

Abstract
System xc- is an amino acid antiporter that typically mediates the exchange of extracellular l-cystine and intracellular l-glutamate across the cellular plasma membrane. Studied in a variety of cell types, the import of l-cystine through this transporter is critical to glutathione production and oxidative protection. The exchange-mediated export of l-glutamate takes on added significance within the CNS, as it represents a non-vesicular route of release through which this excitatory neurotransmitter can participate in either neuronal signalling or excitotoxic pathology. When both the import of l-cystine and the export of l-glutamate are taken into consideration, system xc- has now been linked to a wide range of CNS functions, including oxidative protection, the operation of the blood–brain barrier, neurotransmitter release, synaptic organization, viral pathology, drug addiction, chemosensitivity and chemoresistance, and brain tumour growth. The ability to selectively manipulate system xc-, delineate its function, probe its structure and evaluate it as a therapeutic target is closely linked to understanding its pharmacology and the subsequent development of selective inhibitors and substrates. Towards that goal, this review will examine the current status of our understanding of system xc- pharmacology and the structure–activity relationships that have guided the development of an initial pharmacophore model, including the presence of lipophilic domains adjacent to the substrate binding site. A special emphasis is placed on the roles of system xc- within the CNS, as it is these actions that are among the most exciting as potential long-range therapeutic targets.
Keywords: l-glutamate, l-cystine, transporter, exchanger, glutathione, cystine-cysteine shuttle, glia, transporter pharmacophore, molecular pharmacology
System xc- (SxC-) is an amino acid antiporter that typically mediates the exchange of extracellular l-cystine (l-Cys2) and intracellular l-glutamate (l-Glu) across the cellular plasma membrane. Initial characterizations of this transporter most often focused on the influx of l-Cys2, as in many cells this uptake serves as a rate-limiting step in providing the intracellular l-cysteine (l-CysH) required for the synthesis of glutathione (GSH). Within the context of the central nervous system (CNS), recent attention has shifted to the other half of the exchange reaction, as the efflux of l-Glu through the exchanger carries with it the potential to contribute to excitatory signalling and/or excitotoxic pathology. When both sides of the Sxc--mediated exchange are taken into consideration, the range of CNS processes to which this transport system has now been linked is rather surprising, and includes oxidative protection (Shih et al., 2006a), the operation of the blood brain barrier (Hosoya et al., 2002), neurotransmitter release (Baker et al., 2002), synaptic organization (Augustin et al., 2007), viral pathology (Espey et al., 1998), drug addiction (Baker et al., 2003; Knackstedt et al., 2008; Kalivas, 2009), chemosensitivity and chemoresistance (Huang et al., 2005), and brain tumour growth (Chung et al., 2005; Chen et al., 2009; de Groot and Sontheimer, 2010). As with many transporters, the ability to selectively manipulate SxC- activity, delineate its function, probe its structure and evaluate it as a therapeutic target is closely linked to understanding its pharmacology and the subsequent development of selective inhibitors and substrates. Towards that goal, this review will examine the current status of our understanding of SxC- pharmacology and the structure–activity relationships (SARs) that will likely guide the development of ligands with greater specificity and potency. A special emphasis is placed on the roles of SxC- within the CNS, as it is these actions that are amongst the most exciting as potential long-range therapeutic targets.
Structure
Sxc- is a member of the heteromeric amino acid transporter (HAT) family (a.k.a. glycoprotein-associated amino acid exchangers) (Broer and Wagner, 2002; Verrey et al., 2003; Palacin et al., 2005). These transporters are heterodimers composed of one of two types II N-glycosylated ‘heavy chains’ (4F2hc or rBAT, Solute Carrier SLC3 family) covalently linked via a disulphide bond to one of several hydrophobic, non-glycosylated ‘light chains’ (SLC7 family). In the instance of Sxc-, the heterodimer is composed of 4F2hc (≈80 kDA, a.k.a. CD98 or FRP1) and xCT (SLC7A11, ≈40 kDa). The xCT subunit is credited with the transport activity of the dimer, while 4F2hc acts in the trafficking of the light chain and is required for cell surface expression. Reconstitution studies with the HAT dimer rBAT/b°,+AT (SLC7A9), however, demonstrate that light chains may be fully functional in the absence of its corresponding heavy subunit (Reig et al., 2002). Structural studies, including those based upon cysteine-scanning and thiol-modifying reagent accessibility, indicate that xCT has 12 transmembranes domains (TMDs), intracellular N and C termini, and a re-entrant loop between TMD 2 and 3 that appears to participate in substrate binding and/or permeation (Gasol et al., 2004; Jimenez-Vidal et al., 2004). More recently, the crystallization of a representative APC (amino acid, polyamine, organo-cation) transporter from the thermophile Methanocaldoccus jannaschii has provided much greater insight into HAT structure (Shaffer et al., 2009). ApcT acts as a sodium-independent transporter with a broad-specificity (e.g. substrates include l-Glu, l-alanine, l-serine, l-glutamine and l-phenylalanine). The transporter, which was crystallized in an apo (no substrate bound), inwards-facing form, exhibits an inverted repeat of five TMD helicies reminiscent of the LeuT transporter (Yamashita et al., 2005). Figure 1 shows a homologous structure for xCT that was constructed by threading the human xCT sequence through the ApcT structure (Figure 1).
Figure 1.
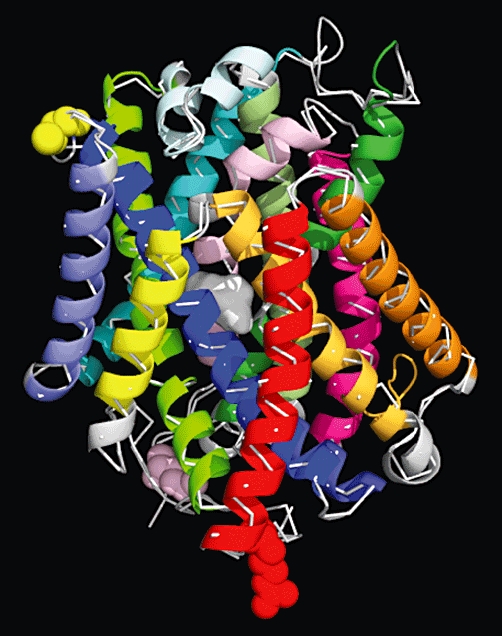
An xCT homology model was constructed by threading the hxCT sequence over the crystal structure of ApcT (RCSB: pdb3GIA) from M. jannaschii (GI:1591319) as reported by Goaux and co-workers (Shaffer et al., 2009) using FASTA (Pearson and Lipman, 1988) followed by multiple sequence alignments with ClustalW (Larkin et al., 2007). The structure of ApcT is shown as a white thread overlayed with the helical ribbons of the 12 TMDs of xCT: TMD 1A/B in light pink; TMD2 in dark pink; IL1 in dark blue; TMD3 in bright blue; TMD4 in light purple; TMD5 in teal, TMD6A/B in dark green; TMD7 in olive green; EL4A/B in silver; TMD8 in light green; TMD9 in yellow; TMD10 in gold; TMD11 in orange and TMD12 in red. Truncated intracellular N (light pink) and C (red) termini are shown with spheres. The cysteine-158 that participates in the disulphide bond with 4F2hc is highlighted with yellow spheres. The protein is shown in its inwardly facing Apo-form. A hypothetical substrate is depicted by the centrally located white surface as predicted by Schaffer et al., based upon the binding site of LeuT.
Localization
Sxc- has been characterized in a wide variety of cells, both outside (e.g. fibroblasts, macrophages, hepatocytes and endothelial cells) (Bannai, 1986; Sato et al., 1999; Hosoya et al., 2002; Sasaki et al., 2002) and inside the CNS [e.g. astrocytes (Cho and Bannai, 1990; Allen et al., 2001; Gochenauer and Robinson, 2001), microglia (Piani and Fontana, 1994), retinal Muller cells (Kato et al., 1993), immature cortical neurons (Murphy et al., 1990), and glioma cell lines (Cho and Bannai, 1990; Ye et al., 1999)]. The characterization of Sxc- distribution by using antibodies to the xCT and 4F2hc subunits show the transporter to be localized to neurons and glia in the CNS. Furthermore, Sxc- was highly expressed at the borders of the CNS and periphery including the vascular endothelial cells, ependymal cells, choroid plexus and the leptomeninges (Burdo et al., 2006). Similarly, expression of xCT and 4F2hc was also detected immunohistochemically in the brush border membranes of the kidney and duodenum. Work focusing on the distribution of mRNA for xCT in the mouse brain complements the antibody labelling studies in that xCT mRNA was highly detected in the meninges and specific regions facing the cerebral ventricles (Sato et al., 2002). It must be noted, however, that the detection of either xCT mRNA or xCT immunoreactivity does not necessarily confirm the expression of functional Sxc- at the level of the plasma membrane (Lim et al., 2007).
Pharmacology
Initial studies characterizing Sxc- focused primarily on non-CNS cells and concentrated on the uptake of l-Cys2; differentiating the specificity, kinetics and ionic dependence of Sxc- from other transport systems. Thus, Sxc- was shown to (i) utilize either l-Glu or l-Cys2 as substrates, with each acting as a competitive inhibitor of the other; (ii) function as an antiporter, mediating the 1:1 exchange of an intracellular and extracellular amino acid; (iii) act in a sodium-independent, chloride-dependent, electroneutral manner; and (iv) transport l-Glu or l-Cys2 in an anionic form (Bannai and Kitamura, 1980, 1981; Waniewski and Martin, 1984; Cho and Bannai, 1990). In most of the cell types examined, the Km values for the uptake of either l-Glu or l-Cys2 were typically in the 50–100 µM range, although more recent studies with differentiated astrocytes yielded lower Km values close to 20 µM (Seib et al., 2011).
