

Abstract
The van der Waals interaction is a weak, long-range correlation, arising from quantum electronic charge fluctuations. This interaction affects many properties of materials. A simple and yet accurate estimate of this effect will facilitate computer simulation of complex molecular materials and drug design. Here we develop a fast approach for accurate evaluation of dynamic multipole polarizabilities and van der Waals (vdW) coefficients of all orders from the electron density and static multipole polarizabilities of each atom or other spherical object, without empirical fitting. Our dynamic polarizabilities (dipole, quadrupole, octupole, etc.) are exact in the zero- and high-frequency limits, and exact at all frequencies for a metallic sphere of uniform density. Our theory predicts dynamic multipole polarizabilities in excellent agreement with more expensive many-body methods, and yields therefrom vdW coefficients C6, C8, C10 for atom pairs with a mean absolute relative error of only 3%.
The van der Waals interaction (1) is a long-range attraction between chemical species, arising from instantaneous charge fluctuations on each. This interaction is present even when there is no chemical bond, and even when each species has no permanent multipole moment. Its significance first came to light when van der Waals (vdW) (2) formulated the equation of state for real gases and liquids in 1873 by taking this correlation effect into consideration. The vdW interaction is much weaker in strength than a normal chemical bond, but it has important effects on the properties of materials (3). For example, it is responsible for many phenomena such as sublimation of iodine, naphthalene, dry ice, and other organic molecules, high interlayer mobility of graphite, foldings of long biomolecular chains such as DNA, RNA, proteins, polymers, etc. The vdW interaction has been extensively studied (4, 5) on account of its ubiquity and importance for soft matter relevant to human activity. Many theoretical methods, including CI (6, 7) (configuration interaction), MBPT (8) (many-body perturbation theory), coupled cluster (9) methods, or their combinations such as CI+MBPT (10, 11), have been developed to estimate this effect. These methods are highly accurate (within about 1%) but computationally expensive. A cheap but usefully accurate method is density functional theory (12) (DFT). While this theory has achieved a high level of sophistication and has become the mainstream electronic structure theory, it often fails in practice to describe vdW systems, because the long-range part of the vdW interaction is absent in commonly used DFT approximations [e.g., (13–15)].
In the large separation limit of two spherically symmetric interacting densities A and B, second-order perturbation theory yields the (nonretarded) potential energy EvdW, which can be expressed as a power series (16),
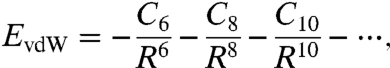 |
[1] |
where R is the separation between centers, C6 describes the instantaneous dipole-dipole interaction, C8 the dipole-quadrupole interaction, and C10 the quadrupole-quadrupole and the dipole-octupole interactions. [Note that, if higher-order perturbation theory is employed, odd-order terms C2k+1R-(2k+1) appear (17) in the power series of Eq. 1 for k≥5.] This series is obviously divergent as the internuclear distance of two atoms tends to zero, where the true vdW (or dispersion) energy remains finite. To fix this problem, each term of the series, C2kR-2k, may be multiplied by a damping function f(R), which suppresses the singularity at R = 0. Several such damping functions have been proposed [e.g., (18–20)]. The intermolecular or atom-pair potential energy is obtained as the sum of two contributions: the large part obtained from ordinary DFT methods and the small part which accounts for the vdW interaction. Therefore, in the simulation of intermolecular potentials, the main task is to find a simple and yet accurate way to evaluate these vdW coefficients C2k. Many efforts [e.g., (21–24)] have been devoted to the study of the leading coefficient C6, while higher-order coefficients C8 and C10 are less investigated. There are two good reasons for this fact. First, unlike C6, these higher-order coefficients essentially cannot be measured directly by experiments (25) and calculation of them is much more difficult (*). Second, it was believed that the higher-order contribution is small in comparison to the leading term [only 20% of the leading term (26)]. So, the vdW potential energy is often truncated to the leading term, -C6R-6. However, a detailed study (23) of vdW effects on the lattice constant and cohesive energies of alkali metals shows that the higher-order contributions can be as large as 50% of the leading term, consistent with an analysis of ref. 27. This study suggests that higher-order contributions cannot be neglected in the evaluation of the vdW effect. A simple and yet accurate evaluation of these higher-order coefficients is of strong interest in many branches of science such as chemistry, physics, materials science, molecular biology, drug design, etc. Several models (28, 29) were proposed to estimate these higher-order coefficients. However, none of these models is satisfactory. For example, an empirical model (29) with the static multipole polarizability as input has achieved good accuracy for higher-order vdW coefficients, but at the cost of introducing additional empirical parameters beyond those used for C6. A disadvantage of empirical models is that the physics behind the models is neither transparent nor transferable from one case to another, due to empirical fitting. A nonempirical model was proposed (28), without any empirical parameter beyond the static dipole polarizability. Although an excellent accuracy was achieved with the model for C6, the errors for C8 and C10 significantly increased (to 20%) from that for C6 (3%).
