

Abstract
A highly combinatorial structure-based protein engineering method for obtaining enantioselectivity is reported that results in a thorough modification of the substrate binding pocket of Candida antarctica lipase A (CALA). Nine amino acid residues surrounding the entire pocket were simultaneously mutated, contributing to a reshaping of the substrate pocket to give increased enantioselectivity and activity for a sterically demanding substrate. This approach seems to be powerful for developing enantioselectivity when a complete reshaping of the active site is required. Screening toward ibuprofen ester 1, a substrate for which previously used methods had failed, gave variants with a significantly increased enantioselectivity and activity. Wild-type CALA has a moderate activity with an E value of only 3.4 toward this substrate. The best variant had an E value of 100 and it also displayed a high activity. The variation at each mutated position was highly reduced, comprising only the wild type and an alternative residue, preferably a smaller one with similar properties. These minimal binary variations allow for an extremely condensed protein library. With this highly combinatorial method synergistic effects are accounted for and the protein fitness landscape is explored efficiently.
Keywords: kinetic resolution, library design, protein design, enzyme catalysis
The employment of enzymes in industrial processes is steadily increasing, which reflects their attractiveness as catalysts. Enzymes show high substrate specificity, good enantioselectivity, and high activity and, furthermore, they can be used under mild reaction conditions. The high enantioselectivity displayed by many enzymes is useful for the preparation of enantiomerically pure compounds, which is increasingly important for the production of pharmaceuticals (1).
A major focus of applied biocatalysis today is to develop enzymes for increased enantioselectivity in organic transformations (2, 3). Various protein engineering methods for improving or altering the catalytic properties of enzymes have been developed during the past two decades (4–10). Directed evolution is a powerful tool in modern protein engineering (4–6) and is based on iterative cycles of mutagenesis and selection. It is a highly useful method to climb stepwise in activity, enantioselectivity, or some other phenotypic property (11). In directed evolution a random mutagenesis approach has often been used (12, 13). A current trend in protein engineering is to move away from methods incorporating random mutations over a whole gene sequence toward more semirational approaches (14) using site-directed mutagenesis, introducing focused mutations at defined sites. In random and semirational methods a library of protein variants is generated, which subsequently is screened for a defined property.
The number of sites changed and the number of amino acids varied at each site are important parameters when deciding what protein engineering strategy to use (15), because it is desirable to keep time-consuming and labor-intensive screening to a minimum (16). Iterative saturation mutagenesis (ISM), a rational based method where saturation mutagenesis is performed iteratively at chosen sites in an enzyme, has been particularly successful in improving properties such as thermostability, substrate acceptance or enantioselectivity (17, 18). These methods generally create small libraries, as only a few amino acid residues are targeted. Protein engineering methods that focus on the active site has a higher chance of influencing catalytic properties (19). Such active-site-focused libraries have been used with good results for the improvement of activity and enantioselectivity (3). Two or three amino acid positions are generally subjected simultaneously to mutagenesis to account for the potential synergistic conformational and electrostatic effects that may appear. This approach is called combinatorial active-site saturation test (CAST) (20) and may also be used in an iterative manner. It has also been shown that enzyme libraries with highly reduced amino acid alphabets can be used for attaining excellent enantioselectivity (21).
Combinatorial mutagenesis with variations at many sites simultaneously has also been used for broadening the protein functional diversity. Typically, each site is substituted with a small set of amino acid residues, and the suggested composition of these sets is usually based on bioinformatics. The degree of amino acid residue conservation derived from a multiple sequence alignment (MSA) can give an indication whether it is permissible to substitute a residue (22). Also, the structure-based MSA 3DM database have been used for suggesting mutational sites and allowed residues (23). Combinatorial libraries with small sets of amino acid residues have been used for the generation of consensus libraries for the development of thermostable enzymes (22, 24, 25).
Candida antarctica lipase A (CALA) is a highly thermostable enzyme displaying unusual properties such as activity toward tertiary alcohols (26), high enantioselectivity toward β-amino acids (27), and sn-2 fatty acid preference of triglycerides (28). The X-ray structure of CALA has been reported (29), and its fold belongs to a mostly unexplored subfamily of lipases (30). We have recently studied the directed evolution of CALA toward esters of α-substituted carboxylic acids (31, 32) using CAST (20) and developed a highly active and enantioselective variant (32). This variant, a triple mutant, showed remarkably broad substrate scope. However, although this variant showed excellent selectivity toward 2-phenylpropanoate esters, we had difficulties in obtaining improved variants for a more sterically demanding analogue, 2-(4-isobutylphenyl)propanoate ester 1 (ibuprofen ester, see Fig. 1). Six libraries with the CAST approach with pairwise mutagenesis led to no significantly improved variants toward hydrolysis of 1. Instead of exploring more pathways, it was decided to use an approach that radically restructures the active-site pocket. Highly combinatorial approaches with binary amino acid sets have previously been used for creating functional diversity of proteins (22, 25, 33). Combinatorial libraries using only alanine as substitution residue was recently used for enabling the acceptance of larger substrates (34). However, structure-based combinatorial mutagenesis employing binary sets has to our knowledge never been used for creating enantioselective enzymes. We therefore decided to test this approach for obtaining enantioselectivity for a case that failed with previously used methods.
