

Abstract
G protein-coupled receptors play a central role in signal transduction and were only known to be activated by agonists. Recently it has been shown that membrane potential also affects the activity of G protein-coupled receptors. For the M2 muscarinic receptor, it was further shown that depolarization induces charge movement. A tight correlation was found between the voltage-dependence of the charge movement and the voltage-dependence of the agonist binding. Here we examine whether depolarization-induced charge movement causes a conformational change in the M2 receptor that may be responsible for the voltage-dependence of agonist binding. Using site-directed fluorescence labeling we show a voltage-dependent fluorescence signal, reflecting a conformational change, which correlates with the voltage-dependent charge movement. We further show that selected mutations in the orthosteric site abolish the fluorescence signal and concomitantly, the voltage-dependence of the agonist binding. Surprisingly, mutations in the allosteric site also abolished the voltage-dependence of agonist binding but did not reduce the fluorescence signal. Finally, we show that treatments, which reduced the charge movement or hindered the coupling between the charge movement and the voltage-dependent binding, also reduced the fluorescence signal. Our results demonstrate that depolarization-induced conformational changes in the orthosteric binding site underlie the voltage-dependence of agonist binding. Our results are also unique in suggesting that the allosteric site is also involved in controlling the voltage-dependent agonist binding.
Keywords: acetylcholine, electrochromic, fluorescence quenching, gating
G protein-coupled receptors (GPCRs) are involved in many signal transduction processes within cells. Although GPCRs span the cell membrane, they were not considered to be affected by voltage. Recently, it was shown that depolarization affects the activity of various GPCRs belonging to classes A and C (1–9).
Depolarization could affect GPCRs’ activity by modifying agonist efficacy and/or its binding. Effect of voltage on efficacy was suggested, but was not directly demonstrated, to explain the ability of depolarization to generate a P2Y1 receptor response in the presence of a competitive antagonist and no agonist (5). However, for the M2 muscarinic receptor (M2R) it was directly shown that voltage affects binding. Specifically, voltage-dependent [3H]ACh binding to M2R was seen in synaptosomes (10) and in synaptoneurosomes (11). In the above studies (10, 11), it was shown that the M2R exhibits two agonist-binding affinity states. These authors suggested that depolarization shifts the high affinity state into a low affinity state.
A relevant question is whether measurements of M2R-induced G protein-activated inwardly rectifying K+ (GIRK) currents can faithfully reflect the effect of voltage on binding. To answer this question, measurements of GIRK currents and of radio-labeled agonist ([3H]ACh) binding were conducted at different membrane potentials in M2R-expressing Xenopus oocytes. It was shown that the apparent affinity of the GIRK currents decreased under depolarization. Additionally, the binding of [3H]ACh was lower upon depolarization (3). The existence of the proposed two affinity states and a shift of the high affinity state into a low one by depolarization (10, 11) were also demonstrated in measurements of GIRK currents (3). The above observations justify the use of GIRK current measurements to assess the effect of voltage on agonist binding.
Previously, the M2R was shown to display depolarization-induced charge movement (1, 2), and a tight correlation was found between the voltage-dependence of the charge movement and the voltage-dependence of the fraction of receptors that shifted to a low affinity state (2) (denoted, for brevity as “change in binding”). Intramembrane charge movement is a mechanism by which biological molecules (e.g., voltage-gated channels) link changes in membrane potential to changes in conformation, leading, in channels, to their opening (12, 13). Similarly, the depolarization-induced charge movement could alter the conformation of the M2R, which in turn could cause a change in its binding. In the present work we have tested this idea, using site-directed fluorescence labeling of tetramethylrhodamine maleimide (TMRM) conjugated to externally available cysteines, and measuring fluorescence signals as reporters of local changes in the M2R conformation under voltage clamp. When a conformational change occurs near the label, the fluorescence intensity may decrease or increase as a result of many possible mechanisms. For example, if the fluorophore gets closer to or farther from a quenching group, the result is that fluorescence will decrease or increase, respectively. Fluorescence can also increase or decrease if the fluorophore enters a different environment that increases or decreases its quantum yield or shifts its spectrum. The fluorescence measurements were done simultaneously with measurements of gating currents, which report charge movement (13) (the terms “gating currents” and “charge movement” will be used interchangeably).
Results
Voltage-Dependent Fluorescence Signal of TMRM-Cys416 Has Two Separable Components.