Nonempirical Model for the Dynamic Multipole Polarizability
We have developed a method for systematic calculations of higher-order vdW coefficients C8, and C10, on the basis of a simple generalization of an approach (23) for C6. The only needed input is the electron density and the static multipole polarizabilities of an atom, without empirical fitting. The contribution from second-order perturbation theory to the vdW coefficient C2k [the whole contribution (17) for C6, C8, and C10]for two spherically-symmetric densities can be expressed (30) exactly as an integral over u of the product of the dynamic multipole polarizabilities αl(iu) for l ≤ k - 2, for each density; i.e.,
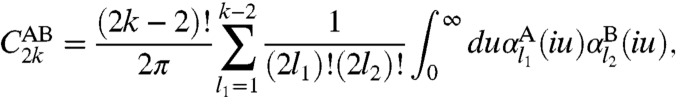 |
[2] |
where l2 = k - l1 - 1, 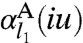 is the
is the 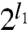 -pole dynamical polarizability of system A, and
-pole dynamical polarizability of system A, and 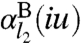 is the
is the 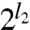 -pole dynamical polarizability of system B: l = 1 (dipole), 2 (quadrupole), 3 (octupole), etc. Here iu is an imaginary argument that replaces the real frequency ω. For C6 , only the dipole polarizabilities are needed, while C8 requires also the quadrupole polarizabilities, etc. For simple frequency dependences, the integral over frequency can be evaluated analytically.
-pole dynamical polarizability of system B: l = 1 (dipole), 2 (quadrupole), 3 (octupole), etc. Here iu is an imaginary argument that replaces the real frequency ω. For C6 , only the dipole polarizabilities are needed, while C8 requires also the quadrupole polarizabilities, etc. For simple frequency dependences, the integral over frequency can be evaluated analytically.
All the properties of a many-electron system are functionals of the ground-state density n(r) (12), although some like the dynamic multipole polarizabilities αl(iu) cannot be found from the density functional for the ground-state total energy. Here we will model the nonlocal density functionals for the dynamic multipole polarizabilities in a way that is exact in the zero- and high-frequency limits. The model interpolates between those limits in a way that is exact for a paradigm density from textbooks on classical electromagnetism: the metal sphere of uniform density. Because the u-dependence of αl(iu) is simple, our formula transfers with little error from the paradigm density to the density of a real atom.