Fig. 1.

α-Methylcarboxylate esters of interest for the project with different steric bulk. CALA-YNG has high enantioselectivity and activity toward 2, 3, and 4, but poor enantioselectivity and activity toward 1.
The best protein engineering strategy is stated to be the one that reaches the desired goal with the least effort (15). Here we describe a powerful, yet simple, semirational combinatorial protein engineering technique customized for the creation of a tailor-made substrate pocket. With this method highly enantioselective variants of CALA for sterically demanding substrate 1 were obtained.
Results
Substrate Walking.
Previously, we have reported on a CALA enzyme variant developed by directed evolution (CASTing). This variant, designated CALA-YNG (32) after the new amino acids introduced, had the following substitutions: Phe149Tyr, Ile150Asn, and Phe233Gly. CALA-YNG was found to be highly active and enantioselective toward various α-substituted esters such as 2-phenylpropanoate esters (e.g., 2) as well as the para-methyl analogue 3 (Fig. 1). Also, 2-methylalkanoate esters such as 4 gave high enantioselectivity. Yet the similar para-isobutyl analogue, ibuprofen ester 1, displayed poor reactivity and essentially no enantioselectivity. It was assumed that an active variant toward ibuprofen could be obtained via directed evolution using CALA-YNG as template. This assumption is reasonable as directed evolution has been used for stepwise enzyme adaptation, where a slightly different substrate was employed in each iteration of the screening compared to the preceding iteration. This approach is called substrate walking (35, 36). Even the best variants isolated from these libraries showed only moderate improvement of activity, and unsatisfying enantioselectivity toward 1.*
Strategy Outline.
Instead of exploring more possibilities along the line of varying two amino acids simultaneously, a highly combinatorial approach for reshaping the substrate pocket was devised. Inspired by the insights from: (i) the strong synergistic effects observed when combining several mutations, (ii) the highly reduced sampling requirements when using condensed libraries, (iii) the mutability of amino acid residues, and (iv) the proximity of amino acid residues to the docked substrate in enzyme models, we have now developed a structure-based protein engineering method with variation of many sites simultaneously for obtaining enantioselective variants toward substrate 1.
The approach is outlined in Fig. 2: (i) First, a close inspection of the substrate binding pocket is performed (Fig. 3) using a computer model where the target substrate is bound into the enzyme. All the surrounding residues close to the substrate are examined by their mutability by comparing the residue conservation (based on MSA). The composition of mutations proposed for each site was the original wild-type (WT) residue and an alternative residue with similar properties, preferably smaller than the WT residue and thereby enabling the possibility of enlarging the substrate pocket (Fig. 3). (ii) Potential mutations are introduced in PCR-formed fragments using degenerate oligonucleotides. (iii) Gene assembly is performed via overlap extension-PCR (OE-PCR) (37), which results in a combinatorial gene fragment. (iv) Homologous recombination of the mutagenized gene fragment and the vector fragment is performed in the yeast host, Pichia pastoris.
Fig. 2.

Flowchart of combinatorial substrate pocket reshaping.
Fig. 3.

The active site of CALA with the tetrahedral intermediate (S)-1 docked in the active site. The nucleophilic Ser184 is covalently bound to the carbonyl of the ester. Surrounding the substrate are the nine sites that were selected for mutagenesis. The residues forming the combinatorial mutagenesis set are displayed with the wild-type residues underlined.
Combinatorial Library Design.
The ibuprofen ester (S)-1 was docked inside the CALA X-ray model in the tetrahedral intermediate form, covalently bound to the nucleophilic Ser184, using molecular dynamics. The model was allowed to settle in a low energy state (Fig. 3). Several residues in the substrate binding pocket were forced to move aside to accommodate the sterically demanding 4-isobutylphenyl group during the energy relaxation. In the selection of mutable amino acids, all residues lining the acyl chain tunnel were considered as potential mutable positions, determined by selecting all residues within 4 Å from the bound ester 1. According to the model, many WT residues within this distance are experiencing sterical clashes with substrate 1. Phe431, located in an active-site flap over the entrance of the substrate tunnel was also included in the set as it has been shown that large hydrophobic bottleneck residues can limit the size range of substrates (38, 39). The degree of residue conservation among related enzymes was determined by MSA of a PSI-BLAST (40) of CALA. Residue sites with strong conservation over a broad family range have fundamental importance for activity or stability (41), and therefore no longer considered as potential targets. After this reduction, the library consisted of nine mutable residues, a manageable number.
