

Abstract
Radiolabeled diacetylbis(4-methylthiosemicarbazonato)copperII [CuII(atsm)] is an effective positron-emission tomography imaging agent for myocardial ischemia, hypoxic tumors, and brain disorders with regionalized oxidative stress, such as mitochondrial myopathy, encephalopathy, and lactic acidosis with stroke-like episodes (MELAS) and Parkinson’s disease. An excessively elevated reductive state is common to these conditions and has been proposed as an important mechanism affecting cellular retention of Cu from CuII(atsm). However, data from whole-cell models to demonstrate this mechanism have not yet been provided. The present study used a unique cell culture model, mitochondrial xenocybrids, to provide whole-cell mechanistic data on cellular retention of Cu from CuII(atsm). Genetic incompatibility between nuclear and mitochondrial encoded subunits of the mitochondrial electron transport chain (ETC) in xenocybrid cells compromises normal function of the ETC. As a consequence of this impairment to the ETC we show xenocybrid cells upregulate glycolytic ATP production and accumulate NADH. Compared to control cells the xenocybrid cells retained more Cu after being treated with CuII(atsm). By transfecting the cells with a metal-responsive element reporter construct the increase in Cu retention was shown to involve a CuII(atsm)-induced increase in intracellular bioavailable Cu specifically within the xenocybrid cells. Parallel experiments using cells grown under hypoxic conditions confirmed that a compromised ETC and elevated NADH levels contribute to increased cellular retention of Cu from CuII(atsm). Using these cell culture models our data demonstrate that compromised ETC function, due to the absence of O2 as the terminal electron acceptor or dysfunction of individual components of the ETC, is an important determinant in driving the intracellular dissociation of CuII(atsm) that increases cellular retention of the Cu.
Keywords: sodium arsenite, energy metabolism, radiopharmaceutical
PET allows noninvasive imaging to assist in diagnosis of disease and monitoring of therapeutic treatments. The technique relies on administration of a compound radiolabeled with a positron-emitting isotope. Although there are several positron-emitting isotopes of copper that are of interest in the development of copper-based PET tracers (copper-60, copper-62, and copper-64), it is essential to selectively and safely deliver the radioactive copper ion to target tissue. An approach to achieve this goal is to incorporate the radioactive copper ion into a coordination complex. The resulting complex is likely to have distinctly different biodistribution when compared to the use of the simple complex ions in aqueous solution.
A family of ligands derived from the condensation of 1,2-diones with substituted thiosemicarbazides collectively known as bis(thiosemicarbazones) form stable, neutral, and lipophilic complexes with CuII. These qualities have led to the use of bis(thiosemicarbazones) as delivery vehicles for radioactive copper isotopes in the development of unique radiopharmaceuticals (1–4). A particular focus is on the use of radiolabeled diacetylbis(4-methylthiosemicarbazonato)copperII [CuII(atsm)] (Fig. 1) as a hypoxia imaging agent. Hypoxia can be associated with aggressive tumors and the effective delineation of degrees of hypoxia is also of interest to clinicians in assessing stroke and myocardial ischemia.
Fig. 1.

Comparative effects of CuCl2, CuII(atsm), and CuII(gtsm). (A) SH-SY5Y cells treated with 10 μM CuCl2, CuII(atsm), or CuII(gtsm) for 1 h before analyzing cells for Cu content by ICP-MS, which does not differentiate between intact CuII(atsm)/CuII(gtsm) and Cu that has dissociated from the respective bis(thiosemicarbazone) scaffolds. (B) Before treating with 500 nM CuCl2, CuII(atsm), or CuII(gtsm) for 6 h, SH-SY5Y cells were transfected with MRE-luciferase construct. Increased bioavailable Cu within the cell upregulates expression of the luciferase reporter, induction of which is measured by luminescence. (C) SH-SY5Y cells treated with 10 μM CuCl2, CuII(atsm), or CuII(gtsm) for 1 h were analyzed for phosphorylated ERK1/2 (pERK1/2). β-Actin levels are shown as a control. (D) Schematic illustrating that although CuII(atsm) and CuII(gtsm) both enter the cell, only CuII(gtsm) is sensitive to the activity of cellular reductants under normal conditions. Increased ERK1/2 phosphorylation is responsive to increases in bioavailable Cu within the cell. Vehicle represents cells treated with the DMSO vehicle used to prepare the Cu compounds. Data are mean values ± SEM, n = 6–12. **P < 0.01 compared to vehicle-treated cells (ANOVA with Tukey’s posttest).
An early study into the potential of 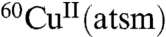 as a hypoxia imaging agent described selective accumulation of
as a hypoxia imaging agent described selective accumulation of 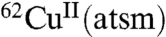 in rat hearts subjected to ischemic insult (5). In control mice only 23% of the injected dose of
in rat hearts subjected to ischemic insult (5). In control mice only 23% of the injected dose of 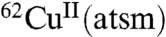 was retained in the heart, whereas 81% was retained in hearts subjected to ischemic insult (5). A subsequent in vivo study using dog models of hypoxic myocardium demonstrated the ability to use PET to monitor selective tissue retention of the 60/64Cu from CuII(atsm) (6), and a preliminary study has indicated the ability to use
was retained in the heart, whereas 81% was retained in hearts subjected to ischemic insult (5). A subsequent in vivo study using dog models of hypoxic myocardium demonstrated the ability to use PET to monitor selective tissue retention of the 60/64Cu from CuII(atsm) (6), and a preliminary study has indicated the ability to use 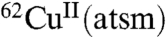 to image the heart in people with coronary heart disease (7). Subsequent studies focused on the potential of radiolabeled CuII(atsm) as an imaging agent of tumor hypoxia. Radiolabeled
to image the heart in people with coronary heart disease (7). Subsequent studies focused on the potential of radiolabeled CuII(atsm) as an imaging agent of tumor hypoxia. Radiolabeled 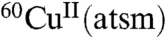 has been investigated as a PET tracer to detect tumor hypoxia in cervical cancer and is currently undergoing human clinical trials (8, 9).
has been investigated as a PET tracer to detect tumor hypoxia in cervical cancer and is currently undergoing human clinical trials (8, 9).