Let us consider a metallic sphere of uniform density, with radius R (small enough to neglect retardation), in empty space. The dynamic multipole polarizability of the sphere is given by (31)
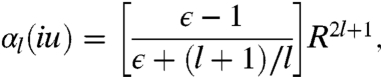 |
[3] |
where ϵ is the dielectric function of the sphere. By setting 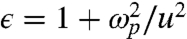 , we find
, we find
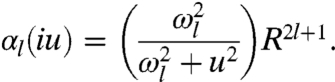 |
[4] |
Here ωl is the multipole resonance frequency of the sphere given by 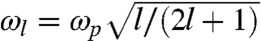 , with
, with 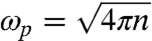 being the plasmon frequency of the extended uniform electron gas and n being the electronic density of the sphere under consideration. A familiar limit of Eq. 4 is the static dipole polarizability R3.
being the plasmon frequency of the extended uniform electron gas and n being the electronic density of the sphere under consideration. A familiar limit of Eq. 4 is the static dipole polarizability R3.
Next we make a nonempirical model for αl(iu) in terms of the electron density, by imposing exact constraints on a simple expression. First, we demand recovery of the correct zero-frequency limit αl(0). Second, we demand recovery of the correct high-frequency (u → ∞) limit (29, 32),
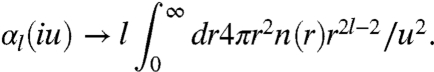 |
[5] |
Third, we make our expression exact (31, 33, 34) for a classical metallic sphere of uniform density inside and zero density outside a radius R [see Eq. 4]. Taking all three conditions into consideration, our model dynamic multipole polarizability of any spherical density is expressed as
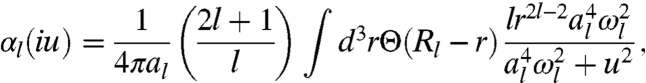 |
[6] |
where Θ(x) is a step function: Θ(x) = 1 for x > 0, and Θ(x) = 0 for x ≤ 0. The metallic sphere has a natural sharp cutoff of all integrals over r , which we extend even to the diffuse density of an atom (†). Unbiased sharp cutoffs are also used in the construction of nonempirical density functionals for the exchange-correlation energy (35–37) and a model dynamic dipole polarizability (38). The parameter al and the cutoff radius Rl are determined by the two imposed constraints. According to the static limit condition, we have
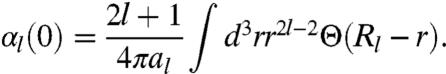 |
[7] |
Without the cutoff, the right-hand side of Eq. 7 would diverge. For spherically-symmetric systems, the radial cutoff is
| [8] |
Imposing the high-frequency limit of [5] leads to
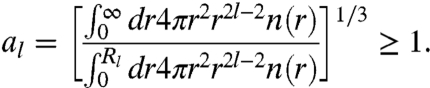 |
[9] |
For a metallic sphere of uniform density with radius R, we find al = 1 and Rl = R. Then Eq. 6 correctly reduces to Eq. 4. Because Eqs. 8 and [9] and are coupled together, they must be solved self-consistently. The results are displayed in Table 1. From Table 1 we see that there is a relationship among the parameters al: a1 < a2 < a3. This ordering arises because, if we keep the same cutoff radius for different order l, the denominator will get smaller relative to the numerator as l increases, due to the cutoff. One desired feature of the present model is that, for l = 1, our expressions Eqs. 6, [7], and [9] reduce to those of our earlier model (23).
Table 1.