Substrate activity
Delineating the specificity of Sxc- was central to these early (as well as more recent) studies and is reviewed in greater detail below. These findings not only provide insight into the SARs that govern Sxc- selectivity and the development of pharmacophore models, the resulting identification of inhibitors (and substrates) can yield needed small molecule probes with which to assess physiological and pathological roles of the transporter. A majority of these investigations have been based upon competitive assays that essentially screen structurally similar small molecules for the ability to block the transport of radiolabelled l-Glu or l-Cys2 under conditions selective for Sxc--mediated uptake. In this manner, the demonstration that none of the class-defining excitatory amino acid (EAA) receptor agonists [e.g. NMDA, kainate (KA), aminomethyl-isoxazole propionic acid (AMPA) and 1-aminocyclopentane-trans-1,3-dicarboxylic acid (trans-ACPD)] nor the well-known excitatory amino acid transporter (EAAT) inhibitors [e.g. l- or d-aspartate, l-trans-2,4-pyrrolidine dicarboxylate (l-trans-2,4-PDC) and dihydrokainate] could block Sxc- served to readily demonstrate that the transporter has a distinct pharmacological profile within the EAA system (Patel et al., 2004; Balazs et al., 2006; Beart and O'Shea, 2007; Bridges and Patel, 2009). While these competition assays shed light on whether or not a compound binds to the transporter, it is important to note that the resulting inhibitory data do not differentiate alternative substrates from non-substrate inhibitors. Indeed, studies on the EAAT's suggest that the chemical requirements for binding and translocation are related, but not necessarily identical to one another (Bridges and Esslinger, 2005). Following substrate activity often requires the synthesis of radiolabelled derivatives or the quantification of a secondary measure, such as the substrate-coupled conduction currents employed to study the electrogenic EAATs (Esslinger et al., 2005) (Sxc- is electroneutral). In the instance of Sxc-, a protocol originally developed to measure the release of l-Glu from synaptosomes (Nicholls et al., 1987) has been modified to quantify Sxc- activity by coupling the efflux of l-Glu through the transporter to its extracellular metabolism by glutamate dehydrogenase and the production of NADPH (Patel et al., 2004; Warren et al., 2004). As will be described in greater detail below, this approach has allowed us to demonstrate that the different classes of compounds that inhibit Sxc- also exhibit a wide range of substrate activities.
Acyclic amino acids (Figure 2)
Figure 2.
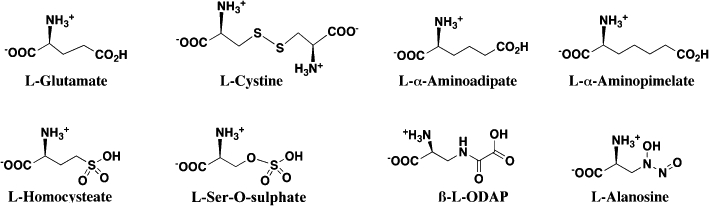
Acyclic amino acid substrates and inhibitors of the system xc- transporter.
The compounds within this series, which also includes l-Glu and l-Cys2, (i) represent some of the most straight forward structural mimics of the endogenous substrates; (ii) were amongst the earliest Sxc- inhibitors identified and (iii) were critical to establishing tolerated limits of stereo-specificity, backbone length and charged functional group substitutions (Bannai and Kitamura, 1980; Waniewski and Martin, 1984; Bannai, 1986; Patel et al., 2004). The transporter exhibits a demonstrable stereo-selective preference for l-amino acids (e.g. d-Glu and d-Cys2 exhibit little or no inhibitory activity), although it is not absolute, as d-serine-O-sulphate is a moderately effective inhibitor (Patel et al., 2004). The inactivity of d- and l-aspartate demonstrates that decreasing the chain length of carboxylic acids results in a loss of inhibitory activity. Given the backbone length of l-Cys2, it is not surprising that dicarboxylic amino acids l-α-aminoadipate and l-α-aminopimelate exhibit inhibitory activities comparable to l-Glu (Bannai, 1986; Patel et al., 2004). These longer dicarboxylate acids appear, however, to be less suitable than l-Cys2 or l-Glu as substrates of Sxc-. The inactivity of l-homocystine and l-djenkolate as inhibitors also demonstrates that there is a limit to the ability of the binding site to accommodate increasing chain length (Patel et al., 2004). Substitutions of the distal carboxylate group on l-Glu analogues further demonstrate that SO3- (e.g. l-homocysteate, S-sulpho-l-cysteine, l-serine-O-sulphate) and SO2- groups (e.g. inhibition by l-homocysteine sulphinate) are tolerated within the binding site, while PO32- groups are not (e.g. lack of inhibition by l-serine-O-phosphate). The extent to which the distal portion of l-Glu analogues can be modified and still retain potent inhibitory activity are illustrated by two novel natural products: the excitotoxin l-β-N-oxalyl-l-α,β-diaminopropionate (β-l-ODAP, Ki≈l-Cys2≈ 40–100 µM) derived from Lathyrus sativus and the antimetabolite, antitumour agent l-alanosine from Streptomyces alanosinicus (Warren et al., 2004; Huang et al., 2005). Interestingly, both of these compounds are also substrates of the transporter, suggesting that portions of their in vivo actions may reflect or be dependent upon intracellular accumulation via Sxc-.
Isoxazoles and related heterocyclics (Figure 3)
Figure 3.
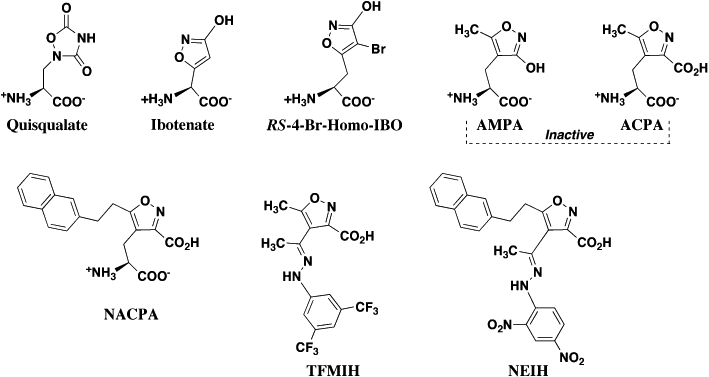
Isoxazole and related heterocyclic substrate and inhibitors of the system xc- transporter.
The recognition that quisqualate (QA) is a potent inhibitor of Sxc-[Ki≈ 5 µM (Patel et al., 2004)] was significant for several reasons. It provided direct evidence that Sxc- was not only present in the CNS but also highlighted the importance of L-Cys2 uptake, GSH production and oxidative protection. Thus, the toxicity of QA in select neuronal cell lines and immature cortical neurons was shown to be attributable to Sxc- inhibition and reduced oxidative protection, rather than EAA receptor-mediated excitotoxicity (Murphy et al., 1989; 1990;). The action of QA also provided a pharmacological link between previously indentified Cl-dependent, l-Glu binding sites on astrocytes and the l-Cys2/l-Glu exchanger (Pin et al., 1984; Bridges et al., 1986; 1987; Kessler et al., 1987). Lastly, the conformationally constrained structure of QA provided early SAR data for pharmacophore modelling, as well as prompted the characterization of isoxazoles as potential inhibitors. In contrast to QA, AMPA [which replaced QA as the defining agonist for AMPA receptors (Monaghan et al., 1989)] is inactive as an Sxc- inhibitor. Other isoxazoles such as ibotenate (Ki≈ 30 µM), and (RS)-4-Br-homoibotenate (Ki≈ 20 µM), proved, however, to be very potent inhibitors of the transporter (Patel et al., 2004). Interestingly, while all the compounds exhibited inhibitory activity, each displayed a different ability to act as substrates. For example, ibotenate is transported by Sxc- as readily as is l-Cys2 (RS)-4-Br-homoibotenate only about half as well, and QA only about a third as well. These findings clearly demonstrate that conformational restriction alone does not necessarily limit the ability of a compound to be transported and set a foundation to begin delineating the ligand characteristics that differentiate binding from translocation. The demonstration that QA was a substrate (albeit only a moderate one) also provided a greater mechanistic understanding of the process of ‘QA sensitization’, where QA applied to brain slices can be sequestered in cells only to be subsequently released through Sxc- and activate EAA receptors (Chase et al., 2001). Collectively, the actions of these isoxazoles (as well a majority of the other compounds described in this review) also serve to highlight a major challenge in the development of selective pharmacological probes of Sxc-, as each is equally well known for its action at other sites within the EAA system. Thus, QA acts as an agonist at AMPA and mGluR receptors, ibotenate as an NMDA receptor and non-selective mGluR agonist (RS)-4-Br-homoibotenate as an AMPA receptor agonist, and (RS)-5-Br-willardiine as a kainate receptor agonist (Monaghan et al., 1989; Balazs et al., 2006).