Recently 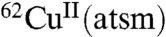 has been used as a PET imaging agent in a single subject with mitochondrial myopathy, encephalopathy, and lactic acidosis with stroke-like episodes (MELAS) (10) and 15 people with Parkinson’s disease (11). Unlike the previous studies that emphasized CuII(atsm) as an agent to image hypoxic tissue, the studies in patients with MELAS and Parkinson’s disease described selective accumulation of Cu from CuII(atsm) in the disease-affected tissue as a method to image regionalized oxidative stress (10, 11). Oxidative stress and hypoxia are both consistent with a cellular environment in which the abundance of cellular reductants relative to O2 is increased above normal conditions, raising the possibility that factors conducive to a perturbed reductant∶O2 balance are a more important determinant of cellular accumulation of the Cu from CuII(atsm) than merely oxygen concentrations or oxidative stress per se. Cell-free studies and experiments using isolated subcellular components support this possibility but have not demonstrated feasibility in whole-cell studies. As a consequence, the cellular conditions that control selective retention of radioactive Cu in disease-affected tissues remain incompletely understood (4). The present study aimed to delineate the role of impaired energy metabolism, oxidative stress, hypoxia, and mitochondrial electron transport chain (ETC) function cellular retention of Cu from CuII(atsm).
has been used as a PET imaging agent in a single subject with mitochondrial myopathy, encephalopathy, and lactic acidosis with stroke-like episodes (MELAS) (10) and 15 people with Parkinson’s disease (11). Unlike the previous studies that emphasized CuII(atsm) as an agent to image hypoxic tissue, the studies in patients with MELAS and Parkinson’s disease described selective accumulation of Cu from CuII(atsm) in the disease-affected tissue as a method to image regionalized oxidative stress (10, 11). Oxidative stress and hypoxia are both consistent with a cellular environment in which the abundance of cellular reductants relative to O2 is increased above normal conditions, raising the possibility that factors conducive to a perturbed reductant∶O2 balance are a more important determinant of cellular accumulation of the Cu from CuII(atsm) than merely oxygen concentrations or oxidative stress per se. Cell-free studies and experiments using isolated subcellular components support this possibility but have not demonstrated feasibility in whole-cell studies. As a consequence, the cellular conditions that control selective retention of radioactive Cu in disease-affected tissues remain incompletely understood (4). The present study aimed to delineate the role of impaired energy metabolism, oxidative stress, hypoxia, and mitochondrial electron transport chain (ETC) function cellular retention of Cu from CuII(atsm).
Results
CuII(atsm) is Resistant to Intracellular Dissociation Under Normal Cellular Conditions.
The chemistry and cellular metabolism of the bis(thiosemicarbazonato)copper(II) complexes is dependent on the backbone substituents of the ligand. The electron donating methyl groups on the atsm ligand lower the CuII/CuI reduction potential for CuII(atsm) when compared to glyoxalbis[N(4)-methylthiosemicarbazonato]CuII [CuII(gtsm)] (Fig. 1) (Em = 0.60 mV and Em = 0.44 mV, respectively, versus SCE where Fc/Fc+ = 0.54 V) (12, 13). CuII(atsm) is therefore more resistant to intracellular reduction of the metal ion and less likely to dissociate under normal cellular conditions. This salient feature of CuII(atsm) compared to CuII(gtsm) is demonstrated in SH-SY5Y cells treated with 10 μM CuCl2, CuII(atsm), or CuII(gtsm) for 1 h. Cells analyzed for Cu content show that both bis(thiosemicarbazonato)-CuII compounds increase cellular Cu levels more efficiently than CuCl2 (Fig. 1A) (14). However, analysis of cell pellets by inductively coupled plasma (ICP)-MS does not provide information on whether the copper ion has dissociated from the ligand within the cell. Intracellular dissociation of the compounds cells was therefore examined using cells transfected with a metal-responsive element (MRE)-luciferase reporter (15, 16) prior to treating with the bis(thiosemicarbazonato)-CuII compounds. The MRE-luciferase reporter responds to treatments that induce an increase in cytosolic levels of bioavailable Zn (16). The increased bioavailable Zn activates the metal-responsive transcription factor 1 (MTF-1) that in turn promotes the expression of genes that contain MREs in their promoter region (17, 18). Increasing bioavailable Zn in cells by treating them with high concentrations of ZnCl2 (40–100 μM for 24 h) promotes expression of the MRE-luciferase construct, as does treating with high concentrations of CuCl2 (40–100 μM for 24 h) (16). The effect induced by treating with Cu is due to the increased bioavailable Cu within the cell displacing Zn from endogenous metallothioneins (16). When treated with 500 nM CuII(gtsm) for 6 h SH-SY5Y cells transfected with the MRE-luciferase reporter showed a threefold induction of the reporter (Fig. 1B), indicating the CuII(gtsm) treatment had increased bioavailable Cu within the cells. By contrast, the same concentration of CuII(atsm), which increased overall Cu content of the cells to a level comparable to the CuII(gtsm) treatment as determined by ICP-MS (Fig. 1A), did not influence the MRE-luciferase (Fig. 1B), indicating no change to bioavailable Cu in the CuII(atsm)-treated cells. Further, when SH-SY5Y cells were treated with the bis(thiosemicarbazonato)-CuII compounds or the CuCl2 control under normal cellular conditions, only the CuII(gtsm) induced an increase in phosphorylation of the signaling kinase ERK1/2. Together with the MRE-luciferase data in Fig. 1B, these data (Fig. 1C) show that cellular responses leading to increased ERK1/2 phosphorylation, induced by treating with bis(thiosemicarbazonato)-CuII compounds, are initiated by the compound increasing bioavailable Cu within the cell.