Each selected position was set to randomize between the wild-type and another smaller residue with similar properties, unless some other prior knowledge indicated that another substituent is more suitable. Phe149Tyr and Ile150Asn were included because we previously found that the 149Tyr/150Asn pair contributed to strong enantioselectivity toward a broad range of substrates (32). These studies also suggested that 233 is an important position (a hot spot) and we wanted to be able to access several amino acids at this site. Glycine was found to be crucial at this site for the acceptance of α-arylpropanoate esters (32). Also, valine and cysteine which are intermediate in size between phenylalanine and glycine were included. Cysteine brings about an intriguing electrostatic environment.
The final set that was used for the mutagenesis is shown in Table 1. The number of possible variants that could be generated with this set is 28 × 41 = 1,024.
Table 1.
Combinatorial substrate pocket library set
| Position | WT residue | Alternative residue(s) |
| 149 | Phe | Tyr |
| 150 | Ile | Asn |
| 215 | Pro | Ala |
| 221 | Thr | Ser |
| 225 | Leu | Val |
| 233 | Phe | Cys/Gly/Val |
| 234 | Ala | Gly |
| 237 | Gly | Ala |
| 431 | Phe | Val |
Mutagenesis and Homologous Recombination.
Because of sequence proximity, the nine mutations were clustered into four primer pairs. Using those, together with primers for the ends of the gene, five fragments partly overlapping with one another were generated in five individual PCR reactions (see Fig. 2 B and C). The five fragments were simultaneously assembled in a modified OE-PCR (42, 43). A vector fragment with ends homologous to the combinatorial library gene fragment was also produced. The template vector pBGP1 is an episomally replicating plasmid, useful for constitutively expressed enzyme libraries (44). The gene and vector fragments were transformed into P. pastoris, taking advantage of the yeasts’ internal homologous recombination system (45) to generate the secretory vector (Fig. 2D). Transformants were sequenced to validate that the diversity of the library was adequate, confirming no observed bias toward wild-type residues.
Screening Toward Ibuprofen Ester 1.
Expression of the generated enzyme variants was followed by screening for increased hydrolytic activity, as described previously (32). According to our previous experience, most of the variants with an increased activity also displayed a significantly improved enantioselectivity (31, 32). The ester (rac)-1 was used as screening substrate for the spectrophotometric activity assay. We assayed 2,400 transformants, which corresponds to a sequence coverage of 90% (46). Of all the screened variants, only a small group displayed good activity toward substrate 1. Two of these enzyme variants showed high enantioselectivity with E values of 53 and 100. Sequencing of the corresponding transformants showed that the former has the four following substitutions: Thr221Ser/Leu225Val/Phe233Cys/Phe431Val. It was designated SV1CV2 after the new amino acids introduced (Table 2). The latter variant was found to have five substitutions of which four were identical to those of the first variant and the fifth was a Gly237Ala substitution. This pentasubstituted variant was designated SV1CAV2. Both variants showed a higher specific activity compared to wild-type CALA, SV1CV2 being approximately four times as active and SV1CAV2 being more than six times as active (Table 2).
Table 2.
Specific activity and enantioselectivity of selected variants from the screening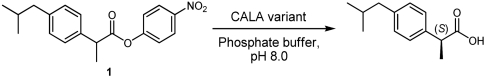
| Variant |
Specific activity (nmol min-1 mg-1) |
E* |
| WT | 21 ± 1 | 3.4 |
| SV1CV2 | 81 ± 0.3 | 53 |
| SV1CAV2 | 133 ± 2 | 100 |
*Enantioselectivity factor, calculated from eep and conversion according to Sih and Wu (47). All variants have (S) enantiopreference.
Functional Diversity of the Library.