Prompted by the dichotomy that some isoxazoles are potent Sxc- inhibitors, while others are not, more recent work continued to examine the isoxazole ring as a scaffold for the further development of inhibitors. Building on the extensive work of Krogsgaard-Larsen and colleagues (Brauner-Osborne et al., 2000), a series of derivatives based upon AMPA, amino-3-carboxy-5-methyl isoxazole propionic acid (ACPA) and non-amino acid biosteres of ACPA acid-hydrazones were synthesized in which additional substitutions were made to the isoxazole core at the 4 and 5 positions of the ring (Patel et al., 2010). While neither AMPA nor ACPA are active as inhibitors, a subset of derivatives containing increasingly larger lipophilic groups, such as napthyl (Ki≈ 50 µM), and trifluoromethyl-phenyl (Ki≈ 60 µM), moieties, were found to block Sxc- with affinities comparable with l-Cys2. The resulting SAR data revealed not only a divergence point that distinguishes the binding sites of Sxc- and GluR2, it also demonstrated the presence of lipophilic binding domains adjacent to the substrate site on the Sxc- transporter. Significantly, exploiting the presence of such lipophilic pockets has been key to developing very potent and selective EAAT inhibitors (Shimamoto et al., 2004; Dunlop et al., 2005). Indeed, the structural alignment of these inhibitors [e.g. S-2-naphthyl-ethyl-ACPA (NACPA) and bis-trifluoromethyl-phenyl-isoxazole-4-hydrazone (TFMIH)] suggested the presence of two distinct lipophilic domains (see pharmacophore section below). That this appears to be the case was confirmed by the inhibitory activity of a ‘hybrid’ isoxazole that included lipophilic substituents at each position, 5-naphthylethyl isoxazole-4-(2,4-dinitrophenol)hydrazone-dinitrophenol (NEIH, Ki≈ 100 µM). Exchange reactions also confirmed that none of these inhibitors could act as Sxc- substrates, suggesting that while the binding site could accommodate the added steric bulk, an effective interaction between the added groups and the lipophilic domains on the transporter precluded translocation (Patel et al., 2010). While the derivatized isolazoles have not yet yielded ligand affinities comparable with what has been achieved for the EAAT system, these blockers have been key to localizing lipophilic domains that can be targeted in the next generation of inhibitors.
Benzoic and related aromatic acids (Figure 4)
Figure 4.
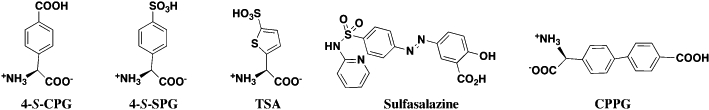
Benzoic acid and aromatic sulphonic acid inhibitors of the system xc- transporter.
Two of the most potent Sxc- inhibitors characterized to date are (S)-4-carboxyphenyglycine (4-S-CPG, Ki≈ 1–5 µM) (Ye et al., 1999; Patel et al., 2004) and sulphasalazine (SSZ; (6-oxo-3-(2-[4-(N-pyridin-2-ylsulphamoyl)phenyl]diazenyl)-cyclohexa-1,4-dienecarboxylic acid), IC50 < 1 µM) (Gout et al., 2001). 4-S-CPG was initially developed and characterized as a group I metabotropic receptor antagonist (Bedingfield et al., 1995), again highlighting the issue of cross-reactivity among Sxc- blockers (Patel et al., 2010). The observation that 4-S-CPG effectively blocked the release of l-Cys2 from glial tumour cells (see pathology section below) provided the critical pharmacological link to Sxc- (Ye et al., 1999). In this manner, S-4-carboxy,3-hydroxyphenylglycine and S-3-carboxy,4-hydroxyphenylglycine were also identified as inhibitors, although less potent that 4-S-CPG. Similar analogues not possessing a second carboxylate group (e.g. 3-S-hydroxyphenylglycine, RS-35-dihydroxyphenylglycine) were essentially inactive. In contrast to ibotenate or QA, 4-S-CPG was essentially inactive as a Sxc- substrate when examined in the fluorometric exchange assay (Patel et al., 2004). More recently, a 4-S-CPG derivative in which the distal carboxylate was replaced with a sulphonic acid (4-S-SPG) was also found to inhibit Sxc- (Etoga et al., 2010). Unlike the analogous anionic substitution between l-Glu and l-homocysteate, 4-S-SPG was markedly less potent than 4-S-CPG, inhibiting uptake even less effectively than l-Cys2. Both R- and S-4-sulphophenylalanine were also shown to lack inhibitory activity at Sxc-. Interestingly, when the phenyl ring of 4-S-SPG was replaced with a thiophene (R,S-4-sulphothienylglycine), inhibitory activity increased to a level comparable with l-Cys2 (Ki≈ 75 µM; Etoga et al., 2010). Similar to 4-S-CPG, SSZ was identified as a Sxc- inhibitor within the context of assessing its action on tumour cells (Gout et al., 2001). Originally developed as a pro-drug containing sulphapyridine and 5-amino salicylic acid, SSZ has been used as an anti-inflammatory agent to treat Chron's disease and inflammatory bowel disorders. SSZ is also a very potent (≈4-S-CPG) inhibitor of Sxc-. Although SSZ possess a benzoic acid moiety common to many of the inhibitors, it stands out amongst the other compounds described in this review in that it lacks the prototypical l-α-amino acid head group. Comparisons with 4-S-CPG also led to synthesis of [(R,S)-4-[4′-carboxyphenyl]-phenylglycine; CPPG] (Etoga et al., 2010), which in some ways might be considered a hybrid between 4-S-CPG and SSZ. While CPPG proved to be as active as l-Cys2 as an inhibitor, it failed to match the potency of either 4-S-CPG or SSZ. As will be discussed below, this may suggest that portions of SSZ interacts effectively enough with the other domains within the substrate binding site of Sxc-, that the interactions with an l-α-amino acid head group are no longer required.
Modelling the binding site
Pharmacophore models provide a strategy to collectively analyse and visualize the SARs that govern ligand-binding site interactions. This approach, which relies on the 3D superposition of low energy conformation of substrates and inhibitors, has been successfully employed in the characterization of the EAATs (Esslinger et al., 2005; Mavencamp et al., 2008). In the instance of Sxc-, a pharmacophore model has been developed (Figure 5) using l-Glu, l-Cys2, QA and 4-S-CPG (panel B) as a training set and aligning these ligands in a manner that maximizes overlap of the α-C, α-COOH, α-NH2 and distal COOH groups or related isosteric moieties (Patel et al., 2010). One of the first postulates to emerge from this comparison of minimized structures was that l-Cys2 may be binding to Sxc- in a pseudo chair-like conformation that maximizes hydrogen bonding between the 4-position sulphur atom and a proton of the C7 terminal amine group (Figure 5A). The resulting ring-like structure is consistent with NMR spectroscopic findings (Edmonds and Summers, 1969; Hunston et al., 1982) and aligns with the central core rings of QA and 4-S-CPG (Figure 5B). Overlaying this conformation of l-Cys2 and l-Glu then leads to the likelihood that the distal carboxylate groups of the two substrates may be interacting with different sub-domains within a larger substrate binding site. The available COOH–COOH distances are also consistent with the inactivity of l-aspartate (too short) and the ability of the substrate site to accommodate l-α-aminoadipate. Furthermore, such an arrangement provides an explanation for the activities of 4-S-CPG and QA in which the positions of the distal carboxylate-mimics are relatively fixed as a consequence of the rigid structure of the inhibitors.
Figure 5.
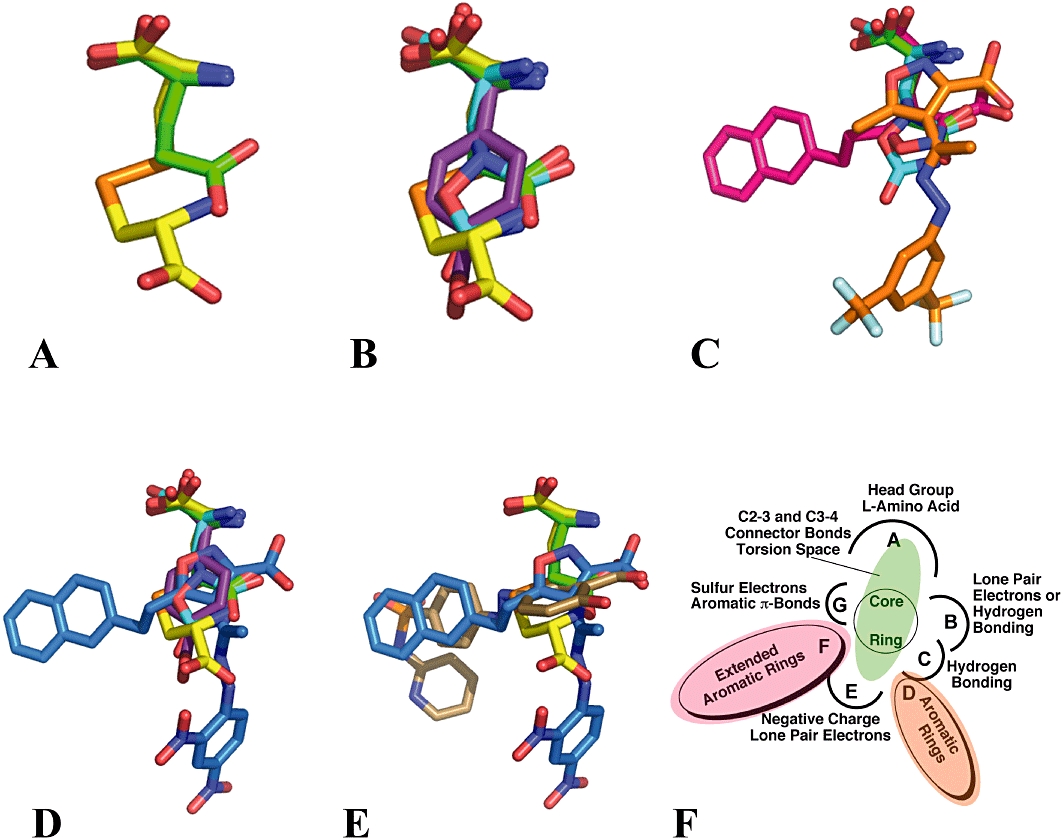
A ligand-based, superposition, 3D pharmacophore model for the substrate binding on Sxc-. A–E. l-Glu in green; l-Cys2 in yellow; QA in teal; 4-S-CPG in purple; NACPA in red; TFMIH in orange; NEIH in blue and SSZ in bronze. F. Pharmacophore binding template.