Hypoxia Impairs Energy Metabolism and Promotes Cellular Retention of Cu from CuII(atsm).
To examine the cellular conditions involved in hypoxia-selective retention of Cu from CuII(atsm), SH-SY5Y cells were subjected to hypoxic conditions prior to treating with CuII(atsm) (10 μM). Hypoxia increased the levels of cellular Cu in CuII(atsm)-treated cells (Fig. 2A) suggesting intracellular dissociation of Cu from CuII(atsm), presumably following reduction of CuII/CuI, and sequestration of CuI by intracellular ligands. Supporting this mechanism, treating control (normoxic) cells with CuII(atsm) did not alter levels of phosphorylated ERK1/2, but treating hypoxic cells with CuII(atsm) significantly increased levels of phosphorylated ERK1/2 (Fig. 2 B and C).
Fig. 2.

Effects of hypoxia on energy metabolism and cellular responses to CuII(atsm) in SH-SY5Y cells. (A) ICP-MS analysis of cellular Cu in normoxic control cells and hypoxic cells after treating with 10 μM CuII(atsm) for 1 h. (B) Western blotting image showing CuII(atsm) promotes phosphorylation of ERK1/2 (pERK1/2) in hypoxic cells. β-Actin levels are shown as a control. (C) Densitometry analysis of Western blotting results. (D) pH of media collected from cells after 2 d in culture. (E) Lactate produced and (F) glucose consumed by cells over 2 d in culture. (G) ATP and (H) NADH content of control cells and hypoxic cells. (I) Reduction of MTT mediated by cell lysates collected from control and hypoxic cells. (J) Relative content of oxidatively modified proteins in control cells and hypoxic cells. Data are mean values ± SEM, n = 3–6. Values shown in A and E–I are expressed per milligram cellular protein. P < 0.01 compared to control cells (t test) except for C where **P < 0.01 compared to vehicle-treated cells (ANOVA with Tukey’s posttest).
To elucidate the mechanisms through which hypoxia promotes cellular retention of Cu from CuII(atsm) a number of analyses were performed to compare hypoxic and control cells. Hypoxic conditions decreased the pH of the media (Fig. 2D) a result likely to be the consequence of increased reliance of the hypoxic cells on glycolytic energy production. Increased glycolysis is supported by data showing the hypoxic cells generated more lactate (Fig. 2E) and consumed more glucose (Fig. 2F) compared to control cells. Glycolysis is a less efficient means of ATP production compared to a functional ETC and the data in Fig. 2G show hypoxic cells contained less ATP. These ATP data are consistent with hypoxic conditions preventing normal function of the ETC by decreasing availability of the terminal electron acceptor O2. An accumulation of NADH, the primary source of electrons for the ETC, also indicates decreased ETC function within hypoxic cells (Fig. 2H).
NADPH-cytochrome P450 reductase and NADH-cytochrome b5 reductase have been reported to contribute to the reduction of CuII(atsm) in subcellular fractions from tumor cells (19). To assess reductase activity in SH-SY5Y cells, cell extracts were incubated with the generic reductase substrate 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT). Cell extracts from hypoxic cells were less effective at reducing MTT compared to extracts from control cells (Fig. 2I), so it is unlikely that increased activity of reductases is responsible for the increased retention of Cu from CuII(atsm) in hypoxic cells (Fig. 2A). Further, an analysis of oxidatively modified proteins in SH-SY5Y cells revealed an absence of hypoxia-induced oxidative damage in this cell culture model of hypoxia (Fig. 2J).
Mitochondrial Xenocybrid Cells Show Increased Cellular Retention of Cu from CuII(atsm).
Data presented in Fig. 2 indicate impaired function of the mitochondrial ETC increases cellular retention of Cu from CuII(atsm). To directly test ETC function in increased retention of Cu from CuII(atsm), without hypoxia being a confounding factor, the cellular retention of Cu from CuII(atsm) was examined in mitochondrial xenocybrid cells. ATP production via the ETC is dependent on the coordinated assembly of protein subunits encoded by nuclear DNA and mitochondrial DNA to form functional multisubunit protein complexes of the ETC. Because of a series of chemical and genetic manipulations (20), xenocybrid cells contain nuclear DNA and mitochondrial DNA from two separate species. This genetic mismatch between the nuclear and mitochondrial encoded subunits compromises the ability of the nuclear and mitochondrial encoded subunits to form multisubunit protein complexes. The result is that normal transfer of electrons through the ETC is impaired. The xenocybrid cells used in our study contained nuclear DNA from Mus musculus and mitochondrial DNA from Rattus norvegicus (21). After treating with CuII(atsm) (10 μM) for 1 h, xenocybrid cells contained more Cu compared to CuII(atsm)-treated control cells (Fig. 3A). The CuII(atsm)-induced increase in phosphorylated ERK1/2 in the xenocybrid cells (Fig. 3 B and C) indicated the increased cellular retention of Cu in the xenocybrid cells involved intracellular dissociation of the compound to increase bioavailable Cu. Supporting this mechanism, xenocybrid cells transfected with the MRE-luciferase reporter displayed a 2.5-fold induction in MRE-luciferase in response to CuII(atsm) (Fig. 3D). By contrast, control cells transfected with the same MRE-luciferase reporter were not affected by the CuII(atsm) treatment (Fig. 3D).