To investigate the quality of the protein library, the activity toward different 4-nitrophenyl esters of a small subset of the library was compared. These esters have different steric demands on the enzyme variants and in this way the functional diversity of the library is explored (Fig. 4). The enzyme variants were screened for hydrolytic activity, using racemic esters 1, 2, and 4. Several interesting aspects were observed: (i) The library had large functional diversity, i.e., it contained variants that were more active than the wild type as well as variants that were less active than the wild type. This functional diversity is an indication that the library was of good quality. (ii) Only a small minority of the library had increased activity toward ibuprofen ester 1. (iii) There seemed to be some correlation between activities toward different substrates. (iv) There were some enzyme variants that deviate from this correlation. Several enzyme variants showed distinguished activity toward one of the esters. One of these variants was the SV1CAV2 variant, which displayed unique strong activity toward 1. At the same time, the enzyme variants that displayed the strongest activity toward 2 and 4 displayed lower activity toward 1. This observation implies it would have been difficult to reach a high activity of 1 by using 2 and 4 as model substrates.
Fig. 4.

Comparison of activity toward different α-methylcarboxylate esters for a small subset (89 transformants) of the combinatorial library. The transformants are ordered after increasing activity toward substrate 2. The highest activity for each substrate in this subset was normalized to 100. P. pastoris X33 supernatants were used as blank. The highest activity toward 1 found in this subset is an SV1CAV2 variant. For comparison, in this subset the initial rates for the best variants toward 1, 2, and 4 are 1.1, 22, and 32 mOD410 min-1, respectively.
Deconvolution of Substitutions.
To explore whether all substitutions in the SV1CAV2 variant are necessary, remutations to the wild-type residues were performed. These experiments are crucial for determining whether some of the substitutions were redundant (17). Five individual variants were created, where each variant had one of the five residue substitutions mutated back to the wild-type amino acid (one of these, SV1CV2, was already accessible; Table 2).
Each remutation showed a decreased activity compared to the SV1CAV2 variant, indicating that each substitution is important for activity (Table 3). Two substitutions Thr221Ser (S) and Leu225Val (V1) are crucial for enantioselectivity based on the fact that the E values of V1CAV2 and SCAV2 dropped from 100 to 4–5. Also Phe233Cys (C) is of importance because SV1AV2 gave an E value of 22, with a significant decrease in activity. However, SV1 and SV1C gave low E values (4.0 and 10.2, respectively). The high enantioselectivity found in the SV1CAV2 variant therefore appears to be a merging of many cooperative effects.
Table 3.
Specific activity and enantioselectivity of remutated variants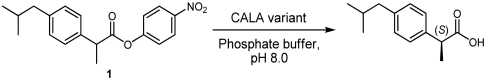
| Variant | Specific activity (nmol min-1 mg-1) | E* |
| WT | 21 ± 1 | 3.4 ± 0.5† |
| SV1CAV2 | 133 ± 2 | 100 ± 5† |
| V1CAV2 | 43 ± 0.3 | 5.2 ± 0.8† |
| SCAV2 | 31 ± 0.1 | 3.9 ± 0.5† |
| SV1AV2 | 11 ± 0 | 22 ± 2† |
| SV1CV2‡ | 81 ± 0.3 | 53 ± 3† |
| SV1CA | 96 ± 1 | 80 ± 4† |
| SV1C | ND§ | 10.2 ± 1.7¶ |
| SV1 | ND§ | 4.0 ± 0.9¶ |
*Enantioselectivity factor, calculated according to Table 2. All variants have (S) enantiopreference. Several independent reactions were performed to validate the reported E values.
†Estimated error.
‡This variant was obtained from the screening (Table 2).
§ND, not determined.
¶Standard error of the mean is reported.
Our initial attempts to find selective CALA variants toward ibuprofen by variation of two amino acid residues at the same time were unsuccessful. An interesting question is whether it would have been possible at all to find the SV1CAV2 (E = 100) variant with iterative CASTing. Success with this approach requires that there are any single or double mutations in SV1CAV2 that have good enough E values for being considered as improvements.
To provide information whether iterative CASTing could have reached SV1CAV2, all possible double mutations as well as all single mutants of this variant, were prepared by site-directed mutagenesis. As can be seen from Table 4 all the double mutants and single mutants of SV1CAV2 showed very poor E values. The conclusion is therefore that with the present screening method it would not have been possible to find the SV1CAV2 variant by iterative CASTing with variation of only two amino acid residues at the same time.
Table 4.
Enantioselectivity of single and double mutants of CALA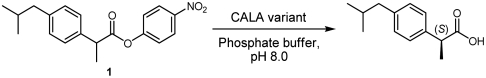
| Variant | E*† | Variant | E*† |
| SV1 | 4.0 ± 0.9 | CV2 | 1.2 ± 0.1 |
| SC | 3.9 ± 0.8 | AV2 | 1.3 ± 0.1 (R) |
| SA | 1.9 ± 0.5 | S | 2.1 ± 0.3 |
| SV2 | 1.4 ± 0.2 | V1 | 1.1 ± 0.1 (R) |
| V1C | 5.1 ± 0.8 | C | 1.3 ± 0 |
| V1A | 1.4 ± 0.3 (R) | A | 1.5 ± 0.2 (R) |
| V1V2 | 2.0 ± 0.2 | V2 | 1.1 ± 0 (R) |
| CA | 1.3 ± 0.1 |
*Enantioselectivity factor, calculated according to Table 2. At least three independent reactions were performed to validate the reported E values. Standard error of the mean is reported.