The pharmacophore model also provides a framework on which to superimpose the inhibitors containing appended steric and lipophilic bulk as a way to identify the relative positions of the corresponding domains within the binding site that interact or accommodate these groups (Patel et al., 2010). As depicted in Figure 5, modelling of NACPA, TFMIH and NEIH led to the identification of previously unrecognized ‘lipophilic pockets’ on the transporter that are adjacent to the substrate binding domains. While the hybrid molecule NEIH clearly establishes the presence of two such pockets, the presence of multiple anionic groups on the substrates and inhibitors makes it more difficult to definitively assign the relative position of these lipophilic substituents with respect to each of the inhibitor COOH groups with the specific α- and distal COOH groups of l-Glu and l-Cys2. In the overlay shown in Figure 5C, the α-C, α-COOH and α-NH2 of NACPA is aligned with the l-α-amino acid head group of l-Glu and l-Cys2. In this configuration, the naphthyl ring of NACPA delineates one of the two lipophilic pockets. In turn, overlaying the naphthyl groups of NACPA and NEIH positions the 3-COOH group extending from the isoxazole ring of NEIH such that it aligns with the distal COOH group of l-Glu and delineates a second lipophilic pocket occupied by the 2,4-dinitrophenyl moiety (Figure 5D and E). Similarly, overlaying the 3,5-bis-trifluoromethyl-phenyl group of TFMIH with the 2,4-dinitrophenyl group of NEIH also aligns the 3-COOH group extending from the isoxazole ring of NEIH to the distal COOH moiety of l-Glu (Figure 5D). The issue of the relative positioning of lipophilic groups also arises in the case of SSZ, as the pyridylsulphamoylphenyl portion of SSZ can be correlated to either of the lipophilic regions depending upon whether the COOH group of the salicyclic moiety of SSZ is aligned with either the α- or distal COOH group of l-Glu. Although the pictured lipophilic group alignments are consistent with one another, it is readily acknowledged that the positions could be reversed, resulting in a switch in the alignments between the COOH groups and related isosteric moieties. Regardless of the exact positioning, however, the activities of NACPA, NEIH and TFMIH highlight that interaction with these lipophilic domains appears significant enough to overcome less than optimal binding of the COOH and NH2 biosteres (evidenced by the inactivity of AMPA and ACPA) or, in the instance of SSZ, the lack of a prototypical l-α-amino acid head group. A pharmacophore template summarizing the modelling results (Figure 5F) depicts the overall binding site as a collection of sub-domains that can interact with an l-amino acid head group, a distal COOH or similar biostere, a hydrogen bonding group equivalent to the distal l-amino acid moiety of l-Cys2 or a second heteroatom of an isoxazole ring, a lone pair electron or π-electron density associated with the sulphur atoms of l-Cys2 or the aromatic ring of 4-S-CPG, and two distinct lipophilic pockets that can accommodate aromatic rings. Importantly, the presently identified group of inhibitors indicates that a ligand needs to interact with a subset, but not necessarily all, of these domains to effectively bind and inhibit Sxc-. The continued advancement of delineating the pharmacology of Sxc- will utilize these models in two ways: (i) developing the next generation of inhibitors to optimize interactions with the lipohlilic domains as a strategy to increase potency and (ii) structurally aligning and docking the ligand-based models to the homology-based protein models of Sxc-.
Physiological and pathological roles
Regulation
Given the functional importance of Sxc- in cell types both within and outside the CNS, it is not surprising that multiple mechanisms exist through which to regulate its expression. Indeed, amino acid deprivation, xenobiotic exposure and oxidative stress have all been shown to trigger the up-regulation of Sxc- (Bannai, 1984; Bannai et al., 1989; Sato et al., 1995; Ishii et al., 2000). The best-characterized of these mechanisms are those linked to the Nrf2-ARE signalling pathway (Figure 6) in which the transcription of a specific gene is governed by the presence of a cis-acting regulatory DNA element (i.e. the antioxidant response element, ARE) in its promoter. Numerous enzymes associated with the synthesis of GSH, the inactivation of reactive oxygen species (ROS) and phase II detoxification contain AREs (Ishii et al., 2000) (Prochaska et al., 1985; Talalay, 1989; Spencer et al., 1991; Limon-Pacheco et al., 2007), and at least four ARE motifs have been identified in the promoter region sequence of mouse xCT (Sasaki et al., 2002). The expression of these ARE-containing genes is up-regulated by the binding of nuclear factor erythroid-2-related factor (Nrf2), a CNC-bZIP transcription factor (Jaiswal, 2004; van Muiswinkel and Kuiperij, 2005; Vargas and Johnson, 2009; Giudice et al., 2010) following its translocation into the nucleus. Electrophilic or oxidative stress can also lead to direct phosphorylation of Nrf2 by kinases in several cascades [e.g. protein kinase C, phosphatidyl-inositol 3-kinase, PKR-like endoplasmic reticulum kinase, JNK, ERK and p38 MAPK (MK14)] that also facilitates translocation into the nucleus (Kobayashi and Yamamoto, 2006; Kandil et al., 2010). The regulatory control provided by these pathways appears to vary among cell types (Sasaki et al., 2002; Qiang et al., 2004), and several studies point to existence of alternate mechanisms of Sxc- regulation (Sasaki et al., 2002; Sato et al., 2004; Lewerenz et al., 2009).
Figure 6.
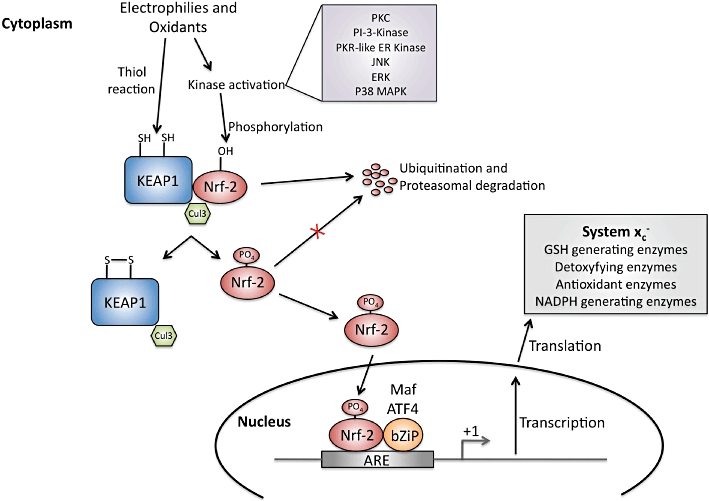
Nrf-2/ARE activation pathway. Under basal conditions Nrf-2 is dimerized with KEAP1 and continuously targeted for degradation due to ubiquitination by Cul3. Electrophilies and oxidants can disrupt the dimerization of Nrf2 and KEAP directly by modifiying cysteine residues in KEAP1 or through phosphorylation of Nrf2 at Ser40 by protein kinases. Nrf2 is then free to translocate into the nucleus, bind with an adaptor protein (e.g. Mafs and ATF4) and increase ARE-driven transcription.
In CNS tissue, treatment of primary astrocytes with dibutyryl cyclic AMP (dbcAMP) has been shown to differentiate the glial cells into a more in vivo-like, activated phenotype that includes the increased expression of several proteins associated with the glutamate system, including: glutamine synthetase, EAATs and Sxc- (Hertz et al., 1998; Gochenauer and Robinson, 2001; Daginakatte et al., 2008). More recently, a closer examination of these dbcAMP-treated astrocytes reveals that Sxc- expression may also directly be regulated by GSH levels in a phenotype dependent manner (Seib et al., 2011). Sxc- activity is markedly up-regulated in dbcAMP-treated astrocytes in which GSH levels have been depleted with synthesis blocker buthionine sulphoximine (BSO), a response not observed in untreated protoplasmic astrocytes. Interestingly, an Nrf2-dependent increase in Sxc- activity is observed when the electrophile tBHQ is added to the protoplasmic astrocytes, but not in differentiated cultures, suggesting a distinct regulatory mechanism. Changes in Sxc- activity induced by BSO also correlate to increases in both xCT mRNA and protein and are temporally linked to GSH levels. Furthermore, the effect of BSO cannot be mimicked by H2O2 treatment or reversed by the addition of general antioxidants, suggesting that it is a change in GSH level that triggers the induction. Consistent with such a mechanism, co-administration of permeable GSH pro-drug GSH-ethylester and BSO prevents the up-regulation of Sxc- activity.
Studies seeking drug candidates to induce the up-regulation of EAAT expression as a potential point of intervention in amyotrophic lateral sclerosis (ALS) demonstrated that some β-lactam antibiotics could increase expression of EAAT2 through an action at the promoter level (Rothstein et al., 2005). One of the more effective of these compounds, Ceftriaxone, was recently reported to also produce an increase in Nrf-2 and xCT expression in HT22 hippocampal cells and fibroblasts, as well as protect these cells from l-Glu-induced exicitotoxicty (Lewerenz et al., 2009). Such an induction has also been postulated to provide a novel therapeutic approach in inflammatory neurodegenerative disease (for review, see Chen and Kunsch, 2004; Vargas and Johnson, 2009). In those instances where an increase in Sxc- activity may be of therapeutic benefit (see physiology and pathology section below), pharmacological strategies targeting enhanced expression provide an alternative to screening for potential allosteric activators.