Fig. 3.

Effects of a genetically impaired mitochondrial electron transport chain on energy metabolism and cellular responses to CuII(atsm). (A) ICP-MS analysis of cellular Cu in control cells and xenocybrid cells after treating with 10 μM CuII(atsm) for 1 h. (B) Western blotting image showing CuII(atsm) promotes phosphorylation of ERK1/2 (pERK1/2) in xenocybrid cells. β-Actin levels are shown as a control. (C) Densitometry analysis of Western blotting results. (D) Before treating with 500 nM CuII(atsm) for 6 h, cells were transfected with MRE-luciferase construct. Treatment-induced increases in bioavailable Cu within the cell upregulates expression of the luciferase reporter, induction of which is measured by luminescence. (E) pH of media collected from cells after 6 d in culture. (F) Lactate produced and (G) glucose consumed by cells over 6 d in culture. (H) ATP and (I) NADH content of control cells and xenocybrid cells. (J) Reduction of MTT mediated by cell lysates collected from control cells and xenocybrid cells. (K) Relative content of oxidatively modified proteins in control cells and xenocybrid cells. Data are mean values ± SEM, n = 3–6. Values in A and E–J are expressed per milligram cellular protein. ∗P < 0.05/∗∗P < 0.01 compared to control cells (t test) except for C and D where *P < 0.01 compared to vehicle-treated cells (ANOVA with Tukey’s posttest).
Consistent with the hypoxic SH-SY5Y cells, the xenocybrid cells acidified their culture media (Fig. 3E) because of increased glycolytic activity (Fig. 3 F and G). The xenocybrid cells also contained lower levels of ATP (Fig. 3H) and elevated levels of NADH (Fig. 3I). Also, consistent with the mechanism of selective Cu retention in CuII(atsm)-treated hypoxic cells, the increased retention of Cu in CuII(atsm)-treated xenocybrid cells did not involve increased reductase activity (Fig. 3J).
Oxidative Stress in the Absence of an Impaired ETC does not Promote Cellular Retention of Cu from CuII(atsm).
Oxidative modification of proteins was elevated in the xenocybrid cells (Fig. 3K). Unlike the hypoxic SH-SY5Y cells, the xenocybrid cells contained an impaired ETC in the presence of available O2. The availability of O2 as an electron acceptor together with electron leakage from the impaired ETC of the xenocybrid cells is the most likely source of the reactive oxygen species (ROS) that will give rise to the increase in oxidative damage observed in xenocybrid cells (Fig. 3K). Accordingly, elevated retention of Cu from CuII(atsm) in the xenocybrid cells supports the notion that CuII(atsm) may be used to image regions of oxidative stress (10, 11). To determine whether oxidative stress in the absence of an impaired ETC could promote cellular retention of Cu from CuII(atsm) we pretreated SH-SY5Y cells with the glutathione synthesis inhibitor buthionine sulfoximine (BSO) (22) before exposing to CuII(atsm). Pretreatment with BSO increased oxidative modification of proteins in SH-SY5Y cells (Fig. S1A), but did not promote cellular retention of the Cu from CuII(atsm) (Fig. S1B). In addition, exposing BSO-treated cells to CuII(atsm) did not alter levels of phosphorylated ERK1/2 (Fig. S1 C and D). These results provide further support that CuII(atsm)-mediated increases in bioavailable Cu within the cell, detectable indirectly by changes to the phosphorylation state of ERK1/2, are needed to promote cellular retention of the Cu from CuII(atsm).
Acidification of the Cell Culture Media in the Absence of an Impaired ETC does not Alter Cellular Responses to CuII(atsm).
Conditions conducive to an impaired ETC promote upregulation of alternate ATP producing pathways, as indicated in both the hypoxic cells (Fig. 2 E and F) and the xenocybrid cells (Fig. 3 F and G) by data showing lactate production and glucose consumption are increased. A consequence of the increased glycolysis is increased acidification of the cell culture media (Fig. 2D, Fig. 3E). To test whether acidification of the culture media contributed to cellular retention of the Cu from CuII(atsm), lactic acid was titrated into the media of SH-SY5Y cells prior to treating with CuII(atsm). Lactic acid was added to the media until the pH reached 6.5, as per the media of hypoxic cells (Fig. 2D). Decreasing the media pH in the absence of an impaired ETC did not promote cellular retention of Cu from CuII(atsm) (Fig. S2A), nor did it increase bioavailable Cu within the CuII(atsm)-treated cells as indicated by unaltered levels of phosphorylated ERK1/2 (Fig. S2 B and C).
Inhibiting the Tricarboxylic Acid (TCA) Cycle does not Alter Cellular Responses to CuII(atsm).
Impairment of the ETC in hypoxic and xenocybrid cells leads to decreased levels of ATP (Fig. 2G, Fig. 3H) and an accumulation of NADH (Fig. 2H, Fig. 3I). To differentiate between impaired ETC function and an overall impediment to ATP production in cellular retention of Cu from CuII(atsm), cells were pretreated with the TCA cycle inhibitor NaAsO2 (23). Treatment with NaAsO2 decreased ATP levels (Fig. S3A) and moderately increased Cu levels in CuII(atsm)-treated cells (Fig. S3B). However, there was no indication of increased bioavailable Cu in CuII(atsm)-treated cells due to the NaAsO2, as indicated indirectly by the lack of changes to phosphorylated ERK1/2 (Fig. S3 C and D).