Conformational changes that lead to changes in agonist binding are likely to take place in the binding site itself or in the nearby allosteric site, which was shown to affect binding of agonist to the orthosteric binding site (14). We thus took advantage of the two endogenous cysteines, Cys413 and Cys416, located in the third extracellular loop (e3), which is part of the M2R allosteric site (14). Two other extracellular cysteines, Cys96 and Cys176, exist in the M2R. We first tested whether they might be labeled by the fluorophore and produce a fluorescence signal. We thus measured, in M2R-expressing Xenopus oocytes, the fluorescence signal obtained when Cys413 and Cys416 were simultaneously mutated to serine. This mutant, where only Cys96 and Cys176 were available for labeling, contained a nearly undetectable fluorescence signal (defined as ΔF/F0, where ΔF is the change in fluorescence during the voltage pulse, and F0 is the basal fluorescence) (Fig. S1, compare with Fig. 1B). This process ensures that Cys96 and Cys176 do not produce substantial fluorescence changes upon TMRM-labeling of the e3 cysteines. We next labeled residues Cys413 and Cys416, one at a time, after mutating the other cysteine to serine. When Cys416 was labeled (denoted TMRM-Cys416; its location is shown in Fig. 1A), a voltage step of 60 ms from a holding potential of −120 mV to +40 mV elicited a robust fluorescence signal comprised of a very fast rise in fluorescence intensity (ΔFfast/F0) followed by a slow decay (ΔFslow/F0) at the “on” and “off” of the pulse (Fig. 1B and Fig. S2A). In contrast, the same pulse elicited only a small and irreproducible fluorescence signal when Cys413 was labeled. In the rest of this article, the fluorescence signal refers to the fluorescence changes of TMRM conjugated to Cys416.
Fig. 1.

The voltage-dependent fluorescence signal of TMRM-Cys416 has two separable components. (A) Side view of the M2R. Indicated are: Cys416 (purple sphere) and the approximate locations of the orthosteric and allosteric binding sites (yellow and purple shades, respectively). The horizontal line represents the boundary between the extracellular solution and the lipid bilayer. TMI and TMII were omitted. (B) Representative fluorescence signal elicited by a depolarizing pulse; the pulse protocol is shown above. ΔFfast/F0 and ΔFslow/F0 are indicated with arrows. Here and in subsequent figures %ΔF/F0 indicates the percentage change in fluorescence relative to the background fluorescence. (C) ΔFfast/F0 follows the time course of the capacitive current integral. Data were sampled at 200 kHz and filtered at 20 kHz. Shown is a representative trace from one oocyte. n = 6 (here and below oocytes were taken from two or more batches). (D) (ΔFfast/F0) − V and (ΔFslow/F0) − V, calculated as described in Fig. S3, each normalized to its value at a step to +40, from one representative oocyte. Data were obtained using the same protocol as in B, but with steps in increments of 20 mV. (E) Representative traces of fluorescence signals from one oocyte. (Upper) Depolarizing pulse from +40 mV to +120 mV. (Lower) Depolarizing pulse from −80 mV to 0 mV. (Insets) Pulse protocol (n = 7). (F) (Inset) Representative fluorescence traces (from the “on” response) at 10 °C and 20 °C from one oocyte (n = 4). The time constant, τ, of ΔFslow/F0 (τΔFslow/F0) was extracted by fitting a single exponent to the decaying phase of the signal (see Materials and Methods). The plot shows the fitted values of τΔFslow/F0 obtained at various voltages from one oocyte at 10 °C and 20 °C. Pulse protocol was as in B. (G) The procedure to extract ΔFslow/F0.
The fast rise and slow decay of the fluorescence signal shown in Fig. 1B for a +40 mV depolarization was also seen for different levels of depolarization, but the fluorescence amplitude and decay time changed with the depolarization level (Fig. S2A). The analysis of the fluorescence signals indicated that the ΔFfast/F0 increasing component and ΔFslow/F0 decreasing component result from two different mechanisms in the fluorescence emission of the label, the first being electrochromic and the second most likely produced by quenching. The evidence for an electrochromic component is as follows: (i) The kinetics of ΔFfast/F0 are very rapid and correlate well with the time integral of the capacitive current (Fig. 1C), which reflects the time course of membrane charging or the time course of the actual membrane potential. It is thus likely that ΔFfast/F0 reflects an electrochromic event (i.e., a shift in the fluorophore emission spectrum because of an interaction of the fluorophore electronic state with the local electric field). The electrochromic effect was shown to be the case in the voltage-gated Shaker K+ channel, conjugated to an ANEP (aminonaphthylethenylpyridinium) fluorophore derivative (15). (ii) ΔFfast/F0 and ΔFslow/F0 differ in their voltage-dependence. The dependence on voltage of ΔFfast/F0 [(ΔFfast/F0) − V] is linear (Fig. 1D), as in the case of the electrochromic response of the ANEP derivative in Shaker (15), but (ΔFslow/F0) − V saturates at extreme voltages (the procedure to separate the two components plotted in Fig. 1D is shown in Fig. S3). In the positive voltage range the magnitude of ΔFslow/F0 saturates. Therefore, we expect that if voltage steps are applied from a holding potential higher than +20 mV, then we should only record ΔFfast/F0. Indeed, when pulsing from +40 mV to +120 mV, the fluorescence signal is comprised of only ΔFfast/F0 (Fig. 1E, Upper). To rule out the possibility that the absence of ΔFslow/F0 is because the pulse step (80 mV) was too small to produce a detectable fluorescence signal, we used the same step magnitude, but from –80 mV to 0 mV, where we expect a ΔFslow/F0 signal. Fig. 1E, Lower, shows that in this case both components, fast and slow, can be clearly seen. (iii) Distinction between ΔFfast/F0 and ΔFslow/F0 can be achieved by yet another way. A mutation in the orthosteric binding site (for full discussion, see below), abolished ΔFslow/F0 but retained ΔFfast/F0 (Fig. S2B). (iv) ΔFfast/F0 and ΔFslow/F0 differ in their kinetics (Fig. 1B). The slower kinetics of ΔFslow/F0 and its saturation at extreme voltages may suggest that this component reflects a depolarization-induced conformational change. If this change is the case, then we expect the kinetics of ΔFslow/F0 to be temperature-dependent. Because of the variability between oocytes, we did not average the results; rather, we present results from one oocyte. Three other oocytes showed similar temperature-dependence. Indeed, the time constant, τ, of ΔFslow/F0, extracted from the decaying phase of the “on” response (Fig. 1F, Inset), is appreciably larger at 10 °C than at 20 °C at all membrane potentials (Fig. 1F). In contrast, if the fast component represents the membrane charging, as suggested above, the kinetics of ΔFfast/F0 should not be affected by temperature; indeed, this is the case (Fig. 1F, Inset).