Static multipolar polarizabilities αl(0) and cutoff radii Rl (both in atomic units), and parameters al of atoms
| Atom | α1(0) | α2(0) | α3(0) | R1 | R2 | R3 | a1 | a2 | a3 |
| H | 4.50(a) | 15.0(a) | 131.25(a) | 1.726 | 1.856 | 2.194 | 1.143 | 1.469 | 1.863 |
| He | 1.38(b) | 2.331(d) | 9.932(d) | 1.155 | 1.268 | 1.508 | 1.116 | 1.405 | 1.784 |
| Ne | 2.67(b) | 7.33(e) | 42.1(e) | 1.409 | 1.545 | 1.794 | 1.047 | 1.200 | 1.422 |
| Ar | 11.1(b) | 51.84(f) | 534.85(f) | 2.249 | 2.277 | 2.564 | 1.025 | 1.180 | 1.364 |
| Kr | 16.8(b) | 98.43(f) | 1269.6(f) | 2.571 | 2.574 | 2.892 | 1.012 | 1.149 | 1.332 |
| Xe | 27.4(b) | 223.3(e) | 3640.6(e) | 3.023 | 3.020 | 3.348 | 1.008 | 1.124 | 1.296 |
| Li | 164.1(c) | 1424(g) | 39688(g) | 5.507 | 4.526 | 4.888 | 1.018 | 1.333 | 1.680 |
| Na | 162.6(c) | 1878(g) | 55518(g) | 5.470 | 4.719 | 5.128 | 1.006 | 1.246 | 1.679 |
| K | 290.2(c) | 5000(g) | 176940(g) | 6.629 | 5.682 | 6.033 | 1.004 | 1.184 | 1.645 |
| Be | 37.8(b) | 299.9(d) | 4765(d) | 3.401 | 3.298 | 3.590 | 1.041 | 1.301 | 1.613 |
| Mg | 71.7(b) | 845.4(d) | 16772(d) | 4.171 | 4.007 | 4.276 | 1.012 | 1.222 | 1.559 |
| Ca | 158.6(b) | 3083(e) | 65170(e) | 5.425 | 5.143 | 5.190 | 1.007 | 1.168 | 1.557 * |
The central idea described in the previous paragraph is parallel to the one that has been used before to construct nonemprical density functionals for the exchange-correlation energy (14, 15, 39): Make the functional exact for a paradigm density, and satisfy other exact constraints for a broader class of densities of interest. We believe that our use of a paradigm density (the metallic sphere or “local density approximation” in the vdW DFT) is what leads to accurate higher-order vdW coefficients without empiricism.
Application to Atom-Atom-Pair Interaction
Dynamic Multipole Polarizability.
We apply the present model to calculate the dynamic multipole polarizabilities of the He atom and compare them to the corresponding reference values (16, 40) from high-level many-body calculations, as shown in Figs. 1–3. We observe from Figs. 1–3 that our model dynamic multipole polarizabilities are in excellent agreement with the reference values within the whole range of frequencies. We also apply the present theory to the H atom for which the exact dynamic multipole polarizabilities are available (41), and find that the deviation of the model dynamic polarizabilities from the exact ones is also indistinguishable (see Figs. S1, S2, S3).
Fig. 1.

Dynamical dipole polarizability α1(iu) (in atomic units where e2 = ℏ = m = 1) of the He atom: reference value (42) (red) and present model (green). ω = iu is the imaginary frequency.
Fig. 2.

The same as Fig. 1, but for quadrupole polarizability. Reference value is taken from ref. 16.
Fig. 3.

The same as Fig. 2, but for octupole polarizability.
vdW Coefficients.
Finally, we evaluate C8 and C10 of 78 diverse atom-atom-pair interactions by substituting the model dynamic multipole polarizabilities Eqs. 6, [8], and [9], into the exact expression [2]. (The explicit expressions for C6, C8, and C10, and detailed numerical results, are given in the SI Text). The atoms selected in this study include rare-gas atoms (He, Ne, Ar, Kr, Xe), hydrogen and alkali-metal atoms (H, Li, Na, K), and alkaline-earth atoms (Be, Mg, Ca). For convenience, the static multipole polarizabilities as input data are tabulated in Table 1. The results are summarized in Table 2. The mean absolute relative error (MARE) is calculated as mean absolute relative deviation of the present theory from the reference values obtained from high-level many-body methods updated from those of ref. 23. Detailed information has been tabulated and displayed in SI Text, Table S1, Table S2, Table S3. From Table 2 we can see that the MARE remains almost the same for C6, C8, and C10. In order to better understand the present theory, a comparison of the cutoff radii on the dipole, quadrupole, and octupole polarizabilities has been made. The results are displayed in Table 1. From Table 1 we observe that the cutoff radii are also close to each other for all three, and to a conventional vdW radii α1(0)1/3, as expected, because all cutoff radii equal the sphere radius R for a metallic sphere. Nearly the same MARE and cutoff radius strongly suggest that the main physics behind the model dynamic dipole polarizability correctly transfers to the model dynamic multipole polarizabilities. The theory described here is not limited to the dynamic multipole polarizabilities and the higher-order vdW coefficients studied here. In principle, the formulas of this paper generate dynamic multipole polarizabilities and vdW coefficients (arising from second-order perturbation theory) to any multipolar order with the same quality, due to the correct transfer of the physics from one order to another, and thus provide a nearly correct account of the vdW interaction. For a recent application of C6, C8, and C10, see ref. 47. The present theory may be generalized to other systems with the static multipole polarizability tensors (42, 48) as input.