CNS GSH production and oxidative protection via Sxc-
The generation and maintenance of GSH levels in all mammalian cells is critical to the detoxification of xenobiotics and the prevention of oxidative stress and damage. This latter effect is especially relevant to the CNS given its high levels of oxygen consumption, the abundance of enzymes and metabolites that can generate ROS and its limited antioxidant capacity (Halliwell, 2006). As a transporter, Sxc- acts as a key component in GSH production by providing l-Cys2 as a synthetic precursor (Figure 7). The combination of micromolar concentrations of l-Cys2 in the CSF and millimolar concentrations of l-Glu in the astrocyte serve to drive the exchange, after which the intracellular l-Cys2 is rapidly reduced to l-CysH and enzymatically incorporated into GSH (Sagara et al., 1993). While both astrocytes and neurons have been shown to express the xCT subunit of Sxc-, mature neurons exhibit little or no Sxc- activity and appear even less capable of directly mediating the uptake l-Cys2. GSH production in neurons is thus dependent upon the uptake of L-CysH that is provided by astrocytes via the ‘cystine/cysteine cycle’ (Dringen et al., 1999; Wang and Cynader, 2000). Essentially, the l-Cys2 that is transported into the astrocytes by Sxc- is reduced and subsequently released as either l-CysH itself or GSH, which can also serve as an l-CysH source following its extracellular metabolism by γ-glutamyl-transpeptidase (GGT) and aminopeptidase N. As previously mentioned, the observation that QA toxicity in immature cortical neurons could be attributed to blocking Sxc- and the resultant oxidative stress (and not EAA receptor mediated excitotoxicity) highlighted the significance of the transporter in these pathways (Murphy et al., 1989; Cho and Bannai, 1990; Shih et al., 2003; 2006a;). Under conditions of primary or secondary oxidative stress, astrocytes also are capable of contributing to neuroprotection by rapidly providing neurons with increased levels of GSH precursors via the Nrf2-dependent induction of the GSH synthesis (Sagara et al., 1993; Dringen et al., 1999; Wang and Cynader, 2000; Guebel and Torres, 2004) (Shih et al., 2006a).
Figure 7.
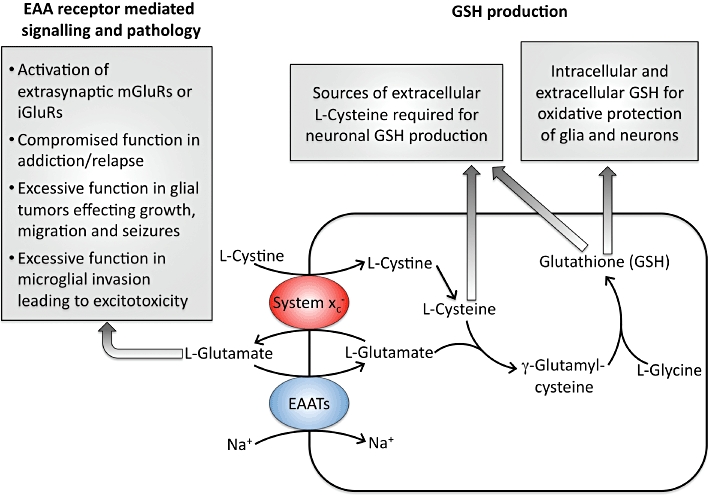
Functional roles of the system xc- transporter in the CNS.
Given that the maintenance of intracellular GSH levels is critical to most cells, those that do not express Sxc-, as is the case with mature neurons, presumably accumulate either l-CysH or l-Cys2 through alternative systems. Other members of SLC7 family are also capable of transporting these precursors. Thus, CysH is a substrate of the 4F2hc-linked SLC7A10 transporter Asc-1. The SLC7A9 transporter b°,+AT (SLC7A9) can mediate the uptake of Cys2, although it is dimerized with rBAT instead of 4F2hc. (Verrey et al., 2003). As its name implies, functionally defined ASC (alanine–serine–cysteine) system members ASCT1 and ASCT2 (SLC1A4 and A5, respectively) transport L-CysH in a sodium-dependent manner (Zerangue and Kavanaugh, 1996a; Kanai and Hediger, 2004). l-CysH is also a substrate of sodium-dependent neutral amino acid transporters SNAT1, SNAT 2 and SNAT4, which are members of the SLC38 family and are typically associated with glutamine and asparagine uptake (Mackenzie and Erickson, 2004). Amongst the alternative routes of entry for l-CysH or l-Cys2 within CNS cells, the most intriguing is associated with the EAATs (Zerangue and Kavanaugh, 1996b; McBean, 2002; Chen and Swanson, 2003). In particular, l-CysH was shown to be transported by the neuronal transporter EAAT3 (EAAC-1) with a maximal flux rate comparable with that of l-Glu (Zerangue and Kavanaugh, 1996b). Consistent with such a role, GSH levels are negatively regulated by the interaction of EAAC-1 with the glutamate transport-associated protein 3–18 (GTRAP3-18) (Watabe et al., 2007) and mice homozygous deficient for EAAT3, exhibit not only increased neurodegeneration with age, but also decreased neuronal levels of GSH concentrations and an increased vulnerability to oxidative stress (Aoyama et al., 2006).
Source for extrasynaptic excitatory signalling
The l-Cys2-coupled export of l-Glu through Sxc- takes on additional significance within the CNS as it represents a non-vesicular route by which this excitatory transmitter can access EAA receptors, particularly those present in extrasynaptic locales. Given the presence of Sxc- on astrocytes, l-Glu released in this manner also represents a novel mechanism for glial-neuronal communication. Work by Kalivas, Baker and co-workers demonstrate that the extracellular l-Glu released through Sxc- has a physiological role in the tonal regulation of extrasynaptic type 2/3 mGluR receptors and the control of neurotransmitter release throughout the CNS (Baker et al., 2002; Xi et al., 2003; Mohan et al., 2011). In both the striatum and nucleus accumbens, extrasynaptic activation of type 2/3 mGluR receptors inhibits the release of L-Glu and/or dopamine. Aberrant changes in Sxc- activity and l-Glu homeostasis at these synapses have been implicated in the pathophysiology of drug addiction (Kalivas et al., 2009; Madayag et al., 2010). In rat models of chronic, self-administered cocaine addiction, a series of neuroadaptations have been identified that underlie the disease including: a decrease in extra-synaptic l-Glu concentrations in the nucleus accumbens, a decrease in l-Cys2 and l-Glu exchange, and an increase in glutamatergic transmission. Restoring Sxc- activity by applying l-CysH or N-acetyl-CysH (NAC) can return extra-synaptic l-Glu to control levels (Baker et al., 2003); restoring normal glutamatergic transmission and in doing so prevents cocaine primed drug seeking and relapse (Baker et al., 2003; Amen et al., 2011). A similar mechanism has been implicated for nicotine addiction and suggests targeting l-Glu homeostasis is a potential method for treating addiction (Knackstedt and Kalivas, 2009; Knackstedt et al., 2009).
The ability of Sxc- to contribute to neuronal signalling through the release of l-Glu is, however, not uniformally accepted. Thus, while a l-Cys2-mediated efflux of l-Glu was shown to be sufficient to activate non-NMDA receptors in cerebellar slices and NMDA receptors in hippocampal slices (Warr et al., 1999; Cavelier and Attwell, 2005), it was concluded that the combination of CSF l-Cys2 levels (typically in the 0.1–0.5 µM range) and the Km for l-Cys2 at Sxc- (typically reported in 50–100 µM range), would preclude Sxc- from contributing to ambient l-Glu levels. Such a conclusion, however, must be tempered somewhat by assay results quantifying the uptake of 35S-l-Cys2 into slices of nucleus accumbens, which yielded Km values in the 2–4 µM range (Baker et al., 2003). More recent studies on primary cultures of differentiated astrocytes also yielded Km values for l-Glu transport that were two- to fivefold lower than observed in many glial cell lines (Seib et al., 2011). l-Cys2 levels in the 0.1–0.3 µM range were able to produce an Sxc--mediated efflux of l-Glu that decreased the synaptic release of l-Glu as a consequence presynaptic mGluR2 activation (Moran et al., 2005). These different findings point to the likelihood that the ability of l-Glu released from Sxc- to activate EAA receptors probably varies among circuits.
Beyond its role in GSH production and oxidative protection within the different cell types of the mammalian eye (Lim et al., 2005; 2007; Li et al., 2007) additional insight into the role of the Sxc- in extrasynaptic signalling comes from a closer examination of retinal circuits (Hu et al., 2008). Electron microscopy studies reveal the ultra-structural localization of Sxc- (based upon xCT expression) to the photoreceptor (rod and cone) ribbon synapse with ON bipolar cells of the outer plexiform layer. Using the cation channel probe agmatine (AGB) as a fluorescent reporter, it was shown that l-Glu released through Sxc- could activate mGluR 6 receptors that, in turn, would close non-selective cation channels on the post synaptic ON bipolar cells. Consistent with the participation of Sxc-, the ability of l-Cys2 to reduce this AGB fluorescence was blocked by co-incubation with 4-S-CPG. The findings suggest that signalling within the photoreceptor synapse incorporates both vesicular release and Sxc--mediated exchange as a source of l-Glu.
Evidence validating Sxc- as a source of ambient l-Glu and its contribution to signalling and plasticity has also come from work with Drosophila melanogaster. Studies on an xCT homologue genderblind in D. melanogaster suggest that the transporter influences courtship behaviour through its regulation of ambient extracellular l-Glu in vivo (Grosjean et al., 2008). The loss of this gene reduces extracellular l-Glu levels by about half. Furthermore, changes in ambient l-Glu levels arising from altered Sxc- activity were shown to influence both the function (i.e. desensitization) and localization (i.e. clustering) of ionotropic glutamate receptors (iGluRs) in the Drosophila CNS (Augustin et al., 2007; Featherstone, 2010).