†Unless otherwise noted the selectivity was in favor of the S enantiomer.
Enzyme Models.
Enzyme models were prepared with the substitutions found in the SV1CAV2 variant introduced, using molecular dynamics, and letting the system settle in a low energy state (Fig. 5). The model shows that several substitutions in the SV1CAV2 variant appear to influence the acceptance of the bulky ibuprofen moiety. The Thr221Ser/Leu225Val (V1) substitutions contributed to a better accommodation for the isobutyl branch. From this model it is more difficult to determine if the Phe431Val (V2) substitution contributes to substrate specificity, as this residue is located on a loop that is most likely to fluctuate dynamically.
Fig. 5.

Models of CALA variants with the tetrahedral intermediate (S)-1 seen in the active site. The variants shown are (A) WT, and (B) SV1CAV2. SV1CAV2 provides more space than WT to accommodate the substrate.
Discussion
Protein Fitness Landscapes.
A protein fitness landscape (11) is a conceptual multidimensional sequence space landscape with topology based on fitness, a variable based on the protein phenotype (48). The fitness is often a property such as thermal stability, activity, or enantioselectivity. Directed evolution is a highly useful method to explore and climb in protein fitness landscapes. One interesting phenomenon observed when combining several mutations in ISM (18) is the high degree of epistatic effects (49). These effects imply that it is possible to bypass valleys in the fitness landscape by utilizing strong synergistic effects (50). If the sequence space close to the starting point (point A in Fig. 6) on the fitness landscape has a topology that is neutral, it is not possible to make an upward fitness climb by traditional directed evolution methods, such as error-prone PCR. In such a situation it is imperative to develop methods that can probe the fitness landscape effectively to discover topologies with higher fitness.
Fig. 6.

A greatly simplified illustration of a protein fitness landscape that is mostly neutral but with a distant peak. Point A represents a starting point with neutral surroundings. B represents a point in a distant patch with increased fitness compared to point A. To be able to directly jump from A to B the fitness landscape has to be probed radically.
Traversing a Neutral Landscape.
The site-directed mutagenesis of CALA using CAST did not result in any improvement for hydrolysis of ibuprofen ester 1. Also, the single and double mutants in Table 4, which simulated an initial walk from the wild-type toward the SV1CAV2 variant, did not display any significant improvement of enantioselectivity. This observation suggests a fitness landscape with a mostly neutral topology (50), but with a distant peak. To be able to reach a far-away peak on the fitness landscape simultaneous variation of many mutations in the substrate pocket is required. The highly combinatorial approach can be described as pole vaulting in the fitness landscape (illustrated by the jump in Fig. 6).
We do not claim that the SV1CAV2 variant represents a fitness peak maximum. Rather, it is most likely that the SV1CAV2 variant is located on a patch with a peak (point B on Fig. 6), with a local maximum somewhere higher up. We propose that this variant can be used as a starting point for a stepwise climb upward, using established directed evolution methods, and this approach will be the subject of a subsequent study.
Conclusions
We have developed a method for radically reshaping the active site of an enzyme for improved activity and enantioselectivity. The method was tested for creation of an enantioselective variant of CALA toward a difficult substrate (ibuprofen ester 1) for which previously used methods for evolving CALA were unsuccessful. Using this unique method, a CALA variant, SV1CAV2, was obtained that has an enlarged substrate pocket and displays highly improved enantioselectivity. We demonstrated that the simultaneous incorporation of mutations to gain synergistic epistatic effects was necessary to find the enantioselective variant SV1CAV2. We propose that this highly combinatorial protein engineering approach can be utilized for the development of useful focused libraries that can reach regions that are hard to access on the fitness landscape. This approach should be validated toward other enzymes to determine its generality to obtain enantioselectivity. We believe that the demonstrated method has a place in the future protein engineering toolbox for obtaining enantioselective enzyme variants.
Experimental Methods
General equipment, chemicals, media recipes, molecular biology techniques, primers, enzyme purification, kinetic analysis, and kinetic resolution are described in SI Text.
Bioinformatics.