As the present work aims to test whether depolarization causes a conformational change, we will focus here only on the slow component. To this end, we will present in the subsequent figures only ΔFslow/F0, which is extracted from the raw signal by subtracting ΔFfast/F0 from ΔF/F0. The procedure for this subtraction is shown in Fig. 1G.
Relationship Between the Charge Movement and the Conformational Change.
Ben-Chaim et al. (2) showed that a good correlation exists between the voltage-dependence of the charge that moved (Q − V, constructed as described in SI Materials and Methods) and the voltage-dependence of the fraction of receptors that underwent a shift in their agonist binding affinity from high to low (RL − V, evaluated as described in ref. 2, where RL is the fraction of low affinity receptors at any voltage).
We expect that if the conformational change reported by ΔFslow/F0 underlies the change in binding, then (ΔFslow/F0) − V should correlate reasonably well with Q − V and RL − V. Perfect correlation is not expected because fluorescence tracks a local conformational change in the vicinity of the fluorophore but the charge movement is a global rearrangement of charge in the electric field. Fig. 2A shows a reasonable correlation between the three curves, suggesting a causal relationship between the charge movement, the conformational change and the change in binding following depolarization. In addition, the voltage-dependence of the time constants (τ) of Q and ΔFslow/F0 should correlate. Indeed, τΔFslow/F0 and τQ exhibit rather similar dependencies on voltage (Fig. 2B) (τΔFslow/F0 and τQ were extracted from the fluorescence signal traces and the time integral of the gating current, respectively; Fig. 2B, Inset). Notice, however, that τQ is appreciably larger than τΔFslow/F0. A possible explanation is that several transitions may couple depolarization to the voltage sensor, and the one that is responsible for the conformational change tracked by the fluorophore is fast. For example, it has been suggested that the M2R possesses several voltage sensors, from which at least two are independent and exhibit different time constants (16). It is possible that the slowest voltage sensor [its time constant is 20-times larger than the other voltage sensor (16)], which dominates τQ, does not contribute much to the conformational change tracked by TMRM-Cys416 fluorescence signal.
Fig. 2.

Relationship between the charge movement and the conformational change. (A) Q − V (mean ± SEM, n = 12), (ΔFslow/F0) − V (mean ± SEM, n = 21) and RL − V (data adapted from ref. 2), each normalized to its value at +40 mV. (B) (Inset) Traces taken from the “on” response of either the ΔFslow/F0 or the charge movement. For the charge movement the trace is the negative value of the integral. To compare the kinetics, both ΔFslow/F0 and charge movement traces were normalized, each to its value at +40 mV. τΔFslow/F0 was calculated as in Fig. 1F, and the time constant of the charge movement (τQ) was calculated by fitting two exponentials (weighted average is shown, see Materials and Methods). Main figure, fitted values of τ for the charge movement, and ΔFslow/F0 for a pulse from a holding potential of −120 mV to the indicated voltages. Each point shows the mean ± SEM (n = 4–6 oocytes). (C) (Inset) Representative traces recorded in TMRM-Cys416, elicited by a depolarizing pulse from −120 mV to +40 mV, showing the effect of increasing ACh concentration on the charge movement (Left, normalized to the maximal charge movement without ACh) and on the fluorescence signal (Right). The plot shows percentage of control Q or ΔFslow/F0 as a function of ACh concentration (n = 4–20). No significant difference between the two datasets. (D) The same as in C, but with Metho, n = 2–10. No significant difference between the two datasets.