Source for excitotoxicity
When the efflux of l-Glu through Sxc- becomes excessive, its character within the CNS switches from that of a potential excitatory transmitter to that of a well-characterized and pathologically relevant excitotoxin (Waxman and Lynch, 2005; Lau and Tymianski, 2010). Early recognition that Sxc- could be a source of excitotoxic l-Glu came from an examination of microglia cells that, owing to a marked need for oxidative protection, express high levels of the transporter (Piani and Fontana, 1994). Thus, under conditions of CNS infection or trauma, activation of microglia from a resting state by pro-inflammatory stimuli, as well as migration into localized areas, not only have beneficial neuroprotective and neurothrophic effects, but the ability to release substantial levels of l-Glu (Espey et al., 1998; Qin et al., 2006; Shih et al., 2006b; Barger et al., 2007). Given the ubiquitous involvement of microglia in CNS disease and pathology, it is not surprising that this ability to exacerbate neurodegeneration through the conversion of oxidative stress to excitotoxic stress via Sxc- has been linked to a variety of disorders, including: Alzheimer's disease (Barger and Basile, 2001), Parkinson's disease, AIDS (Zeng et al., 2010), bacterial infection/LPS (Taguchi et al., 2007) and multiple sclerosis (Domercq et al., 2007). Ironically, the observations that microglia express high levels of Sxc- and that transporter-mediated efflux of l-Glu provides a non-synaptic pathway of mGluR signalling also prompted the examination of this mechanism within the non-neural cells of the immune system. These studies revealed that l-Glu released by dendritic cells acts through mGluRs on T cells to modulate T-cell activation and that this release occurs through Sxc- (Pacheco et al., 2006; 2007;).
The specific up-regulation of Sxc−- in astrocytes by interleukin-1β (IL-1β) and subsequent activation of mGluR1 receptors has also been demonstrated to enhance hypoxic neuronal injury (Fogal et al., 2005; 2007;). Neurons co-cultured with astrocytes were found to be more susceptible to hypoxic neuronal cell death after treatment of cultures with IL-1β, an affect that was mediated through increased expression and efflux of l-Glu through Sxc-. Confirming the role of Sxc-, the IL-1β-induced increase in l-Cys2 uptake (and l-Glu efflux) could be blocked with 4-S-CPG and was absent in astrocytes prepared from mice carrying a mutation in the SLC7A11 gene (Jackman et al., 2010).
Probably the strongest links between the Sxc--mediated efflux of l-Glu and CNS pathology have been made during the course of investigation on brain tumours, where Sxc- is a major contributor to the growth, survival and expansion of malignant gliomas (de Groot and Sontheimer, 2010). The high levels of xCT expression in astrocytomas and gliomas, compared with normal astrocytes, likely reflects the increased growth and metabolic rates associated with tumour cells (Ye et al., 1999). More specifically, however, the import of l-Cys2 through Sxc- is needed to meet the increasing demand for cellular GSH as the tumour outgrows its blood supply, becomes hypoxic and is challenged by the accompanying build up of ROS and NO (Ogunrinu and Sontheimer, 2010). From a therapeutic perspective, the increased GSH levels can also represent an enhanced ability of glioma cells to protect themselves from both radiation and chemotherapeutic agents (Dai et al., 2007; Liu et al., 2007). Thus, while not yet induced specifically by the inhibition of Sxc-, depletion of GSH using the synthesis inhibitor l- BSO rendered gliomas more sensitive to 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)-based chemotherapy and d-54 human glioma xenografts to radiotherapy (Lippitz et al., 1990; Allalunis-Turner et al., 1991). Even greater attention has focused on the Sxc--mediated efflux of l-Glu from the glial tumours and its impact on brain tumour aetiology. At one level, the released l-Glu can act as an excitotoxin through the over activation of ionotropic EAA receptors, induce neurodegeneration and clear the area for tumour expansion (Ye et al., 1999). Thus, both glioma cultures in vitro and implanted glioma cells in vivo have been shown to release l-Glu (Ye and Sontheimer, 1999; Behrens et al., 2000; Takano et al., 2001). Co-cultures of glioma and neuronal cells demonstrated that the released l-Glu was sufficient to induce excitotoxic damage and that the neuronal loss could be reduced by either by preventing its efflux through Sxc- (4-S-CPG or SSZ) or inhibiting its action at NMDA receptors (MK-801).
Even if the l-Glu does not reach concentrations that are excitotoxic, the excessive activation of these receptors in peritumoural regions provides an explanation for the seizures commonly observed in the early stages of the disease (Sontheimer, 2008). Lastly, the released l-Glu also is capable of mediating an autocrine/paracrine signal that enhances invasive and migratory properties through the activation of Ca2+ permeable AMPA receptors on the glioma cells themselves (Lyons et al., 2007; Lastro et al., 2008). All of these relationships have highlighted the potential of Sxc- as a therapeutic target in tumour cells, particularly gliomas. Blockers could be envisioned as acting by any number of mechanisms, some of which might be synergistic, including: metabolic inhibition, increasing oxidative stress, radio-sensitization, decreasing chemoresistance, reducing invasiveness or preventing peritumoural seizures. Indeed, SSZ (Chart 3), which was originally developed for anti-inflammatory applications before its Sxc- inhibitory actions were discovered (see Pharmacology section), has been shown to reduce GSH and tumour proliferation in both gliomas (Lyons et al., 2007) and small-cell lung cancer (Guan et al., 2009). Inhibition of Sxc- activity also inhibited cell metastasis in esophageal squamous carcinoma cells through caveolin-1/β-catenin pathway (Chen et al., 2009). Treatment of astrocytoma cells with SSZ depleted GSH levels and induced caspase-mediated apoptosis (Chung et al., 2005) Importantly, the antitumour effects of SSZ have been shown to be exclusively via inhibition of Sxc--mediated L-Cys2 uptake and GSH production as opposed to the reported anti-inflammatory effects of SSZ acting via NfkB (Chung and Sontheimer, 2009). Unfortunately SSZ, as well as most of the other Sxc- inhibitors described earlier in this review, suffer from problems related to specificity, potency and pharmacokinetic properties. Progress on mapping out how SSZ is interacting with the domains on the Sxc- binding site should, however, provide insight into optimizing new derivatives.
Conclusion
In summary, considerable progress has been made in the last few years detailing the physiological and pathological significance of both the import of l-Cys2 and the export of l-Glu through the Sxc- antiporter, particularly as associated with the CNS. Not surprisingly, this greater appreciation for the functional importance of Sxc- has also created a demand for pharmacological reagents with which to selectively manipulate its activity. What is also appealing about this challenge is that there is a need for both specific inhibitors (e.g. to reduce Sxc- function in glioma) and substrates (e.g. to increase Sxc- in addiction). A survey of presently available Sxc- ligands, unfortunately, reveals that the most potent blockers tend to lack specificity, while the more recently developed inhibitors that hold a promise for increased selectivity have yet to exhibit the desired affinity. Fortunately, all of these analogues have been of value in the development of an Sxc- pharmacophore model that is beginning to delineate the requisite domains on the transporter, including lipophilic pockets, that can be exploited in the synthesis future ligands to the benefit both specificity and potency.
Acknowledgments
This manuscript is dedicated to the memory of Professor C. Sean Esslinger. The authors wish to thank Dr Michael Braden and UM's Molecular Core Computational Facility for assistance with the structural modelling, as well as Drs John Gerdes, Michael Kavanaugh and Charles Thompson for their valued discussion. Support for much of the described research was from NIH NS R21NS067466 (RB and NN), NIH P20 RR15583 (RB, NN and SP) and the Montana MBRCT programme.
Glossary
Abbreviations
- 4-S-CPG
(S)-4-carboxyphenyglycine
- 4-S-SPG
sulphonic acid phenylglycine
- ACPA
amino-3-carboxy-5-methyl isoxazole propionic acid
- trans-ACPD
1-aminocyclopentane-trans-1,3-dicarboxylic acid
- ApcT
amino acid polyamine organo-cation transporter
- ARE
antioxidant response element
- ASC
alanine–serine–cysteine uptake systems
- β-l-ODAP
L-β-N-oxalyl-l-a,β-diaminopropionate
- BSO
buthionine sulphoximine
- CPPG
(R,S)-4-[4′-carboxyphenyl]-phenylglycine
- dbcAMP
dibutyryl cyclic AMP
- EAA
excitatory amino acid
- EAAT
excitatory amino acid transporter
- GSH
glutathione
- HAT
heteromeric amino acid transporter family
- iGluR
ionotropic glutamate receptor
- KA
kainate
- l-CysH
l-cysteine
- l-Cys2
l-cystine
- l-trans-2
4-PDC, l-trans-2,4-pyrrolidine dicarboxylate
- mGluR
metabotropic glutamate receptor
- NAC
N-acetyl-cysteine
- NACPA
S-2-naphthyl-ethyl-ACPA
- NEIH
5-naphthylethyl isoxazole-4-(2,4-dinitrophenol)hydrazone-dinitrophenol
- Nrf2
nuclear factor erythroid-2-related factor
- QA
quisqualate
- ROS
reactive oxygen species
- SAR
structure–activity-relationship
- SLC
solute carrier gene families, SSZ, sulphasalazine, aka 6-oxo-3-(2-[4-(N-pyridin-2-ylsulphamoyl)phenyl]diazenyl)cyclohexa-1,4-dienecarboxylic acid
- SxC-
System xc-
- TMDs
transmembrane domains
- TSA
R,S-4-sulphothienylglycine
- xCT
transporters subunit of Sxc- in the SLC7A11 superfamily
Conflict of interest
The authors Bridges, Natale and Patel have no conflicts of interests.