Discussion
After 2-keto-3-ethoxybutyraldehydebis(thiosemicarbazonato)CuII [CuII(kts)] enters cells the Cu is reduced from CuII to CuI and dissociates from the ligand; H2kts diffuses back out of the cell while the Cu is, at least initially, retained inside the cell (24, 25). Increased cellular retention of Cu from bis(thiosemicarbazonato)CuII compounds is dictated, in part, by intracellular reduction of the Cu followed by Cu dissociation from the ligand inside the cell and there is a correlation between CuII/CuI reduction potential and hypoxia selectivity (13, 26, 27). An elevated Cu content was evident in CuII(atsm)-treated hypoxic and xenocybrid cells compared to their relevant CuII(atsm)-treated control cells (Fig. 2A, Fig. 3A), and this increase in Cu content was paralleled by increases in ERK phosphorylation (Fig. 2 B and C, Fig. 3 B and C). Increased phosphorylation of cell-signaling kinases after treating with bis(thiosemicarbazonato)CuII compounds is dependent on the relative CuII/CuI reduction potential of the compounds (28, 29). Increased kinase phosphorylation in response to bis(thiosemicarbazonato)CuII treatment is therefore likely to involve intracellular dissociation of the compound giving rise to increased intracellular levels of bioavailable Cu.
In the present study cells transfected with the MRE-luciferase reporter construct were used to monitor cellular conditions that affect the intracellular dissociation of Cu from CuII(atsm). Although the initial event required to upregulate expression of the luciferase reporter gene is Cu release from the atsm ligand, the MRE component of the construct only detects an increase in bioavailable Zn, which occurs subsequent to Cu displacement of Zn from endogenous metallothioneins (16). Observed changes to ERK1/2 phosphorylation (Fig. 2 B and C, Fig. 3 B and C) are likely to be the consequence of multiple overlapping cell signaling cascades in which increased Zn bioavailability plays a promiscuous but central role. Zn bioavailability regulates multiple components of cell-signaling cascades (30–32). Collectively, the changes to ERK1/2 phosphorylation in CuII(atsm)-treated hypoxic cells (Fig. 2 B and C) and xenocybrid cells (Fig. 3 B and C), together with data indicating that increased ERK1/2 phosphorylation in CuII(atsm) or CuII(gtsm)-treated cells is only detectable in cells that also show increased expression of the MRE-luciferase reporter (Fig. 1B, Fig. 3D), indicate that increased cellular retention of Cu from CuII(atsm) requires an initial intracellular dissociation of the Cu from the bis(thiosemicarbazone) ligand. Although the mechanisms by which increased bioavailable Cu within the cell leads to increased ERK1/2 phosphorylation remain to be fully elucidated, the inhibition of phosphatase activity is a possibility (33). Phosphatase inhibition may involve direct inhibition by Cu or involve Cu-mediated displacement of Zn from metallothioneins, as both Cu and Zn are capable of inhibiting phosphatase activity (30, 34).
Hypoxic cells and xenocybrid cells possess an impaired mitochondrial ETC. The origin of impaired ETC function in hypoxic cells is decreased availability of the terminal electron acceptor O2. The cell culture model of hypoxia showing increased retention of Cu from CuII(atsm) (Fig. 2A) is consistent with increased retention of radiolabeled CuII(atsm) in hypoxic tumors and myocardial ischemia, but these data cannot eliminate the possibility that decreased ETC function is responsible for the increased cellular retention of the Cu. The use of xenocybrid cells enabled us to determine the effects of impaired ETC function without needing to deprive the cells of O2 or to expose them to toxic exogenous inhibitors of the ETC, such as rotenone (1,2,6,6a,12,12a-hexahydro-2-isopropenyl-8,9-dimethoxychromeno[3,4-b]furo(2,3-h)chromen-6-one) or paraquat (1,1′-Dimethyl-4,4′-bipyridinium dichloride). Consistency in the data shown for hypoxic cells (Fig. 2) and the mitochondrial xenocybrids (Fig. 3) provides strong support for an impaired mitochondrial ETC in cellular retention of Cu from CuII(atsm).