An iterative PSI-BLAST (40) was performed using 2VEO (29) as template. Four iterations were performed with a cutoff of 500 entries. Sequences were collected and an MSA was generated using CLUSTAL-W (51). Conservation was determined using WebLogo (52) and conservation was ranked for all sites that were considered for mutagenesis.
Molecular Modeling.
Molecular dynamics (MD) simulations were performed using Moloc computational package (53). The used crystal structure [Protein Data Bank (PDB) ID code 2VEO] is crystallized in a closed conformation and therefore the enzyme was allowed to equilibrate by performing a 10-ns MD simulation with the intention to create a more open conformation. After the equilibration the enzyme was considerably more open in the substrate entrance pocket and thus was able to accommodate the 4-nitrophenyl moiety. The acid part of the substrate was modeled using the polyethylene glycol available in the crystal structure as reference.
Selection of Residues for Mutation.
The selection of residues was determined as follows: (i) All residues lining the acyl tunnel close to the potential substrate were considered, and formed a set of potential mutable residues. This selection was done by choosing all amino acids with some part of the side chain (or Cα) within 4 Å from the acyl moiety. The following 12 residues became part of the set: Asp95, Phe149, Ile150, Ser184, Pro215, Thr221, Leu225, Phe233, Ala234, Gly237, Ile336, and His366. (ii) Phe431, located in the entrance of the substrate tunnel, was included in the set as it has been shown that large hydrophobic bottleneck residues can limit the size range of substrates (38). (iii) Amino acids that are crucial for the catalytic machinery were removed from the set. The catalytic residues Ser184 and His366, as well as the oxyanion hole contributor Asp95 were removed. (iv) Conserved residues were removed from the set. Only Ile336 was removed from the set.
Selection of Mutagenic Residue Set.
The selection of the compositions of the residue sets is described in Results. Set suggestions are included in the SI Text.
Supplementary Material
Acknowledgments.
We thank Prof. K. Hult for fruitful discussions. Financial support from the Swedish Research Council, the Swedish Foundation for Strategic Research and the Knut and Alice Wallenberg Foundation is gratefully acknowledged.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1111537108/-/DCSupplemental.
*Details regarding the design of these libraries are included in the SI Text.
References
- 1.Tang WL, Zhao H. Industrial biotechnology: Tools and applications. Biotechnol J. 2009;4:1725–1739. doi: 10.1002/biot.200900127. [DOI] [PubMed] [Google Scholar]
- 2.Turner NJ. Directed evolution drives the next generation of biocatalysts. Nat Chem Biol. 2009;5:567–573. doi: 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- 3.Reetz MT. Laboratory evolution of stereoselective enzymes: A prolific source of catalysts for asymmetric reactions. Angew Chem Int Edit. 2011;50:138–174. doi: 10.1002/anie.201000826. [DOI] [PubMed] [Google Scholar]
- 4.Arnold FH, Georgiou G, editors. Directed evolution library creation: Methods and protocols. Methods in molecular biology. Vol 231. Totowa, NJ: Humana Press; 2003. [Google Scholar]
- 5.Arnold FH. Design by directed evolution. Acc Chem Res. 1998;31:125–131. [Google Scholar]; Farinas ET, Bulter T, Arnold FH. Directed enzyme evolution. Curr Opin Biotechnol. 2001;12:545–551. doi: 10.1016/s0958-1669(01)00261-0. [DOI] [PubMed] [Google Scholar]
- 6.Reetz MT. Directed evolution of enantioselective enzymes: An unconventional approach to asymmetric catalysis in organic chemistry. J Org Chem. 2009;74:5767–5778. doi: 10.1021/jo901046k. [DOI] [PubMed] [Google Scholar]
- 7.Reetz MT. Enzyme engineering by directed evolution. In: Baltz RH, Davies JE, Demain AL, editors. Manual of Industrial Microbiology and Biotechnology. 3rd Ed. Washington, DC: American Society for Microbiology; 2010. pp. 466–479. [Google Scholar]
- 8.Brakmann S, Johnsson K, editors. Directed Molecular Evolution of Proteins: Or How to Improve Enzymes for Biocatalysis. New York: Wiley; 2002. [Google Scholar]
- 9.Svendsen A, editor. Enzyme Functionality. New York: Marcel Dekker; 2004. [Google Scholar]