References
- Allalunis-Turner MJ, Day RS, 3rd, McKean JD, Petruk KC, Allen PB, Aronyk KE, et al. Glutathione levels and chemosensitizing effects of buthionine sulfoximine in human malignant glioma cells. J Neurooncol. 1991;11:157–164. doi: 10.1007/BF02390175. [DOI] [PubMed] [Google Scholar]
- Allen JW, Shanker G, Aschner M. Methylmercury inhibits the in vitro uptake of the glutathione precursor, cystine, in astrocytes, but not in neurons. Brain Res. 2001;894:131–140. doi: 10.1016/s0006-8993(01)01988-6. [DOI] [PubMed] [Google Scholar]
- Amen SL, Piacentine LB, Ahmad ME, Li SJ, Mantsch JR, Risinger RC, et al. Repeated N-acetyl cysteine reduces cocaine seeking in rodents and craving in cocaine-dependent humans. Neuropsychopharmacology. 2011;36:871–878. doi: 10.1038/npp.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, et al. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Augustin H, Grosjean Y, Chen K, Sheng Q, Featherstone D. Nonvesicular release of glutamate by glial xCT transporters supresses glutamate receptor clustering in vivo. J Neurosci. 2007;27:111–123. doi: 10.1523/JNEUROSCI.4770-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Hui S, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, et al. Neuroadaptations in the cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- Balazs R, Bridges RJ, Cotman CW. Excitatory Amino Acid Transmission in Health and Disease. Edn. New York: Oxford University Press; 2006. [Google Scholar]
- Bannai S. Induction of cystine and glutamate transport activity in human fibroblasts by diethyl maleate and other electrophilic agents. J Biol Chem. 1984;259:2435–2440. [PubMed] [Google Scholar]
- Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261:2256–2263. [PubMed] [Google Scholar]
- Bannai S, Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. 1980;255:2372–2376. [PubMed] [Google Scholar]
- Bannai S, Kitamura E. Role of proton dissociation in the transport of cystine and glutamate in human diploid fibroblasts in culture. J Biol Chem. 1981;256:5770–5772. [PubMed] [Google Scholar]
- Bannai S, Sato H, Ishii T, Sugita Y. Induction of cystine transport activity in human fibroblasts by oxygen. J Biol Chem. 1989;264:18480–18484. [PubMed] [Google Scholar]
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beart PM, O'Shea RD. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedingfield JS, Kemp MC, Jane DE, Tse HW, Roberts PJ, Watkins JC. Structure-activity relationships for a series of phenylglycine derivatives acting at metabotropic glutamate receptors (mGluRs) Br J Pharmacol. 1995;116:3323–3329. doi: 10.1111/j.1476-5381.1995.tb15142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens PF, Langemann H, Strohschein R, Draeger J, Hennig J. Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral microdialysis study. J Neurooncol. 2000;47:11–22. doi: 10.1023/a:1006426917654. [DOI] [PubMed] [Google Scholar]
- Brauner-Osborne H, Egebjerg J, Nielsen EO, Madsen U, Krogsgaard-Larsen P. Ligand for glutamate receptors: design and therpeutic prospects. J Med Chem. 2000;43:2609–2645. doi: 10.1021/jm000007r. [DOI] [PubMed] [Google Scholar]
- Bridges RJ, Esslinger CS. The excitatory amino acid transporters: pharmacological insights on substrate and inhibitor specificity of EAAT subtypes. Pharmacol Therap. 2005;107:271–285. doi: 10.1016/j.pharmthera.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Bridges R, Patel S. Pharmacology of glutamate transport in the CNS: substrates and inhibitors of excitatory amino acid transporters (EAATs) and the glutamate/cystine exchanger system xc. In: Napier S, editor. Topics in Med Chem. New York: Springer; 2009. pp. 187–222. [Google Scholar]
- Bridges RJ, Hearn TJ, Monaghan DT, Cotman CW. A comparison of 2-amino-4-phosphonobutyric acid (AP4) receptors and [3H]AP4 binding sites in the rat brain. Brain Res. 1986;375:204–209. doi: 10.1016/0006-8993(86)90977-7. [DOI] [PubMed] [Google Scholar]
- Bridges RJ, Kesslak JP, Nieto SM, Broderick JT, Yu J, Cotman CW. A L-[3H]glutamate binding site on glia: an autoradiographic study on implanted astrocytes. Brain Res. 1987;415:163–168. doi: 10.1016/0006-8993(87)90281-2. [DOI] [PubMed] [Google Scholar]
- Broer S, Wagner CA. Structure-function relationships of heterodimeric amino acid transporters. Cell Biochem Biophys. 2002;36:155–168. doi: 10.1385/CBB:36:2-3:155. [DOI] [PubMed] [Google Scholar]
- Burdo J, Dargusch R, Schubert D. Distribution of the cystine/glutamate antiporter system x-c in the brain, kidney, and duodenum. J Histochem Cytochem. 2006;54:549–557. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol. 2005;564:397–410. doi: 10.1113/jphysiol.2004.082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase LA, Roon RJ, Wellman L, Beitz AJ, Koerner JF. L-Quisqualic acid transport into hippocampal neurons by a cystine-sensitive carrier is required for the induction of quisqualate sensitization. Neurosci. 2001;106:287–301. doi: 10.1016/s0306-4522(01)00278-0. [DOI] [PubMed] [Google Scholar]
- Chen XL, Kunsch C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: a new therapeutic approach for the treatment of inflammatory diseases. Curr Pharm Des. 2004;10:879–891. doi: 10.2174/1381612043452901. [DOI] [PubMed] [Google Scholar]
- Chen Y, Swanson RA. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J Neurochem. 2003;84:1332–1339. doi: 10.1046/j.1471-4159.2003.01630.x. [DOI] [PubMed] [Google Scholar]
- Chen RS, Song YM, Zhou ZY, Tong T, Li Y, Fu M, et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1/beta-catenin pathway. Oncogene. 2009;28:599–609. doi: 10.1038/onc.2008.414. [DOI] [PubMed] [Google Scholar]
- Cho Y, Bannai S. Uptake of glutamate and cystine in C-6 glioma cells and in cultured astrocytes. J Neurochem. 1990;55:2091–2097. doi: 10.1111/j.1471-4159.1990.tb05800.x. [DOI] [PubMed] [Google Scholar]
- Chung WJ, Sontheimer H. Sulfasalazine inhibits the growth of primary brain tumors independent of nuclear factor-kappaB. J Neurochem. 2009;110:182–193. doi: 10.1111/j.1471-4159.2009.06129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daginakatte GC, Gadzinski A, Emnett RJ, Stark JL, Gonzales ER, Yan P, et al. Expression profiling identifies a molecular signature of reactive astrocytes stimulated by cyclic AMP or proinflammatory cytokines. Exp Neurol. 2008;210:261–267. doi: 10.1016/j.expneurol.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Dai Z, Huang Y, Sadee W, Blower P. Chemoinformatics analysis identifies cytotoxic compounds susceptible to chemoresistance mediated by glutathione and cystine/glutamate transport system xc. J Med Chem. 2007;50:1896–1906. doi: 10.1021/jm060960h. [DOI] [PubMed] [Google Scholar]
- Domercq M, Sanchez-Gomez MV, Sherwin C, Etxebarria E, Fern R, Matute C. System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol. 2007;178:6549–6556. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop J, McIlvain B, Carrick T, Jow B, Lu Q, Kowal DM, et al. Characterization of novel aryl-ether, biaryl, and fluorene aspartic acid and diaminopropionic acid analogs as potent inhibitors of the high-affinity glutamate transporter EAAT2. Mol Pharmacol. 2005;68:974–982. doi: 10.1124/mol.105.012005. [DOI] [PubMed] [Google Scholar]
- Edmonds DT, Summers CP. 14N Pure quadropole resonance in solid amino acids. J Mag Res. 1969;12:134–142. [Google Scholar]
- Espey MG, Kustova Y, Sei Y, Basile AS. Extracellular glutamate levels are chronically elevated in the brains of LP-BM5-infected mice: a mechanism of retrovirus-induced encephalopathy. J Neurochem. 1998;71:2079–2087. doi: 10.1046/j.1471-4159.1998.71052079.x. [DOI] [PubMed] [Google Scholar]
- Esslinger CS, Agarwal S, Gerdes JM, Wilson PA, Davies ES, Awes AN, et al. The substituted aspartate analogue L-beta-threo-benzyl-aspartate preferentially inhibits the neuronal excitatory amino acid transporter EAAT3. Neuropharmacology. 2005;49:850–861. doi: 10.1016/j.neuropharm.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Etoga JL, Ahmed SK, Patel S, Bridges RJ, Thompson CM. Conformationally-restricted amino acid analogues bearing a distal sulfonic acid show selective inhibition of system x(c)(-) over the vesicular glutamate transporter. Bioorg Med Chem Lett. 2010;20:2680–2683. doi: 10.1016/j.bmcl.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE. Glial solute carrier transporters in drosophila and mice. Glia. 2010 doi: 10.1002/glia.21085. Nov. DOI: 10.1002/glia.21085 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Fogal B, Hewett J, Hewett S. Interleukin-1beta potentiates neuronal injury in a variety of injury models involving energy deprivation. J Neuroimmunol. 2005;161:93–100. doi: 10.1016/j.jneuroim.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Fogal B, Li J, Lobner D, McCullough L, Hewett S. System x(c)- activity and astrocytes are necessary for interleukin-1beta-mediated hypoxic neuronal injury. J Neurosci. 2007;27:10094–10105. doi: 10.1523/JNEUROSCI.2459-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasol E, Jimenez-Vidal M, Chillaron J, Zorzano A, Palacin M. Membrane topology of system xc-light subunit reveals a re-entrant loop with substrate-restricted accessibility. J Bioll Chem. 2004;279:31228–31236. doi: 10.1074/jbc.M402428200. [DOI] [PubMed] [Google Scholar]