A mechanism in which cellular retention of Cu from CuII(atsm) is promoted by cellular conditions that increase NADH within the cell is a possibility. The present experiments show that conditions conducive to sustained (or potentially accelerated) metabolic flux through the TCA cycle in the presence of an impaired ETC drives accumulation of NADH (Fig. 2H, Fig. 3I). An accumulation of NADH has been proposed to facilitate reduction of CuII(atsm) (5), and the accumulation of NADH shown in hypoxic cells and xenocybrid cells is likely to provide the increased cellular reduction potential required to promote reduction of Cu in CuII(atsm) from CuII to CuI, thereby promoting subsequent intracellular dissociation of CuI from the ligand and increased cellular retention of Cu from CuII(atsm). In an earlier study submitochondrial particles from mouse brains were exposed to CuII(atsm), then levels of CuII(atsm) reduction measured using electron spin resonance spectrometry (5). CuII(atsm) reduction by the submitochondrial particles was promoted by NADH, and although isolated mitochondria supported only marginal (3.4%) reduction of CuII(atsm), the reduction was increased to 14.7% when the mitochondria were treated with the ETC inhibitor rotenone (5). These data validated previous studies where reduction of the closely related pyruvaldehyde bis(4-methylthiosemicarbazonato)CuII [CuII(ptsm)] by mitochondrial preparations was shown to be dependent on NADH and could be induced by treating mitochondrial preparations with inhibitors of the ETC (35, 36). However, none of the previous studies with CuII(atsm) (5) and CuII(ptsm) (35, 36) used whole-cell systems to measure whether Cu content of bis(thiosemicarbazonato)CuII-treated cells was increased by the cellular conditions that elevated NADH levels via impairment of the ETC. Increased cellular retention of Cu from CuII(atsm) in xenocybrid cells (Fig. 3A) are unique data from whole cells showing increased cellular retention of Cu from CuII(atsm) in cells with an impaired mitochondrial ETC. The data showing elevated ERK1/2 phosphorylation (Fig. 2 B and C, Fig. 3 B and C) are consistent with the increased cellular retention of Cu from CuII(atsm) involving intracellular dissociation of the CuII(atsm), and the elevated levels of NADH (Fig. 2H, Fig. 3I) are consistent with elevated reduction potential in cells with an impaired ETC being the mechanism by which CuII(atsm) reduction is promoted. Our experiments that utilized NaAsO2 to inhibit TCA cycle activity revealed that after exposing to CuII(atsm) the overall Cu content of NaAsO2-treated cells was increased (Fig. S3B). However, this increase was moderate compared to the increase observed in hypoxic cells (Fig. 2A) and xenocybrid cells (Fig. 3A). As per the hypoxic cells (Fig. 2G) and the xenocybrid cells (Fig. 3H) the ATP content of the NaAsO2-treated cells was decreased (Fig. S3A), but the absence of changes to ERK1/2 phosphorylation (Fig. S3 C and D) indicated that decreasing ATP production at the level of the TCA cycle did not induce intracellular CuII(atsm) dissociation. Together, these data indicate cellular retention of CuII(atsm) as an intact compound may be affected by cellular ATP levels, but enhanced cellular retention of the Cu requires intracellular dissociation of the compound.
Recent studies have described CuII(atsm) as a PET imaging agent to identify regionalized areas of oxidative stress that can occur when cellular levels of reductants are in excess relative to O2 (10, 11). Oxidative stress via increased levels of reactive oxygen species will occur when electrons inefficiently handled by the ETC are able to interact directly with O2. This occurrence is particularly true in conditions where O2 availability is unaltered as demonstrated by the increased oxidative modification of proteins in the xenocybrid cells (Fig. 3K). However, the fundamental requirement for O2 in the generation of reactive oxygen species means that an excess of reductants relative to O2 due to decreased O2 alone, as occurs in hypoxic tissues, will not increase levels of reactive oxygen species or generate oxidative stress. This phenomenon is supported by data showing oxidative modification of proteins is not increased by hypoxia (Fig. 2J). Together, these data demonstrate that although selective retention of Cu from CuII(atsm) will be evident in disease-affected tissues where oxidative stress is the result of a dysfunctional ETC, as occurs in MELAS and Parkinson’s disease (37, 38), CuII(atsm) will not necessarily be suitable for PET detection of generic oxidative stress. Supporting this possibility, the induction of oxidative stress in the absence of any direct impediment to normal function of the ETC (39) did not promote cellular retention of Cu from CuII(atsm) (Fig. S2).
CuII(atsm) has excellent potential as a PET imaging agent because of selective retention of the radiolabeled Cu in tissues affected by hypoxia either in tumors or as a result of ischemia. Furthermore, recent studies have supported the application of CuII(atsm) in imaging MELAS and Parkinson’s disease. The present study is unique to use whole cells to examine the mechanisms that govern cellular retention of Cu from CuII(atsm). The data presented indicate an accumulation of NADH due to impaired activity of the mitochondrial ETC is the primary mechanism that drives cellular retention of the Cu from CuII(atsm), and that intracellular dissociation of the compound correlates with increased retention of the Cu. Hypoxic tumors, ischemia, Parkinson’s disease, and MELAS are all pathological conditions that involve dysfunction of the mitochondria (40–43). This work suggests the potential for CuII(atsm) as a PET imaging agent may extend to other mitochondrial diseases.
Materials and Methods
Full details of the materials and methods are presented in the SI Materials and Methods. The mitochondrial xenocybrid cells used were M. musculus cells (with M. musculus nuclear DNA) containing R. novegicus mitochondrial DNA (21). Mitochondrial xenocybrid cells are a unique and powerful tool for studying cells with an endogenous impedement to the ETC and are described in detail elsewhere (20). The cybrids were produced by fusing enucleated mitochondrial donor cells with mouse ρ0 cells, which lack mitochondrial DNA (44). Control cells in the xenocybrid experiments were M. musculus cybrids containing the same M. musculus nuclear DNA but with M. musculus mitochondrial DNA. The control cells were therefore cybrids that contained nuclear and mitochondrial DNA from the same species (M. musculus) whereas the xenoxybrid cells contained nuclear and mitochondrial DNA from two different species (M. musculus and R. novegicus, respectively). The xenocybrid cells were previously shown to express defective oxidative phosphorylation complex I, III, and IV activities, and a deficit in ATP generation (4).
Supplementary Material
Acknowledgments.
The metal-responsive element-luciferase and Renilla-luciferase constructs were kindly provided by Dr. Leo Klomp, Department of Metabolic and Endocrine Diseases, University Medical Center Utrecht, The Netherlands. The research was supported by funds from the Australian National Health and Medical Research Council and the Australian Research Council.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1116227108/-/DCSupplemental.
