labeled with 14C or fluoroscein by copolymerizing with 5 mol% of acrylamide [1-14C] or 1 mol% of fluoroscein o-acrylate. The biodistribution of 14C-labeled NP 9 30 min after administration revealed that NPs accumulated mainly in the liver and kidney (Fig. 4C), indicating that the NPs were recognized by macrophages in the liver similar to the fate of other nano objects of similar size (11–14). A histological study of a section of liver showed that both fluorescein labeled NP 9 and Cy5-melittin were found together in the same cells in the liver 70 min after injection of melittin and NP 9 (Fig. 4D).
These results reveal that melittin is captured by NP 9 in the blood stream and subsequently removed from the blood by macrophages in the liver as a complex with NP 9.
Discussion
Binding Capacity, Affinity, and Mechanism of Melittin In Vitro.
Confirming our earlier observations, polymer NPs that capture target toxin by both hydrophobic and electrostatic interactions were prepared by choosing a combination of functional monomers from the library of multifunctional polymer NPs. Binding capacity and affinity were further maximized by optimizing the feed ratio of the functional monomers. The optimized NPs showed a much higher apparent binding constant (6.6 × 107 M-1) than NPs reported earlier (1.6 × 106 M-1) (6) and had an extremely high binding capacity (180 μmol g-1) that is greater than 10 fold larger than IgG (13 μmol g-1). Our results revealed the greater capacity is due to the higher surface to volume ratio of the nano-size materials than the bulk polymer films (5, 26). Melittin might also diffuse into and be captured in the interior of the NP since the particles are hydrogels consisting of > 70% water (19). Binding stoichiometry between functional groups on the particles and the target peptide were quantified to learn more about the binding mechanism. Melittin affinity requires both hydrophobic and electrostatic interactions. The data also indicate that binding affinity increases dramatically from multipoint electrostatic interactions between carboxylic anions and the positive charges on the peptide target.
Detoxification of Melittin In Vivo.
Although all NPs that contain both AAc and TBAm neutralized the hemolytic activity of melittin in vitro, only the NPs with sufficient binding capacity and affinity detoxified melittin in vivo. Although, detoxification capacity of the NPs in vivo was about five times smaller than the binding capacity determined in vitro, it was still greater (2X) than a melittin IgG. Moreover, the detoxification efficiency was more significant than particles prepared by a molecular imprinting strategy (3), nevertheless the imprinted particles exhibited higher affinity for melittin. This result emphasizes the importance of maximizing NP binding capacity as well as binding affinity for in vivo applications.
A biodistribution study revealed that the particles accelerate clearance of melittin. Both NPs and melittin accumulated in the same cells in the liver. Generally, foreign objects such as nanoparticles are sequestered from the blood stream by the immune cells that are part of the mononuclear phagocytic system (MRS) (8, 9). We conclude from those facts that melittin was captured by NPs while circulating in the blood, and then the MRS in the liver subsequently cleared the melittin•NP complexes from the blood. As a result of binding and removal of melittin by NPs, in vivo toxicity of the melittin was significantly diminished.
The difference between detoxification capacity in vitro and in vivo was notable: detoxification capacity of the NPs in vivo was approximately five times smaller than the binding capacity determined in vitro. Since the concentration of melittin in vivo (800 μM) is much higher (more than 1,000 times) than the dissociation equilibrium constant (15 nM—the reciprocal of the binding constant; 660 × 105 M-1) and sufficient NPs to capture all the melittin were injected, more melittin in the blood should have been neutralized if the system was at equilibrium. We believe the system is not at equilibrium due in part to competitive exchange of bound melittin by plasma proteins and phagocytosis of NPs by MRS. To the extent that the rate of exchange of the plastic antidote can be reduced by further optimization, it should be possible to prepare NPs with higher neutralization capacities in vivo.
Biocompatibility of Multifunctional Polymer NPs.