- 10.Shivange AV, Marienhagen J, Mundhada H, Schenk A, Schwaneberg U. Advances in generating functional diversity for directed protein evolution. Curr Opin Chem Biol. 2009;13:19–25. doi: 10.1016/j.cbpa.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 11.Romero PA, Arnold FH. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol. 2009;10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen KQ, Arnold FH. Directed evolution of a thermostable esterase. Proc Natl Acad Sci USA. 1993;90:5618–5622. [Google Scholar]
- 13.Reetz MT, Zonta A, Schimossek K, Liebeton K, Jaeger KE. Creation of enantioselective biocatalysts for organic chemistry by in vitro evolution. Angew Chem Int Ed. 1997;36:2830–2832. [Google Scholar]
- 14.Otten LG, Hollmann F, Arends I. Enzyme engineering for enantioselectivity: From trial-and-error to rational design? Trends Biotechnol. 2010;28:46–54. doi: 10.1016/j.tibtech.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Kazlauskas RJ, Bornscheuer UT. Finding better protein engineering strategies. Nat Chem Biol. 2009;5:526–529. doi: 10.1038/nchembio0809-526. [DOI] [PubMed] [Google Scholar]
- 16.Reetz MT, Kahakeaw D, Lohmer R. Addressing the numbers problem in directed evolution. ChemBioChem. 2008;9:1797–1804. doi: 10.1002/cbic.200800298. [DOI] [PubMed] [Google Scholar]
- 17.Reetz MT, Prasad S, Carballeira JD, Gumulya Y, Bocola M. Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: Rigorous comparison with traditional methods. J Am Chem Soc. 2010;132:9144–9152. doi: 10.1021/ja1030479. [DOI] [PubMed] [Google Scholar]
- 18.Reetz MT, Carballeira JD. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc. 2007;2:891–903. doi: 10.1038/nprot.2007.72. [DOI] [PubMed] [Google Scholar]
- 19.Morley KL, Kazlauskas RJ. Improving enzyme properties: When are closer mutations better? Trends Biotechnol. 2005;23:231–237. doi: 10.1016/j.tibtech.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Reetz MT, Bocola M, Carballeira JD, Zha D, Vogel A. Expanding the range of substrate acceptance of enzymes: Combinatorial active-site saturation test. Angew Chem Int Ed. 2005;44:4192–4196. doi: 10.1002/anie.200500767. [DOI] [PubMed] [Google Scholar]
- 21.Reetz MT, Wu S. Greatly reduced amino acid alphabets in directed evolution: Making the right choice for saturation mutagenesis at homologous enzyme positions. Chem Commun. 2008:5499–5501. doi: 10.1039/b813388c. [DOI] [PubMed] [Google Scholar]
- 22.Amin N, et al. Construction of stabilized proteins by combinatorial consensus mutagenesis. Protein Eng Des Sel. 2004;17:787–793. doi: 10.1093/protein/gzh091. [DOI] [PubMed] [Google Scholar]
- 23.Kourist R, et al. The α/β-hydrolase fold 3DM database (ABHDB) as a tool for protein engineering. ChemBioChem. 2010;11:1635–1643. doi: 10.1002/cbic.201000213. [DOI] [PubMed] [Google Scholar]
- 24.Jäckel C, Bloom JD, Kast P, Arnold FH, Hilvert D. Consensus protein design without phylogenetic bias. J Mol Biol. 2010;399:541–546. doi: 10.1016/j.jmb.2010.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamamatsu N, et al. Biased mutation-assembling: An efficient method for rapid directed evolution through simultaneous mutation accumulation. Protein Eng Des Sel. 2005;18:265–271. doi: 10.1093/protein/gzi028. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt M, et al. Enzymatic removal of carboxyl protecting groups. 1. Cleavage of the tert-butyl moiety. J Org Chem. 2005;70:3737–3740. doi: 10.1021/jo050114z. [DOI] [PubMed] [Google Scholar]
- 27.Liljeblad A, Kanerva LT. Biocatalysis as a profound tool in the preparation of highly enantiopure β-amino acids. Tetrahedron. 2006;62:5831–5854. [Google Scholar]
- 28.Dominguez de Maria P, et al. Biotechnological applications of Candida antarctica lipase A: State-of-the-art. J Mol Catal B Enzym. 2005;37:36–46. [Google Scholar]
- 29.Ericsson DJ, et al. X-ray structure of Candida antarctica lipase A shows a novel lid structure and a likely mode of interfacial activation. J Mol Biol. 2008;376:109–119. doi: 10.1016/j.jmb.2007.10.079. [DOI] [PubMed] [Google Scholar]
- 30.Widmann M, Juhl PB, Pleiss J. Structural classification by the lipase engineering database: A case study of Candida antarctica lipase A. BMC Genomics. 2010;11:123. doi: 10.1186/1471-2164-11-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandström AG, Engström K, Nyhlén J, Kasrayan A, Bäckvall J-E. Directed evolution of Candida antarctica lipase A using an episomaly replicating yeast plasmid. Protein Eng Des Sel. 2009;22:413–420. doi: 10.1093/protein/gzp019. [DOI] [PubMed] [Google Scholar]