References
- 1.Green MA. A potential copper radiopharmaceutical for imaging the heart and brain: Copper-labeled pyruvaldehyde bis(N4-methylthiosemicarbazone) Int J Radiat Appl Instrum Part B. 1987;14:59–61. doi: 10.1016/0883-2897(87)90162-0. [DOI] [PubMed] [Google Scholar]
- 2.Green MA, Klippenstein DL, Tennison JR. Copper(II) bis(thiosemicarbazone) complexes as potential tracers for evaluation of cerebral and myocardial blood flow with PET. J Nucl Med. 1988;29:1549–1557. [PubMed] [Google Scholar]
- 3.Bonnitcha PD, Vavere AL, Lewis JS, Dilworth JR. In vitro and in vivo evaluation of bifunctional bisthiosemicarbazone 64Cu-complexes for the positron emission tomography imaging of hypoxia. J Med Chem. 2008;51:2985–2991. doi: 10.1021/jm800031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paterson BM, Donnelly PS. Copper complexes of bis(thiosemicarbazones): From chemotherapeutics to diagnostic and therapeutic radiopharmaceuticals. Chem Soc Rev. 2011;40:3005–3018. doi: 10.1039/c0cs00215a. [DOI] [PubMed] [Google Scholar]
- 5.Fujibayashi Y, et al. Copper-62-ATSM: A new hypoxia imaging agent with high membrane permeability and low redox potential. J Nucl Med. 1997;38:1155–1160. [PubMed] [Google Scholar]
- 6.Lewis JS, et al. Delineation of hypoxia in canine myocardium using PET and copper(II)-diacetyl-bis(N(4)-methylthiosemicarbazone) J Nucl Med. 2002;43:1557–1569. [PubMed] [Google Scholar]
- 7.Takahashi N, et al. Copper-62 ATSM as a hypoxic tissue tracer in myocardial ischemia. Ann Nucl Med. 2001;15:293–296. doi: 10.1007/BF02987849. [DOI] [PubMed] [Google Scholar]
- 8.Dehdashti F, et al. Assessing tumor hypoxia in cervical cancer by PET with 60Cu-labeled diacetyl-bis(N4-methylthiosemicarbazone) J Nucl Med. 2008;49:201–205. doi: 10.2967/jnumed.107.048520. [DOI] [PubMed] [Google Scholar]
- 9.Dehdashti F, et al. Assessing tumor hypoxia in cervical cancer by positron emission tomography with 60Cu-ATSM: Relationship to therapeutic response, a preliminary report. Int J Radiat Oncol Biol Phys. 2003;55:1233–1238. doi: 10.1016/s0360-3016(02)04477-2. [DOI] [PubMed] [Google Scholar]
- 10.Ikawa M, et al. PET imaging of redox and energy states in stroke-like episodes of MELAS. Mitochondrion. 2009;9:144–148. doi: 10.1016/j.mito.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Ikawa M, et al. Evaluation of striatal oxidative stress in patients with Parkinson’s disease using [62Cu]ATSM PET. Nucl Med Biol. 2011;38:945–951. doi: 10.1016/j.nucmedbio.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Dearling JL, Blower PJ. Redox-active metal complexes for imaging hypoxic tissues: Structure activity relationships in copper(II) bis(thiosemicarbazone) complexes. Chem Commun. 1998:2531–2532. [Google Scholar]
- 13.Xiao Z, Donnelly PS, Zimmermann M, Wedd AG. Transfer of copper between bis(thiosemicarbazone) ligands and intracellular copper-binding proteins. Insights into mechanisms of copper uptake and hypoxia selectivity. Inorg Chem. 2008;47:4338–4347. doi: 10.1021/ic702440e. [DOI] [PubMed] [Google Scholar]
- 14.Dearling JL, et al. Design of hypoxia-targeting radiopharmaceuticals: Selective uptake of copper-64 complexes in hypoxic cells in vitro. Eur J Nucl Med. 1998;25:788–792. doi: 10.1007/s002590050283. [DOI] [PubMed] [Google Scholar]
- 15.Materia S, Cater MA, Klomp LW, Mercer JF, La Fontaine S. Clusterin (apolipoprotein J), a molecular chaperone that facilitates degradation of the copper-ATPases ATP7A and ATP7B. J Biol Chem. 2011;286:10073–10083. doi: 10.1074/jbc.M110.190546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Berghe PV, et al. Human copper transporter 2 is localized in late endosomes and lysosomes and facilitates cellular copper uptake. Biochem J. 2007;407:49–59. doi: 10.1042/BJ20070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmiter RD. Regulation of metallothionein genes by heavy metals appears to be mediated by a zinc-sensitive inhibitor that interacts with a constitutively active transcription factor, MTF-1. Proc Natl Acad Sci USA. 1994;91:1219–1223. doi: 10.1073/pnas.91.4.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B, et al. Activity of metal-responsive transcription factor 1 by toxic heavy metals and H2O2 in vitro is modulated by metallothionein. Mol Cell Biol. 2003;23:8471–8485. doi: 10.1128/MCB.23.23.8471-8485.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obata A, et al. Retention mechanism of hypoxia selective nuclear imaging/radiotherapeutic agent cu-diacetyl-bis(N4-methylthiosemicarbazone) (Cu-ATSM) in tumor cells. Ann Nucl Med. 2001;15:499–504. doi: 10.1007/BF02988502. [DOI] [PubMed] [Google Scholar]
- 20.Trounce IA, Pinkert CA. Cybrid models of mtDNA disease and transmission, from cells to mice. Curr Top Dev Biol. 2007;77:157–183. doi: 10.1016/S0070-2153(06)77006-5. [DOI] [PubMed] [Google Scholar]
- 21.McKenzie M, Trounce I. Expression of Rattus norvegicus mtDNA in Mus musculus cells results in multiple respiratory chain defects. J Biol Chem. 2000;275:31514–31519. doi: 10.1074/jbc.M004070200. [DOI] [PubMed] [Google Scholar]
- 22.Griffith OW, Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine) J Biol Chem. 1979;254:7558–7560. [PubMed] [Google Scholar]