Table 2.
Mean absolute relative error of C6, C8, and C10 based on 78 atom-atom-pair interactions calculated with the present theory
| vdW coefficients | C6 | C8 | C10 |
| Mean absolute relative error (%) | 3.2 | 3.0 | 3.1 |
Conclusions and Outlook
We have formulated a unified theory for the dynamic multipole polarizabilities and vdW coefficients, and have achieved the same accuracy as for the leading-order coefficient C6. Consistent accuracy is significantly important, because it guarantees that our model should also be able to describe other many-body vdW interactions (such as Axilrod-Teller-Muto three-body terms) as well. While the present theory considers only systems of spherical density, it can serve as a good starting point for extension to nonspherical densities, due to its excellent accuracy. One such extension would use the static multipole polarizability tensors (42) for the real nonspherical density as input to a generalization of our expressions that is exact for a finite metal of uniform density and similar nonspherical shape. Another such extension would write the real nonspherical density as a sum of effective atom densities (21, 22). For these effective atoms, only the densities and not the orbitals are directly available. Our expressions have the advantage that they require only the densities and not the orbitals. The second extension could in principle describe the vdW interaction between two separated molecules, or between two distant pieces of the same molecule (e.g., a polymer). Because the only needed input for the present theory is the electronic density and the static polarizabilities, which are accessible from the ground-state DFT (49, 50), we have in principle a true DFT approach for the dynamic multipole polarizabilities and vdW coefficients, and therefore may save a significant amount of computer time, compared to other methods including time-dependent DFT (51). Even without extension, the theory proposed here may be applicable to fullerenes (52) and other nearly spherical nanoparticles (53).
Supplementary Material
Acknowledgments.
J.P.P. and A.R. thank J.M. Pitarke, O.A. Vydrov, and G.I. Csonka for helpful discussions. We thank A.D. Becke and W. Yang for comments and suggestions on the manuscript. This material is based in part upon work supported by the National Science Foundation under the NSF Cooperative Agreement No. EPS-1003897 with additional support from the Louisiana Board of Regents. J.T. thanks G.E. Scuseria, L. Bytautas, T. Henderson, and R.L. Martin for discussions, and acknowledges support from two grants to G.E. Scuseria (NSF No. CHE-0807194, and Welch Foundation Grant No. C-0036), and from the Department of Energy under Contract No. DE-AC52-06NA25396 at Los Alamos National Laboratory, where part of the work was done. J.P.P. acknowledges support from NSF under Grant No. DMR-0854769.
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected in 2011.
The authors declare no conflict of interest.
*Accurate calculation of higher-order coefficients beyond C10 is particularly difficult even with wave function-based MBPT, because they not only involve a second-order contribution, but also have other higher-order contributions.
†The static multipole polarizability αl(0) is well defined, but the static polarizability density [e.g., the integrand in Eq. 6] is not. This gauge-dependent quantity can be altered by adding a function that integrates to zero.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118245108/-/DCSupplemental.