In this study, the biocompatibility of polymer NPs (approximately 50 nm) comprised of combinations of hydrophobic, negatively charged, and positively charged monomers, were systematically investigated. Both the cytotoxicity assay against cultured cells and the stability test in plasma revealed that hydrophobic NPs are not suitable for in vivo applications. Positive charge was found to enhance the cytotoxicity and instability of NPs in plasma, but negative charge diminishes these deficiencies.
Design Rational, Potential and Possible Limitations of Synthetic Polymer NPs as Antidotes.
Our results indicate that optimization of NP functionally is critical for the design of an effective plastic antidote. Simultaneously, surface functionality is crucial for biocompatibility; hydrophobic NPs without negative charge are cytotoxic and form aggregates in plasma since they associate with cell surfaces and plasma proteins. Thus, designing plastic antidotes for negatively charged hydrophobic toxins such as lipopoly saccharide and shiga-like toxins would be far more challenging. The capture of such toxins would likely require positively charged hydrophobic NPs, a combination that would challenge the immune system. Incorporation of ”custom” monomers, designed to bind to specific amino acid or peptide sequences by hydrogen bonding and/or van der Waals interactions, (9), and by applying molecular imprinting (19, 29) and/or affinity purification (20) might offer a more fruitful direction to create a synthetic polymer antidote for such toxins. Surface modification of the hydrophobic particles by hydrophilic polymers such as polyethylene glycol would also help to improve biocompatibility and enhance stability of NPs in plasma, although, the modification might also prevent target toxins to be captured by NPs due to steric hindrance.
Despite possible limitations of target selection, we believe these results provide guidelines for the synthesis of plastic antidotes against a number of toxic proteins and peptides. The majority of toxins, especially membrane damaging toxins, share amphipathic amino acid sequences with net positive charge (30, 31). By expanding the diversity of functional monomers and optimizing the combinations and ratios of monomers, plastic antidotes with the capability of capturing and neutralizing a wide range of target toxins in vivo should now be accessible.
Materials and Methods
For further details, please see supporting information (SI Text).
Analysis of Composition of Functional Groups in NPs by 1H-NMR and Inverse-Gated 13C-NMR.
In order to determine the ratio of TBAm and NIPAm in the polymer, both 1H NMR and inverse-gated 13C NMR spectroscopy were utilized as described (SI). To determine incorporation of AAc, 13C-enriched AAc was synthesized from vinyl magnesium bromide and 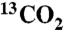 (SI). Then incorporation of 13C-enriched AAc in NPs were quantified by inverse-gated 13C NMR spectroscopy.
(SI). Then incorporation of 13C-enriched AAc in NPs were quantified by inverse-gated 13C NMR spectroscopy.
Determination of Hemolytic Activity Neutralization Capacity of NPs In Vitro.
Melittin-neutralization constants of NPs were calculated as we described (SI Text, 6).
Determination of Binding Affinity between NPs and Melittin by 27-MHz QCM.
Melittin-neutralization constants of NPs were calculated as we described (SI Text, 6, 19, 32).
In Vitro Biocompatibility Test.
HT-1080 human fibrosarcoma cells (ATCC, Manassas, VA) were cultured with various concentrations of the NPs for 24 h. Then, 10 μL/well of Alamar Blue® was added and incubated for 4 h. Viable cells were determined by the standard Alamar Blue® assay (SI Text).
Stability of NPs in Plasma Solution.
DNS-labeled NPs (0.1 mg mL-1) were incubated with mouse plasma (50% Alsever’s solution) for 1 h at 37 °C and centrifuged (15,000 × G). The amount of NPs that were aggregated with plasma was quantified from the difference of fluorescent intensity before and after the incubation-centrifugation process (SI Text).
Biocompatibility of NPs In Vivo.
All animal experiments were reviewed, approved, and supervised by the Institutional Animal Care and Use Committee at the University of Shizuoka. To examine in vivo toxicity, body mass of the mice was monitored and the sections of kidney and liver tissues harvested from the mice 2 weeks after injection were examined by a pathologist as described (3, SI Text).
In Vivo Neutralization Assay.