- 23.Chacin J, et al. Effect of Krebs cycle intermediates and inhibitors on toad gastric mucosa. Am J Physiol. 1979;236:E692–E700. doi: 10.1152/ajpendo.1979.236.6.E692. [DOI] [PubMed] [Google Scholar]
- 24.Booth K, Larkin K, Maddocks I. Agranulocytosis coincident with amodiaquine therapy. Br Med J. 1967;3:32–33. doi: 10.1136/bmj.3.5556.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crim JA, Petering HG. The antitumor activity of Cu(II)KTS, the copper (II) chelate of 3-ethoxy-2-oxobutyraldehyde bis(thiosemicarbazone) Cancer Res. 1967;27:1278–1285. [PubMed] [Google Scholar]
- 26.Cowley AR, Dilworth JR, Donnelly PS, Labisbal E, Sousa A. An unusual dimeric structure of Cu(I) bis(thiosemicarbazone) complex; implications for the mechanism of hypoxic selectivity of Cu(II) derivatives. J Am Chem Soc. 2002;124:5270–5271. doi: 10.1021/ja012668z. [DOI] [PubMed] [Google Scholar]
- 27.Holland JP, Green JC, Dilworth JR. Probing the mechanism of hypoxia selectivity of copper bis(thiosemicarbazonato) complexes: DFT calculation of redox potentials and absolute acidities in solution. Dalton Trans. 2006:783–794. doi: 10.1039/b512656h. [DOI] [PubMed] [Google Scholar]
- 28.Crouch PJ, et al. Increasing Cu bioavailability inhibits Aβ oligomers and tau phosphorylation. Proc Natl Acad Sci USA. 2009;106:381–386. doi: 10.1073/pnas.0809057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donnelly PS, et al. Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer’s disease amyloid-β peptide. J Biol Chem. 2008;283:4568–4577. doi: 10.1074/jbc.M705957200. [DOI] [PubMed] [Google Scholar]
- 30.Beyersmann D, Haase H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. BioMetals. 2001;14:331–341. doi: 10.1023/a:1012905406548. [DOI] [PubMed] [Google Scholar]
- 31.Crouch PJ, et al. The Alzheimer’s therapeutic PBT2 promotes amyloid-beta degradation and GSK3 phosphorylation via a metal chaperone activity. J Neurochem. 2011;119:220–230. doi: 10.1111/j.1471-4159.2011.07402.x. [DOI] [PubMed] [Google Scholar]
- 32.Price KA, et al. Activation of epidermal growth factor receptor by metal-ligand complexes decreases levels of extracellular amyloid beta peptide. Int J Biochem Cell Biol. 2008;40:1901–1917. doi: 10.1016/j.biocel.2008.01.033. [DOI] [PubMed] [Google Scholar]
- 33.Price KA, et al. Sustained activation of glial cell epidermal growth factor receptor by bis(thiosemicarbazonato) metal complexes is associated with inhibition of protein tyrosine phosphatase activity. J Med Chem. 2009;52:6606–6620. doi: 10.1021/jm9007938. [DOI] [PubMed] [Google Scholar]
- 34.Kim JH, Cho H, Ryu SE, Choi MU. Effects of metal ions on the activity of protein tyrosine phosphatase VHR: Highly potent and reversible oxidative inactivation by Cu2+ ion. Arch Biochem Biophys. 2000;382:72–80. doi: 10.1006/abbi.2000.1996. [DOI] [PubMed] [Google Scholar]
- 35.Fujibayashi Y, et al. Mitochondria-selective reduction of 62Cu-pyruvaldehyde bis(N4-methylthiosemicarbazone) (62Cu-PTSM) in the murine brain; a novel radiopharmaceutical for brain positron emission tomography (PET) imaging. Biol Pharm Bull. 1993;16:146–149. doi: 10.1248/bpb.16.146. [DOI] [PubMed] [Google Scholar]
- 36.Taniuchi H, et al. Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone) (Cu-PTSM), a metal complex with selective NADH-dependent reduction by complex I in brain mitochondria: A potential radiopharmaceutical for mitochondria-functional imaging with positron emission tomography (PET) Biol Pharm Bull. 1995;18:1126–1129. doi: 10.1248/bpb.18.1126. [DOI] [PubMed] [Google Scholar]
- 37.DiMauro S. Mitochondrial diseases. Biochim Biophys Acta. 2004;1658:80–88. doi: 10.1016/j.bbabio.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 38.Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochim Biophys Acta. 2010;1802:29–44. doi: 10.1016/j.bbadis.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 39.Heales SJ, Menzes A, Davey GP. Depletion of glutathione does not affect electron transport chain complex activity in brain mitochondria: Implications for Parkinson’s disease and postmortem studies. Free Radical Biol Med. 2011;50:899–902. doi: 10.1016/j.freeradbiomed.2010.11.032. [DOI] [PubMed] [Google Scholar]
- 40.Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: What is so special about them? Trends Cell Biol. 2008;18:165–173. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 41.Moro MA, Almeida A, Bolanos JP, Lizasoain I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radical Biol Med. 2005;39:1291–1304. doi: 10.1016/j.freeradbiomed.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 42.Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 43.Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: Basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008;1142:133–158. doi: 10.1196/annals.1444.011. [DOI] [PubMed] [Google Scholar]
- 44.Trounce I, et al. Cloning of neuronal mtDNA variants in cultured cells by synaptosome fusion with mtDNA-less cells. Nucleic Acids Res. 2000;28:2164–2170. doi: 10.1093/nar/28.10.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.