References
- 1.Buckingham AD, Fowler PW, Hudson JM. Chem Rev. Vol. 88. Washington, D.C.: 1988. Theoretical studies of van der Waals molecules and intermolecular forces; pp. 963–988. [Google Scholar]
- 2.van der Waals JD. On the gaseous and the liquid state. The Netherlands: University of Leiden; 1873. Dissertation. [Google Scholar]
- 3.Maitland GC, Rigby M, Smith EB, Wakeham WA. Intermolecular Forces, Their Origin and Determination. Oxford: Clarendon Press; 1981. [Google Scholar]
- 4.Stone AJ. The Theory of Intermolecular Forces. Oxford: Clarendon Press; 1996. [Google Scholar]
- 5.Kaplan IG. Intermolecular Interactions: Physical Picture, Computational Methods and Model Potentials. England: John Wiley and Sons Ltd; 2006. [Google Scholar]
- 6.Yan Z-C, Babb JF, Dalgarno A, Drake GWF. Variational calculations of dispersion coefficients for interactions among H, He, and Li atoms. Phys Rev A. 1996;54:2824–2833. doi: 10.1103/physreva.54.2824. [DOI] [PubMed] [Google Scholar]
- 7.Bytautas L, Ruedenberg K. Ab initio potential energy curve of F2. IV. Transition from the covalent to the van der Waals region: competition between multipolar and correlation forces. J Chem Phys. 2009;130:204101-1–204101-14. doi: 10.1063/1.3139114. [DOI] [PubMed] [Google Scholar]
- 8.Thakkar AJ, Hettema H, Wormer PES. Ab initio dispersion coefficients for interactions involving rare-gas atoms. J Chem Phys. 1992;97:3252–3257. [Google Scholar]
- 9.Rice JE, Taylor PR, Lee TJ, Almlöf J. The determination of accurate dipole polarizabilities α and γ for the noble gases. J Chem Phys. 1991;94:4972–4979. [Google Scholar]
- 10.Dzuba VA, Flambaum VV, Kozlov MG. Combination of the many-body perturbation theory with the configuration-interaction method. Phys Rev A. 1996;54:3948–3959. doi: 10.1103/physreva.54.3948. [DOI] [PubMed] [Google Scholar]
- 11.Porsev SG, Derevianko A. High-accuracy relativistic many-body calculations of van der Waals coefficients C6 for alkaline-earth-metal atoms. Phys Rev A. 2002;65:020701-1–020701-4. (R) [Google Scholar]
- 12.Parr RG, Yang W. Density Functional Theory of Atoms and Molecules. New York: Oxford University Press; 1989. [Google Scholar]
- 13.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 14.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 15.Tao J, Perdew JP, Staroverov VN, Scuseria GE. Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys Rev Lett. 2003;91:146401-1–146401-4. doi: 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- 16.Dalgarno A, Victor GA. Time-dependent Hartree-Fock theory and long-range interactions. Proceedings of the Physical Society. 1967;90:605–608. [Google Scholar]
- 17.Ovsiannikov VD, Mitroy J. Regular approach for generating van der Waals Cs coefficients to arbitrary orders. J Phys B: At Mol Opt. 2006;39:159–187. [Google Scholar]
- 18.Tang KT, Toennies JP. An improved simple model for the van der Waals potential based on universal damping functions for the dispersion coefficients. J Chem Phys. 1983;80:3726–3741. [Google Scholar]
- 19.Wu Q, Yang W. Empirical correction to density functional theory for van der Waals interactions. J Chem Phys. 2002;116:515–524. [Google Scholar]