- 32.Engström K, Nyhlén J, Sandström AG, Bäckvall J-E. Directed evolution of an enantioselective lipase with broad substrate scope for hydrolysis of α-substituted esters. J Am Chem Soc. 2010;132:7038–7042. doi: 10.1021/ja100593j. [DOI] [PubMed] [Google Scholar]
- 33.Treynor TP, Vizcarra CL, Nedelcu D, Mayo SL. Computationally designed libraries of fluorescent proteins evaluated by preservation and diversity of function. Proc Natl Acad Sci USA. 2007;104:48–53. doi: 10.1073/pnas.0609647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis JC, et al. Combinatorial alanine substitution enables rapid optimization of cytochrome P450BM3 for selective hydroxylation of large substrates. ChemBioChem. 2010;11:2502–2505. doi: 10.1002/cbic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen ZL, Zhao HM. Rapid creation of a novel protein function by in vitro coevolution. J Mol Biol. 2005;348:1273–1282. doi: 10.1016/j.jmb.2005.02.070. [DOI] [PubMed] [Google Scholar]
- 36.Savile CK, et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science. 2010;329:305–309. doi: 10.1126/science.1188934. [DOI] [PubMed] [Google Scholar]
- 37.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes—gene-splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 38.Schliessmann A, Hidalgo A, Berenguer J, Bornscheuer UT. Increased enantioselectivity by engineering bottleneck mutants in an esterase from Pseudomonas fluorescens. ChemBioChem. 2009;10:2920–2923. doi: 10.1002/cbic.200900563. [DOI] [PubMed] [Google Scholar]
- 39.Pavlova M, et al. Redesigning dehalogenase access tunnels as a strategy for degrading an anthropogenic substrate. Nat Chem Biol. 2009;5:727–733. doi: 10.1038/nchembio.205. [DOI] [PubMed] [Google Scholar]
- 40.Altschul SF, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schueler-Furman O, Baker D. Conserved residue clustering and protein structure prediction. Proteins Struct Funct Genet. 2003;52:225–235. doi: 10.1002/prot.10365. [DOI] [PubMed] [Google Scholar]
- 42.An YF, et al. A rapid and efficient method for multiple-site mutagenesis with a modified overlap extension PCR. Appl Microbiol Biotechnol. 2005;68:774–778. doi: 10.1007/s00253-005-1948-8. [DOI] [PubMed] [Google Scholar]
- 43.Peng RH, Xiong AS, Yao QH. A direct and efficient PAGE-mediated overlap extension PCR method for gene multiple-site mutagenesis. Appl Microbiol Biotechnol. 2006;73:234–240. doi: 10.1007/s00253-006-0583-3. [DOI] [PubMed] [Google Scholar]
- 44.Lee CC, Williams TG, Wong DW, Robertson GH. An episomal expression vector for screening mutant gene libraries in Pichia pastoris. Plasmid. 2005;54:80–85. doi: 10.1016/j.plasmid.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 45.Cregg JM, Cereghino JL, Shi J, Higgins DR. Recombinant protein expression in Pichia pastoris. Mol Biotechnol. 2000;16:23–52. doi: 10.1385/MB:16:1:23. [DOI] [PubMed] [Google Scholar]
- 46.Bosley AD, Ostermeier M. Mathematical expressions useful in the construction, description and evaluation of protein libraries. Biomol Eng. 2005;22:57–61. doi: 10.1016/j.bioeng.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 47.Sih CJ, Wu SH. Resolution of enantiomers via biocatalysis. Top Stereochem. 19:63–125. [Google Scholar]
- 48.Smith JM. Natural selection and concept of a protein space. Nature. 1970;225:563–564. doi: 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- 49.Reetz MT, Sanchis J. Constructing and analyzing the fitness landscape of an experimental evolutionary process. ChemBioChem. 2008;9:2260–2267. doi: 10.1002/cbic.200800371. [DOI] [PubMed] [Google Scholar]
- 50.Poelwijk FJ, Kiviet DJ, Weinreich DM, Tans SJ. Empirical fitness landscapes reveal accessible evolutionary paths. Nature. 2007;445:383–386. doi: 10.1038/nature05451. [DOI] [PubMed] [Google Scholar]
- 51.Chenna R, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerber PR. Charge distribution from a simple molecular orbital type calculation and non-bonding interaction terms in the force field MAB. J Comput Aid Mol Des. 1998;12:37–51. doi: 10.1023/a:1007902804814. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.