Melittin were injected into BALB/c mice slowly via tail vein. Then, NP were injected slowly via tail vein 20 ± 5 s after injection of the melittin solution (3, SI Text).
Histological Analysis of Inflammation.
The severity of inflammation observed on the peritoneum was quantified as described (3, SI Text).
In Vivo and Ex Vivo Fluorescent Imaging of fluorescent-labeled Melittin.
BALB/c nu/nu mice were fixed in the IVIS 200 imaging system under isoflurane anesthesia. Cy-7 melittin were injected into mice slowly via tail vein. Then, NPs was injected slowly via tail vein 20–5 s after injection of melittin solution. Mice were imaged every 5 min. For ex vivo imaging, the organs of BALB/c nu/nu mice injected with Cy-5 melittin followed with NPs were harvested 10 min after injection, and imaged using the Cy5 filters (3, SI Text).
In Vivo Distribution Study of 14C Labeled NPs.
14C labeled NPs were prepared as described (SI, 6). Distribution of the 14C labeled NPs in mice was quantified as described (SI Text, 6).
Confocal Microscopy Imaging and Analysis of Cy5-melittin and Fluorescein-NPs.
Cy5-melittin were injected into BALB/c mice slowly via tail vein. Then, fluorescein-NPs was injected slowly via tail vein 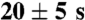 after injection of melittin solution. The section of a liver harvested from the mice 70 min after injection. The liver sections were fluorescently observed with confocal microscope (SI Text).
after injection of melittin solution. The section of a liver harvested from the mice 70 min after injection. The liver sections were fluorescently observed with confocal microscope (SI Text).
Supplementary Material
ACKNOWLEDGMENTS.
Financial supports from National Institutes of Health (GM080506), Ministry of Education, Culture, Sports, Science & Technology in Japan (MEXT) (23111716), Japan Society for the Promotion of Science (23750193), the Ogasawara Foundation, and the Kao Foundation for Arts and Sciences are greatly appreciated.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1112828109/-/DCSupplemental.
References
- 1.Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt M, Pei L. Synthetic toxicology: Where engineering meets biology and toxicology. Toxicol Sci. 2011;120(suppl 1):S204–S224. doi: 10.1093/toxsci/kfq339. [DOI] [PubMed] [Google Scholar]
- 3.Hoshino Y, et al. Recognition, neutralization, and clearance of target peptides in the bloodstream of living mice by molecularly imprinted polymer nanoparticles: A plastic antibody. J Am Chem Soc. 2010;132:6644–6645. doi: 10.1021/ja102148f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leroux J-C. Injectable nanocarriers for biodetoxification. Nat Nano. 2007;2:679–684. doi: 10.1038/nnano.2007.339. [DOI] [PubMed] [Google Scholar]
- 5.Oya T, et al. Reversible molecular adsorption based on multiple-point interaction by shrinkable gels. Science. 1999;286:1543–1545. doi: 10.1126/science.286.5444.1543. [DOI] [PubMed] [Google Scholar]
- 6.Hoshino Y, et al. Design of synthetic polymer nanoparticles that capture and neutralize a toxic peptide. Small. 2009;5:1562–1568. doi: 10.1002/smll.200900186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De M, You C-C, Srivastava S, Rotello VM. Biomimetic interactions of proteins with functionalized nanoparticles: A thermodynamic study. J Am Chem Soc. 2007;129:10747–10753. doi: 10.1021/ja071642q. [DOI] [PubMed] [Google Scholar]
- 8.Dernedde J, et al. Dendritic polyglycerol sulfates as multivalent inhibitors of inflammation. Proc Natl Acad Sci USA. 2010;107:19679–19684. doi: 10.1073/pnas.1003103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koch SJ, Renner C, Xie X, Schrader T. Tuning linear copolymers into protein-specific hosts. Angew Chem Int Ed. 2006;45:6352–6355. doi: 10.1002/anie.200601161. [DOI] [PubMed] [Google Scholar]