To further substantiate the link between the charge movement and the conformational change we examined whether treatments that affect the gating currents will likewise affect the fluorescence signal. ACh was shown to reduce the gating currents in M2R-expressing oocytes (17). We therefore tested whether ACh also reduces ΔFslow/F0. Fig. 2C (Inset) shows samples of the gating currents’ integral (Left Inset) and the fluorescence signal (Right Inset) at three ACh concentrations, where it is clearly seen that ACh inhibits both the charge movement and the fluorescence signal. Full dose-inhibition curves show a tight correlation between the dependence of Q and ΔFslow/F0 on ACh concentration (Fig. 2C). Although these results strengthen the notion of causal relationship between charge movement and conformational change, another explanation should be considered. It is possible that binding of ACh and depolarization produce similar conformational changes. Under such conditions, the depolarization-induced conformational change is expected to gradually decline as the ACh concentration increases. It is interesting to note that Gurung et al. (5) demonstrated such a behavior. In particular, these authors have shown that the greatest effect of depolarization on signaling via the P2Y1 receptor was observed at the lowest effective agonist concentrations. As the agonist concentrations were increased, the relative effect of depolarization was decreased.
To discern between the possibility of a causal relationship between charge movement and conformational change, and the possibility of a similar conformational change by ACh and depolarization, we repeated the experiment of Fig. 2C, but instead of ACh we used methoctramine (Metho), an M2R-specific competitive antagonist. As Metho does not lead to G-protein activation, it is not expected to produce a conformational change, at least not the same one as that produced by ACh. However, Metho was shown to inhibit, like ACh, the charge movement in a dose-dependent manner (17) (Fig. 2D, Left Inset). If Metho will reduce the conformational change as did ACh, it will be compatible with the suggestion of a causal relationship. Fig. 2D (Right Inset) shows that this is indeed the case. Another possibility to explain the results with Metho is that it prevents the voltage-dependent conformational change that is induced in the presence or absence of ACh. Although possible, the observation that the reduction in charge movement upon ligand application tightly correlated with the reduction in ΔFslow/F0 (Fig. 2D), combined with the different binding natures and actions of these ligands, strengthens the conclusion that it is the charge movement which leads to the conformational change tracked by fluorescence.
Mutations in the Orthosteric and Allosteric Binding Sites Abolish Voltage-Dependent Binding and Alter the Fluorescence Signal.
The reasonable correlation between Q − V, (ΔFslow/F0) − V, and RL − V suggests that the fluorophore reports on a conformational change that is largely induced by the charge movement, and that this conformational change is relevant for the voltage-dependent binding. Thus, to gain insight into the mechanism by which charge movement affects agonist binding, we examined which residues modulate the fluorescence signal as the conformational change takes place. To do so, we searched for residues that satisfy three criteria: (i) might serve as quenchers of TMRM (18); (ii) are located in the vicinity of the fluorophore, so that their putative movement relative to the fluorophore may be detected by the signal intensity; and (iii) are located in the region of the orthosteric site, to which ACh binds, or in the allosteric site, which is known to affect the binding affinity of ACh, as well as of other M2R agonists (14). The residues that are located close to the fluorophore were identified using a homology model of the M2R (19). These residues are not necessarily adjacent to Cys416, to which the fluorophore is attached, because the linker to the fluorophore dipole is 12–14 Å in length.
The residues that satisfied the above criteria are: Tyr403 at transmembrane domain (TM) 6 and Tyr426 at TM7, both part of the orthosteric site (20); Trp99 at TM3, at the interface between the allosteric and orthosteric sites (21); and Trp422 at e3, part of the allosteric site (22) (Fig. 3A). These residues were mutated to alanine (using the Cys416 construct as a template) and the effect of the substitution was evaluated by recording the fluorescence signal. Fig. 3B and the sample traces in Fig. 3 D and E (Upper Insets) show that mutating the two residues in the orthosteric site (TMRM-Cys416 Tyr403Ala and TMRM-Cys416 Tyr426Ala) abolished the fluorescence signal (compare with the native TMRM-Cys416: Fig. 3B and sample trace in Fig. 3C, Upper Inset), indicating that these residues serve as quenchers. The abolition of the signal cannot be attributed to reduction in the charge movement, as the latter was not affected appreciably by the mutations (see traces of gating currents in Fig. 3 D and E, Lower Insets, and compare with Fig. 3C, Lower Inset). Mutating Trp99 did not alter the fluorescence signal (TMRM-Cys416 Trp99Ala) (Fig. 3 B and F, Upper Inset), suggesting that it does not serve as a quencher of the TMRM-Cys416. Mutating Trp422 even increased the signal (TMRM-Cys416 Trp422Ala) (Fig. 3 B and G, Upper Inset). A possible explanation could be that following this mutation, the quenching residues Tyr403 and Tyr426 get closer to the fluorophore.
Fig. 3.