- 20.Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J Comput Chem. 2011;32:1456–1465. doi: 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- 21.Johnson ER, Becke AD. A post-Hartree-Fock model of intermolecular interactions. J Chem Phys. 2005;123:024101-1–024101-7. doi: 10.1063/1.1949201. [DOI] [PubMed] [Google Scholar]
- 22.Tkatchenko A, Scheffler M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys Rev Lett. 2009;102:073005-1–073005-4. doi: 10.1103/PhysRevLett.102.073005. [DOI] [PubMed] [Google Scholar]
- 23.Tao J, Perdew JP, Ruzsinszky A. Long-range van der Waals attraction and alkali-metal lattice constants. Phys Rev B. 2010;81:233102-1–233102-4. [Google Scholar]
- 24.Vydrov OA, Van Voorhis T. Dispersion interactions from a local polarizability model. Phys Rev A. 2010;81:062708-1–062708-6. [Google Scholar]
- 25.Tang KT, Toennies JP. A simple theoretical model for the van der Waals potential at intermediate distances. I. Spherically symmetric potentials. J Chem Phys. 1977;66:1496–1507. [Google Scholar]
- 26.Rehr JJ, Zaremba E, Kohn W. van der Waals forces in the noble metals. Phys Rev B. 1975;12:2062–2066. [Google Scholar]
- 27.Mitroy J, Bromley MWJ. Higher-order Cn disperion coefficients for the alkali-metal atoms. Phys Rev A. 2005;71:042701-1–042701-7. [Google Scholar]
- 28.Becke AD, Johnson ER. Exchange-hole dipole moment and the dispersion interaction revisited. J Chem Phys. 2007;127:154108-1–154108-6. doi: 10.1063/1.2795701. [DOI] [PubMed] [Google Scholar]
- 29.Lima NA. Van der Waals density functional from multipole dispersion interactions. J Chem Phys. 2010;132:014110-1–014110-4. doi: 10.1063/1.3282265. [DOI] [PubMed] [Google Scholar]
- 30.Patil SH, Tang KT. Multipolar polarizabilities and two- and three-body dispersion coefficients for alkali isoelectronic sequences. J Chem Phys. 1997;106:2298–2305. [Google Scholar]
- 31.Kelly KL, Coronado E, Zhao LL, Schatz GC. The optical properties of metal nanoparticles: The influence of size, shape, and dielectric environment. J Phys Chem B. 2003;107:668–677. [Google Scholar]
- 32.Mitroy J, Bromley MWJ. Semiempirical calculation of van der Waals coefficients for alkali-metal and alkaline-earth-metal atoms. Phys Rev A. 2003;68:052714-1–052714-16. (2005) ibid. 71: 019902-1-019902-2. [Google Scholar]
- 33.Ferrell TL, Echenique PM. Generation of surface excitations on dielectric spheres by an external electron beam. Phys Rev Lett. 1985;55:1526–1529. doi: 10.1103/PhysRevLett.55.1526. [DOI] [PubMed] [Google Scholar]
- 34.Pitarke JM, Silkin VM, Chulkov EV, Echenique PM. Theory of surface plasmons and surface-plasmon polaritons. Rep Prog Phys. 2007;70:1–87. [Google Scholar]
- 35.Langreth DC, Mehl MJ. Beyond the local-density approximation in calculations of ground-state electronic properties. Phys Rev B. 1983;28:1809–1834. [Google Scholar]
- 36.Perdew JP, Wang Y. Accurate and simple density functional for the electronic exchange energy: generalized gradient approximation. Phys Rev B. 1986;33:8800–8802. doi: 10.1103/physrevb.33.8800. [DOI] [PubMed] [Google Scholar]
- 37.Perdew JP, Burke K, Wang Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys Rev B. 1996;54:16533–16539. doi: 10.1103/physrevb.54.16533. [DOI] [PubMed] [Google Scholar]
- 38.Andersson Y, Langreth DC, Lundqvist BI. van der Waals interactions in density-functional theory. Phys Rev Lett. 1996;76:102–105. doi: 10.1103/PhysRevLett.76.102. [DOI] [PubMed] [Google Scholar]