- 10.Cabaleiro-Lago C, et al. Inhibition of amyloid β protein fibrillation by polymeric nanoparticles. J Am Chem Soc. 2008;130:15437–15443. doi: 10.1021/ja8041806. [DOI] [PubMed] [Google Scholar]
- 11.Tabata Y, Ikada Y. Effect of the size and surface charge of polymer microspheres on their phagocytosis by macrophage. Biomaterials. 1988;9:356–362. doi: 10.1016/0142-9612(88)90033-6. [DOI] [PubMed] [Google Scholar]
- 12.Sun X, et al. An assessment of the effects of shell cross-linked nanoparticle size, core composition, and surface PEGylation on in vivo biodistribution. Biomacromolecules. 2005;6:2541–2554. doi: 10.1021/bm050260e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Owens Iii DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cedervall T, et al. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci USA. 2007;104:2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gan D, Lyon LA. Synthesis and protein adsorption resistance of PEG-modified poly(N-isopropylacrylamide) core/shell microgels. Macromolecules. 2002;35:9634–9639. [Google Scholar]
- 17.Dobrovolskaia MA, Germolec DR, Weaver JL. Evaluation of nanoparticle immunotoxicity. Nat Nano. 2009;4:411–414. doi: 10.1038/nnano.2009.175. [DOI] [PubMed] [Google Scholar]
- 18.Nel AE, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 19.Hoshino Y, Kodama T, Okahata Y, Shea KJ. Peptide imprinted polymer nanoparticles: A plastic antibody. J Am Chem Soc. 2008;130:15242–15243. doi: 10.1021/ja8062875. [DOI] [PubMed] [Google Scholar]
- 20.Hoshino Y, et al. Affinity purification of multifunctional polymer nanoparticles. J Am Chem Soc. 2010;132:13648–13650. doi: 10.1021/ja1058982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Habermann E. Bee and wasp venoms. Science. 1972;177:314–322. doi: 10.1126/science.177.4046.314. [DOI] [PubMed] [Google Scholar]
- 22.Wang R, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 23.Wardenburg JB, Bae T, Otto M, DeLeo FR, Schneewind O. Poring over pores: [alpha]-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. 2007;13:1405–1406. doi: 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- 24.Gold BS, Dart RC, Barish RA. Bites of venomous snakes. N Engl J Med. 2002;347:347–356. doi: 10.1056/NEJMra013477. [DOI] [PubMed] [Google Scholar]
- 25.Torres AM, Kuchel PW. The β-defensin-fold family of polypeptides. Toxicon. 2004;44:581–588. doi: 10.1016/j.toxicon.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 26.Nishino H, Huang C-S, Shea KJ. Selective protein capture by epitope imprinting. Angew Chem Int Ed. 2006;45:2392–2396. doi: 10.1002/anie.200503760. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Yang H-H, You Q-H, Zhuang Z-X, Wang X-R. Protein recognition via surface molecularly imprinted polymer nanowires. Anal Chem. 2006;78:317–320. doi: 10.1021/ac050802i. [DOI] [PubMed] [Google Scholar]
- 28.Voet D, Voet JG. Biochemistry. New York: Wiley; 1995. [Google Scholar]
- 29.Zeng Z, Hoshino Y, Rodriguez A, Yoo H, Shea KJ. Synthetic polymer nanoparticles with antibody-like affinity for a hydrophilic peptide. ACS Nano. 2010;4:199–204. doi: 10.1021/nn901256s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Epand RM, Shai Y, Segrest JP, Anantharamiah GM. Mechanisms for the modulation of membrane bilayer properties by amphipathic helical peptides. Biopolymers. 1995;37:319–338. doi: 10.1002/bip.360370504. [DOI] [PubMed] [Google Scholar]
- 31.White SH, Wimley WC. Membrane protein folding and stability: Physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 32.Hoshino Y, Kawasaki T, Okahata Y. Effect of ultrasound on DNA polymerase reactions: Monitoring on a 27-MHz quartz crystal microbalance. Biomacromolecules. 2006;7:682–685. doi: 10.1021/bm050738e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.