Effect of mutations in the orthosteric and allosteric binding sites on the fluorescence signal and on the voltage-dependent agonist binding. (A) Side view of the M2R model. Residue Cys416 and the mutated residues are shown as colored spheres (as identified in the figure) and the approximate locations of the orthosteric and allosteric binding sites are shown as yellow and purple shades, respectively. TMI and TMII were omitted. (B) (ΔFslow/F0) − V of the different mutants: Cys416, purple asterisks; Tyr403Ala, green circles; Tyr426Ala, red squares; Trp99Ala, brown triangles; Trp422Ala, blue diamonds. Each point shows mean ± SEM n = 2–5. (C–G) Dose-response curves of GIRK currents following ACh application of the various mutants at −80 mV (black squares) and +40 mV (open triangles). Each point shows mean ± SEM n = 3–7. (Upper Insets) Sample traces of ΔFslow/F0 from each mutant as indicted above each trace. (Lower Insets) Sample traces of gating currents, elicited by a depolarizing pulse, from −120 mV to +40 mV. (H–J) Dose-response curves of GIRK currents following Oxo application at −80 mV (black squares) and +40 mV (open triangles). (H) Cys416, (I) Tyr426Ala, (J) Trp422Ala. Each point shows mean ± SEM n = 3–10.
The above described mutations were examined to determine their effect on the voltage-dependence of the agonist binding. In our previous studies (3, 4), we corroborated our conclusion that the voltage dependence of the GIRK currents is a result of voltage-dependent binding by conducting direct binding experiments. Binding experiments are not, however, possible in the present case, as the binding affinity of the mutants is very low. However, recalling the considerations discussed earlier, we used GIRK current measurements to test whether the mutations affect the voltage-dependence of agonist binding. We thus constructed, as was done previously (2), dose-response curves of M2R-induced GIRK currents at two holding potentials. The dose-response curves of the native Cys416 (Fig. 3C) were compared with those of the various mutants. Fig. 3 D and E show that the two mutations at the orthosteric site, Tyr403Ala and Tyr426Ala, that abolished the fluorescence signal, likewise abolished the voltage dependence of agonist binding; the dose-response curves at −80 mV and +40 mV overlapped (compare with Fig. 3C). Surprisingly, the mutations of Trp99 and of Trp422 that did not reduce the fluorescence signal (Fig. 3B), also abolished the voltage-dependence of binding: the dose-response curves at –80 mV and +40 mV overlapped (Fig. 3 F and G; see also ref. 1 for Trp99Ala). These results suggest that although Trp99 and Trp422 are required for the voltage-dependent binding, this binding dependence is not achieved by the conformational change that is detected by TMRM-Cys416.
The mutations in the orthosteric and allosteric sites shifted the dose-response curves to a very low apparent affinity. Consequently, it could be argued that the lack of voltage-dependence observed in the mutants (Fig. 3 D–G) is not genuine, but rather that even if voltage-dependence exists, it cannot be detected experimentally. To clarify this point we used the high affinity agonist oxotremorine (Oxo) and took one mutation from the orthosteric site (Tyr426Ala) and the mutation from the allosteric site (Trp422Ala) as representatives. Fig. 3H shows the dose-response curve of the native Cys416 with Oxo; the voltage-dependence is clearly apparent. As seen, even with the high affinity agonist, the voltage-dependence of binding was abolished in the two mutants (Fig. 3 I and J).
Reduced Magnitude of ΔFslow/F0 in the ELALL Mutant and Following Pertussis Toxin Treatment.
The cumulative results of Figs. 2 and 3 are compatible with the notion of linkage between charge movement, change in conformation in the orthosteric site, and change in agonist binding. An obvious question to ask is: What is the domain and mechanism by which the voltage sensor relays its signal, charge movement, to a change in binding? Ben-Chaim et al. (2) suggested that the third intracellular loop (i3), which associates with the G protein, is the domain, and the probability of its association with the G protein is the mechanism by which the voltage sensor is coupled to a change in binding. This hypothesis is based on the observation that treatment with pertussis toxin (PTX), thereby preventing association of Gi/o-coupled GPCRs (as the M2R is) to their G protein, abolished the voltage-dependence of [3H]ACh binding to M2R-expressing oocytes (3). Furthermore, mutation of five residues in i3 of the M2R (ELALL mutant, notice that when first described in ref. 2, this mutant was denoted ELAAL; however, the correct name, according to the residues that were exchanged, is the notation ELALL, used here) abolished the voltage-dependence of the M2R-activated GIRK currents (2).
By measuring the change in conformation we were able to directly test the hypothesis that i3 relays the charge movement to a change in binding. If correct, we predict that the fluorescence signal will be greatly reduced following PTX treatment and in the ELALL mutant. Fig. 4 shows that this prediction is correct. We first repeated the experiments of Ben-Chaim et al. (3) and measured binding of [3H]ACh to M2R-expressing oocytes at two voltages, −88 mV and +5 mV, with and without PTX treatment. Following PTX treatment, no M2R-induced GIRK currents were detected, ensuring that the PTX was active. As seen, without PTX treatment, binding is voltage-dependent (Fig. 4A, Inset, control). In contrast, following PTX treatment, the voltage-dependence is abolished; the difference between the binding at −88 mV and +5 mV is not significant (Fig. 4A, P > 0.05; see also Fig. S4). To measure the effect of PTX on the fluorescence signal, PTX was injected to TMRM-Cys416–expressing oocytes and the fluorescence signal was measured 10–20 h after injection. Fig. 4B shows that PTX reduced the voltage-dependent fluorescence signal to about 50% of the signal in the control TMRM-Cys416. The remaining fluorescence signal seen here and in Fig. 4E (described below) may reflect local conformational changes that are not translated to changes in binding. The reduction in the fluorescence signal is not the result of PTX inhibiting the gating currents, as these remained unaltered after PTX injection (Fig. 4C; compare with the control Cys416 in Inset).