- 39.Perdew JP, Ruzsinszky A, Csonka GI, Constantin A, Sun J. Workhorse semilocal density functional for condensed matter physics and quantum chemistry. Phys Rev Lett. 2009;103:026403-1–026403-4. doi: 10.1103/PhysRevLett.103.026403. [DOI] [PubMed] [Google Scholar]
- 40.Bishop DM, Pipin J. Calculation of the polarizability and hyperpolarizability tensors, at imaginary frequency, for H, He, and H2 and the dispersion polarizability coefficients for interactions between them. J Chem Phys. 1992;97:3375–3381. [Google Scholar]
- 41.Tang AZ, Chan FT. Dynamic multipole polarizability of atomic hydrogen. Phys Rev A. 1986;33:3671–3678. doi: 10.1103/physreva.33.3671. [DOI] [PubMed] [Google Scholar]
- 42.Schwerdtfeger P. In: Atomic static dipole polarizabilities. Atoms, Molecules and Clusters in Electric Fields. Theoretical approaches to the calculation of electric polarizability. Maroulis G, editor. vol 1. London: World Scientific; 2006. pp. 1–32. [Google Scholar]
- 43.Derevianko A, Johnson WR, Safronova MS, Babb IF. High-precision calculations of dispersion coefficients, static dipole polarizabilities, and atom-wall interaction constants for alkali-metal atoms. Phys Rev Lett. 1999;82:3589–3592. [Google Scholar]
- 44.Figari G, Musso GF, Magnasco V. London dispersion coefficients from static multipole polarizabilities. Mol Phys. 1983;50:1173–1187. [Google Scholar]
- 45.Spelsberg D, Meyer W. Dynamic multipole polarizabilities of the rare-gas atoms Ne, Ar, and Kr and long-range interaction coefficients of the rare-gas atoms and with H2. J Phys Chem. 1996;100:14637–14642. [Google Scholar]
- 46.Marinescu M, Sadeghpour HR, Dalgarno A. Dispersion coefficients for alkali-metal dimers. Phys Rev A . 1994;49:982–988. doi: 10.1103/physreva.49.982. [DOI] [PubMed] [Google Scholar]
- 47.Steinmann SN, Csonka G, Corminboeuf C. Unified inter- and intramolecular dispersion correction formula for generalized gradient approximation density functional theory. J Chem Theory Comput. 2009;5:2950–2958. doi: 10.1021/ct9002509. [DOI] [PubMed] [Google Scholar]
- 48.Zatsarinny O, Bartschat K, Miltroy J, Zhang J-Y. Multipole polarizabilities and long-range interactions of the fluorine atom. J Chem Phys. 2009;130:124310-1–124310-6. doi: 10.1063/1.3098320. [DOI] [PubMed] [Google Scholar]
- 49.Sekino H, Maeda Y, Kamiya M, Hirao K. Polarizability and second hyperpolarizability evaluation of long molecules by the density functional theory with long-range correction. J Chem Phys. 2007;126:014107-1–014107-6. doi: 10.1063/1.2428291. [DOI] [PubMed] [Google Scholar]
- 50.Henderson TM, Izmaylov AF, Scalmani G, Scuseria GE. Can short-range hybrids describe long-range-dependent properties? J Chem Phys. 2009;131:044108-1–044108-9. doi: 10.1063/1.3185673. [DOI] [PubMed] [Google Scholar]
- 51.Mahan GD, Subbaswamy KR. Local Density Theory of Polarizability. New York: Plenum Pub Corp; 1990. [Google Scholar]
- 52.Jensen L, Astrand P-O, Milkkelsen KV. The static polarizability and second hyperpolarizability of fullerenes and carbon nanotubes. J Phys Chem A. 2004;108:8795–8800. [Google Scholar]
- 53.Cao G. Nanostructures and Nanomaterials: Synthesis, Properties and Applications. London: Imperial College Press; 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.