Fig. 4.

Reduced voltage dependence of ΔFslow/F0 following PTX treatment and in the ELALL mutant. (A and D) [3H]ACh binding to PTX-injected Cys416-expressing oocytes (A) or ELALL-Cys416 (D) at −88 mV and +5 mV. Results are given as mean ± SEM (n = 6–16 for A and 9–14 for D). (Inset) [3H]ACh binding to control Cys416 (n = 19–28). (B and E) (ΔFslow/F0) − V curves for control TMRM-Cys416 and PTX-injected TMRM-Cys416 (B) or ELALL TMRM-Cys416 (E). Results are given as mean ± SEM, (n = 11–23 for B and 10–23 for E). (Insets) Representative fluorescence traces from one oocyte obtained by stepping from a holding potential of −120 mV to +40 mV in control TMRM-Cys416 and in the PTX-injected TMRM-Cys416 (B) or ELALL TMRM-Cys416 (E). (C and F) Sample traces of gating currents elicited by a depolarizing pulse from −120 mV to +40 mV, measured in PTX-injected oocytes (C) or in ELALL TMRM-Cys416–expressing oocytes (F). (Inset in C) Sample trace of gating currents in the control Cys416, same protocol.
We next turned to the ELALL mutant. Fig. 4D shows that binding of [3H]ACh to oocytes expressing the ELALL mutant is also voltage-independent (P > 0.05), and concomitantly, the ELALL TMRM-Cys416 fluorescence signal is reduced by about 50% with respect to the native TMRM-Cys416 (Fig. 4E). Here too, the reduction in the fluorescence signal is not because of inhibition of the gating currents, as the gating currents in the ELALL mutant are similar to those in the native one (compare Fig. 4 F and C, Inset, and see ref. 2). The results of Fig. 4 support the hypothesis (2, 3) that i3 is involved as a domain that, by associating to the G protein, relays the charge movement in the voltage sensor to a conformational change, resulting in a change in agonist binding.
Discussion
The main findings and conclusions of this study are: (i) A reasonable correlation exists between the voltage-dependence of the charge movement, the conformational change, as reflected by the fluorescence signal, and the fraction of receptors that shifted to a low binding affinity (Fig. 2A). This correlation suggests that the fluorophore detects the main conformational change that is responsible for the change in binding. (ii) Eliminating two aromatic residues (Tyr403 and Tyr426) in the orthosteric binding site abolished simultaneously the fluorescence signal and the voltage-dependence of ACh binding, suggesting that the signal itself is the result of quenching because of the fluorophore dipole and the aromatic groups getting closer during the conformational change. Findings i and ii suggest that the major conformational change that leads to a change in binding takes place in the orthosteric site in the region of residues Tyr403 and Tyr426. (iii) PTX treatment and mutation of i3 abolished the voltage-dependency of ACh binding and concomitantly reduced the fluorescence signal. Findings i, ii, and iii directly reinforce the suggestion (2, 3) that charge movement in the voltage sensor is relayed to the orthosteric binding site via i3 and its association with the G protein. An additional result is that mutations of Trp422 in the allosteric site and Trp99 in the interface between the allosteric and orthosteric sites also abolished the voltage-dependency of ACh binding (Fig. 3 F and G). These results are unique in suggesting that the allosteric site may be involved in the mechanism by which voltage affects binding of ACh to the orthosteric site.
In this study we were able to gain insight into the relationship between charge movement in the voltage sensor and the orthosteric site. Specifically, we showed evidence that charge movement induces conformational changes in the orthosteric site, which likely produce a change in agonist binding. Further studies are required to unravel the mechanism by which the allosteric site contributes to the voltage-dependent change in agonist binding.
The electrochromic signal (ΔFfast/F0) was not addressed in this study. However, the existence of a robust electrochromic signal is indicative of a strong electric field in the neighborhood of the orthosteric site, favoring a possible location for a voltage sensor. Further studies are required to unravel if this is indeed the case.
Several GPCRs have been shown to display voltage-dependence (1–11). GPCRs are the largest family of cell-surface receptors and are the target for numerous therapeutic agents. We hypothesize that the findings and conclusions described in this work are general to other GPCRs. To date, only two of them, the M2R and the M1 muscarinic receptor, were shown to exhibit gating currents (2). Future studies will probably reveal that other GPCRs in which voltage affects binding or activity [for example: M1 muscarinic receptor (2, 3), the dopamine D2 receptor (8), the P2Y1 receptor (5, 6), and the metabotropic glutamate mGluR1 and mGluR3 receptors (4)] also exhibit depolarization-induced charge movement that induces a conformational change.
Finally, it is important to emphasize that the conformational change sensed by the fluorophore (ΔFslow/F0), which leads to a change in binding, is in the time range of an action potential's duration (Fig. 2B). Therefore, our results suggest that GPCR-mediated signal transduction in excitable tissues (neurons, heart, muscles) can be efficiently modulated by the brief action potential, making it relevant under physiological conditions. One such case had been already demonstrated, regarding the P2Y receptor that shows an opposite voltage-dependence compared with the M2R: a train of action potentials at a frequency of 1 Hz or 0.2 Hz (imitating a cardiac action potential) enhanced the cellular response to the agonist (6).
Materials and Methods
Xenopus oocytes were prepared, injected, and maintained as previously described (3). Constructs were linearized and transcribed as previously described (2). All mutants were constructed using QuikChange site-directed mutagenesis kit (Stratagene).
Oocyte Labeling for Fluorescence Recordings.
Oocytes were incubated for 1 h in ND96/0.1 mM DTT, and were transferred to ND96/10 μM TMRM (Invitrogen) for 15 min at 4 °C. The oocytes were washed and kept in the dark until being used.
Gating Currents and Fluorescence Recordings.
Fluorescence and gating currents were recorded under voltage-clamp conditions, 3–5 d after RNA injection, using the cut-open voltage clamp technique (23) implemented for epifluorescence measurements (13), with modifications as previously described (24). The external recording solution contained: 120 mM N-methyl-d-glucamine, 10 mM Hepes, and 2 mM CaCl2, pH 7.4 with methanesulfonic acid. The internal solution was similar but did not contain CaCl2 and contained 2 mM EGTA. Data were sampled at 50 kHz and filtered at 1–5 kHz, unless otherwise specified. Symmetric capacitive currents were compensated by analog circuitry and subtracted on-line by using a depolarizing pulse [the P/8 protocol (25)], from a holding potential of +40 mV. Data acquisition and analysis were done with in-house built hardware and software.
Data Analysis.
The time constants of the fluorescence signal (τΔFslow/Fo) and the charge movement (τQ) were analyzed by fitting the fluorescence traces or gating currents integral, respectively, with a single [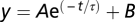 ], or a double [
], or a double [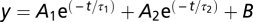 ] exponential equation, where A1 and A2 indicate the amplitude of the two exponential components, τ1 and τ2 indicate the time constants of each component, and B is an offset value. For the charge movement traces fitted with a double exponential, τ was calculated with the following equation:
] exponential equation, where A1 and A2 indicate the amplitude of the two exponential components, τ1 and τ2 indicate the time constants of each component, and B is an offset value. For the charge movement traces fitted with a double exponential, τ was calculated with the following equation: 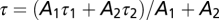 , thus obtaining a single time constant representing the weighted average of the two components.
, thus obtaining a single time constant representing the weighted average of the two components.
Statistical Evaluation.
Significance was checked by student's two-tailed t test. Significance was assumed for P < 0.05.
Supplementary Material
Acknowledgments
We thank Prof. Arthur Christopoulos and Dr. Nathan Hall for providing us with the M2R model coordinates, based on the model published by Gregory et al. (19), and additional information regarding that model; and Dr. K. Stein and the Goldie-Anna Foundation for continuous support. This work was supported by National Institutes of Health Grant GM030376 (to F.B.), and the Pritzker Fellowship in the Neurosciences (to M.F.P.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1119424109/-/DCSupplemental.
References
- 1.Navarro-Polanco RA, et al. Conformational changes in the M2 muscarinic receptor induced by membrane voltage and agonist binding. J Physiol. 2011;589:1741–1753. doi: 10.1113/jphysiol.2010.204107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ben-Chaim Y, et al. Movement of ‘gating charge’ is coupled to ligand binding in a G-protein-coupled receptor. Nature. 2006;444:106–109. doi: 10.1038/nature05259. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Chaim Y, Tour O, Dascal N, Parnas I, Parnas H. The M2 muscarinic G-protein-coupled receptor is voltage-sensitive. J Biol Chem. 2003;278:22482–22491. doi: 10.1074/jbc.M301146200. [DOI] [PubMed] [Google Scholar]
- 4.Ohana L, Barchad O, Parnas I, Parnas H. The metabotropic glutamate G-protein-coupled receptors mGluR3 and mGluR1a are voltage-sensitive. J Biol Chem. 2006;281:24204–24215. doi: 10.1074/jbc.M513447200. [DOI] [PubMed] [Google Scholar]
- 5.Gurung IS, Martinez-Pinna J, Mahaut-Smith MP. Novel consequences of voltage-dependence to G-protein-coupled P2Y1 receptors. Br J Pharmacol. 2008;154:882–889. doi: 10.1038/bjp.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Pinna J, Tolhurst G, Gurung IS, Vandenberg JI, Mahaut-Smith MP. Sensitivity limits for voltage control of P2Y receptor-evoked Ca2+ mobilization in the rat megakaryocyte. J Physiol. 2004;555:61–70. doi: 10.1113/jphysiol.2003.056846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ong BH, et al. G protein modulation of voltage-sensitive muscarinic receptor signalling in mouse pancreatic acinar cells. Pflugers Arch. 2001;441:604–610. doi: 10.1007/s004240000459. [DOI] [PubMed] [Google Scholar]
- 8.Sahlholm K, Nilsson J, Marcellino D, Fuxe K, Arhem P. Voltage-dependence of the human dopamine D2 receptor. Synapse. 2008;62:476–480. doi: 10.1002/syn.20509. [DOI] [PubMed] [Google Scholar]
- 9.Mahaut-Smith MP, Martinez-Pinna J, Gurung IS. A role for membrane potential in regulating GPCRs? Trends Pharmacol Sci. 2008;29:421–429. doi: 10.1016/j.tips.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Ilouz N, Branski L, Parnis J, Parnas H, Linial M. Depolarization affects the binding properties of muscarinic acetylcholine receptors and their interaction with proteins of the exocytic apparatus. J Biol Chem. 1999;274:29519–29528. doi: 10.1074/jbc.274.41.29519. [DOI] [PubMed] [Google Scholar]
- 11.Cohen-Armon M, Sokolovsky M. Depolarization-induced changes in the muscarinic receptor in rat brain and heart are mediated by pertussis-toxin-sensitive G-proteins. J Biol Chem. 1991;266:2595–2605. [PubMed] [Google Scholar]
- 12.Armstrong CM, Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature. 1973;242:459–461. doi: 10.1038/242459a0. [DOI] [PubMed] [Google Scholar]
- 13.Cha A, Bezanilla F. Structural implications of fluorescence quenching in the Shaker K+ channel. J Gen Physiol. 1998;112:391–408. doi: 10.1085/jgp.112.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christopoulos A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nat Rev Drug Discov. 2002;1:198–210. [Google Scholar]
- 15.Asamoah OK, Wuskell JP, Loew LM, Bezanilla F. A fluorometric approach to local electric field measurements in a voltage-gated ion channel. Neuron. 2003;37:85–97. doi: 10.1016/s0896-6273(02)01126-1. [DOI] [PubMed] [Google Scholar]
- 16.Zohar A, Dekel N, Rubinsky B, Parnas H. New mechanism for voltage induced charge movement revealed in GPCRs—Theory and experiments. PLoS ONE. 2010;5:e8752. doi: 10.1371/journal.pone.0008752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kupchik YM, et al. A novel fast mechanism for GPCR-mediated signal transduction—Control of neurotransmitter release. J Cell Biol. 2011;192:137–151. doi: 10.1083/jcb.201007053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blunck R, Cordero-Morales JF, Cuello LG, Perozo E, Bezanilla F. Detection of the opening of the bundle crossing in KcsA with fluorescence lifetime spectroscopy reveals the existence of two gates for ion conduction. J Gen Physiol. 2006;128:569–581. doi: 10.1085/jgp.200609638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregory KJ, Hall NE, Tobin AB, Sexton PM, Christopoulos A. Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J Biol Chem. 2010;285:7459–7474. doi: 10.1074/jbc.M109.094011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hulme EC, Lu ZL, Saldanha JW, Bee MS. Structure and activation of muscarinic acetylcholine receptors. Biochem Soc Trans. 2003;31:29–34. doi: 10.1042/bst0310029. [DOI] [PubMed] [Google Scholar]
- 21.Leach K, Davey AE, Felder CC, Sexton PM, Christopoulos A. The role of transmembrane domain 3 in the actions of orthosteric, allosteric, and atypical agonists of the M4 muscarinic acetylcholine receptor. Mol Pharmacol. 2011;79:855–865. doi: 10.1124/mol.111.070938. [DOI] [PubMed] [Google Scholar]
- 22.Prilla S, Schrobang J, Ellis J, Höltje HD, Mohr K. Allosteric interactions with muscarinic acetylcholine receptors: Complex role of the conserved tryptophan M2422Trp in a critical cluster of amino acids for baseline affinity, subtype selectivity, and cooperativity. Mol Pharmacol. 2006;70:181–193. doi: 10.1124/mol.106.023481. [DOI] [PubMed] [Google Scholar]
- 23.Stefani E, Bezanilla F. Cut-open oocyte voltage-clamp technique. Methods Enzymol. 1998;293:300–318. doi: 10.1016/s0076-6879(98)93020-8. [DOI] [PubMed] [Google Scholar]
- 24.Villalba-Galea CA, Sandtner W, Starace DM, Bezanilla F. S4-based voltage sensors have three major conformations. Proc Natl Acad Sci USA. 2008;105:17600–17607. doi: 10.1073/pnas.0807387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.