- Giudice A, Arra C, Turco MC. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol Biol. 2010;647:37–74. doi: 10.1007/978-1-60761-738-9_3. [DOI] [PubMed] [Google Scholar]
- Gochenauer GE, Robinson MB. Dibutyryl-cAMP (dbcAMP) upregulates astrocytic chloride-dependent L-[3H] transport and expression of both system xc- subunits. J Neurochem. 2001;78:276–286. doi: 10.1046/j.1471-4159.2001.00385.x. [DOI] [PubMed] [Google Scholar]
- Gout P, Buckley A, Simms C, Bruchovsky N. Sulfasalazine, a potent supressor of lymphoma growth by inhibition of the xc- cystine transporter: a new action of an old drug. Leukemia. 2001;15:1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- de Groot J, Sontheimer H. Glutamate and the biology of gliomas. Glia. 2010;59:1181–1189. doi: 10.1002/glia.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean Y, Grillet M, Augustin H, Ferveur JF, Featherstone DE. A glial amino-acid transporter controls synapse strength and courtship in Drosophila. Nat Neurosci. 2008;11:54–61. doi: 10.1038/nn2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Lo M, Dockery P, Mahon S, Karp CM, Buckley AR, et al. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: use of sulfasalazine. Cancer Chemother Pharmacol. 2009;64:463–472. doi: 10.1007/s00280-008-0894-4. [DOI] [PubMed] [Google Scholar]
- Guebel DV, Torres NV. Dynamics of sulfur amino acids in mammalian brain: assessment of the astrocytic-neuronal cysteine interaction by a mathematical hybrid model. Bioch Biophys Acta. 2004;1674:12–28. doi: 10.1016/j.bbagen.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L, Lai JC. Functional studies in cultured astrocytes. Methods. 1998;16:293–310. doi: 10.1006/meth.1998.0686. [DOI] [PubMed] [Google Scholar]
- Hosoya K, Tomi M, Ohtsuki S, Takanaga H, Saeki S, Kanai Y, et al. Enhancement of L-cystine transport activity and its relation to xCT gene induction at the blood-brain barrier by diethyl maleate treatment. J Pharmacol Exp Ther. 2002;302:225–231. doi: 10.1124/jpet.302.1.225. [DOI] [PubMed] [Google Scholar]
- Hu RG, Lim J, Donaldson PJ, Kalloniatis M. Characterization of the cystine/glutamate transporter in the outer plexiform layer of the vertebrate retina. Eur J Neurosci. 2008;28:1491–1502. doi: 10.1111/j.1460-9568.2008.06435.x. [DOI] [PubMed] [Google Scholar]
- Huang Y, Dai Z, Barbacioru C, Sadée W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005;65:7446–7454. doi: 10.1158/0008-5472.CAN-04-4267. [DOI] [PubMed] [Google Scholar]
- Hunston R, Gerothanassis I, Lauterwein J. Oxygen-17 nuclear magnetic resonance studies of some enriched amino acids. Org Mag Res. 1982;18:120–121. [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- Jackman NA, Uliasz TF, Hewett JA, Hewett SJ. Regulation of system x(c)(-)activity and expression in astrocytes by interleukin-1beta: implications for hypoxic neuronal injury. Glia. 2010;58:1806–1815. doi: 10.1002/glia.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Rad Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- Jimenez-Vidal M, Gasol E, Zorzano A, Nunes V, Palacin M, Chillaron J. Thiol modification of cysteine 327 in the eighth transmembrane domain of the light subunit xCT of the heteromeric cystine/glutamate antiporter suggests close proximity to the substrate binding site/permeation pathway. J Biol Chem. 2004;279:11214–11221. doi: 10.1074/jbc.M309866200. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Lalumiere RT, Knackstedt L, Shen H. Glutamate transmission in addiction. Neuropharmacology. 2009;56(Suppl. 1):169–173. doi: 10.1016/j.neuropharm.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA. The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 2004;447:469–479. doi: 10.1007/s00424-003-1146-4. [DOI] [PubMed] [Google Scholar]
- Kandil S, Brennan L, McBean GJ. Glutathione depletion causes a JNK and p38MAPK-mediated increase in expression of cystathionine-gamma-lyase and upregulation of the transsulfuration pathway in C6 glioma cells. Neurochem Int. 2010;56:611–619. doi: 10.1016/j.neuint.2010.01.004. [DOI] [PubMed] [Google Scholar]
- Kato S, Ishita S, Sugawara K, Mawatari K. Cystine/glutamate antiporter expression in retinal muller glial cells: implications for DL-alpha-aminoadipate toxicity. Neuroscience. 1993;57:473–482. doi: 10.1016/0306-4522(93)90080-y. [DOI] [PubMed] [Google Scholar]
- Kessler M, Baudry M, Lynch G. Use of cystine to distinguish glutamate binding from glutamate binding sequestration. Neurosci Lett. 1987;81:221–226. doi: 10.1016/0304-3940(87)91002-0. [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, Kalivas PW. Glutamate and reinstatement. Curr Opin Pharmacol. 2009;9:59–64. doi: 10.1016/j.coph.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Larowe S, Mardikian P, Malcolm M, Upadhyaya H, Hedden S, et al. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2008;65:841–845. doi: 10.1016/j.biopsych.2008.10.040. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, LaRowe S, Mardikian P, Malcolm R, Upadhyaya H, Hedden S, et al. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65:841–845. doi: 10.1016/j.biopsych.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lastro M, Kourtidis A, Farley K, Conklin DS. xCT expression reduces the early cell cycle requirement for calcium signaling. Cell Signal. 2008;20:390–399. doi: 10.1016/j.cellsig.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, et al. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. 2009;111:332–343. doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- Li L, Lim J, Jacobs MD, Kistler J, Donaldson PJ. Regional differences in cystine accumulation point to a sutural delivery pathway to the lens core. Invest Ophthalmol Vis Sci. 2007;48:1253–1260. doi: 10.1167/iovs.06-0861. [DOI] [PubMed] [Google Scholar]
- Lim J, Lam YC, Kistler J, Donaldson PJ. Molecular characterization of the cystine/glutamate exchanger and the excitatory amino acid transporters in the rat lens. Invest Opthalmol Vis Sci. 2005;46:2869–2877. doi: 10.1167/iovs.05-0156. [DOI] [PubMed] [Google Scholar]
- Lim J, Li L, Kistler J, Donaldson PJ. Mapping of glutathione and its precursor amino acids reveals a role for GLYT2 in glycine uptake in the lens core. Invest Ophth Vis Sci. 2007;48:5142–5151. doi: 10.1167/iovs.07-0649. [DOI] [PubMed] [Google Scholar]
- Limon-Pacheco JH, Hernandez NA, Fanjul-Moles ML, Gonsebatt ME. Glutathione depletion activates mitogen-activated protein kinase (MAPK) pathways that display organ-specific responses and brain protection in mice. Free Rad Biol Med. 2007;43:1335–1347. doi: 10.1016/j.freeradbiomed.2007.06.028. [DOI] [PubMed] [Google Scholar]
- Lippitz BE, Halperin EC, Griffith OW, Colvin OM, Honore G, Ostertag CB, et al. L-buthionine-sulfoximine-mediated radiosensitization in experimental interstitial radiotherapy of intracerebral D-54 MG glioma xenografts in athymic mice. Neurosurgery. 1990;26:255–260. doi: 10.1097/00006123-199002000-00012. [DOI] [PubMed] [Google Scholar]
- Liu R, Blower PE, Pham AN, Fang J, Dai Z, Wise C, et al. Cystine-glutamate transporter SLC7A11 mediates resistance to geldanamycin but not to 17-(allylamino)-17-demethoxygeldanamycin. Mol Pharmacol. 2007;72:1637–1646. doi: 10.1124/mol.107.039644. [DOI] [PubMed] [Google Scholar]
- Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67:9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBean GJ. Cerebral cystine uptake: a tale of two transporters. TIPS. 2002;23:299–302. doi: 10.1016/s0165-6147(02)02060-6. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch. 2004;447:784–795. doi: 10.1007/s00424-003-1117-9. [DOI] [PubMed] [Google Scholar]
- Madayag A, Kau KS, Lobner D, Mantsch JR, Wisniewski S, Baker DA. Drug-induced plasticity contributing to heightened relapse susceptibility: neurochemical changes and augmented reinstatement in high-intake rats. J Neurosci. 2010;30:210–217. doi: 10.1523/JNEUROSCI.1342-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavencamp TL, Rhoderick JF, Bridges RJ, Esslinger CS. Synthesis and preliminary pharmacological evaluation of novel derivatives of L-β-threo-benzylaspartate as inhibitors of the neuronal glutamate transporter EAAT3. Bioorg Med Chem. 2008;16:7740–7748. doi: 10.1016/j.bmc.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan A, Pendyam S, Kalivas PW, Nair SS. Molecular diffusion model of neurotransmitter homeostasis around synapses supporting gradients. Neural Comput. 2011;23:984–1014. doi: 10.1162/NECO_a_00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- Moran M, McFarland K, Melendez RI, Seamans JK. Cystine/Glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25:6389–6393. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Muiswinkel FL, Kuiperij HB. The Nrf2-ARE signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord. 2005;4:267–281. doi: 10.2174/1568007054038238. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- Nicholls DG, Sihra TS, Sanches-Prieto J. Calcium-dependent and -independent release of glutamate from synaptosomes monitored by continuous fluorometry. J Neurochem. 1987;1987:50–57. doi: 10.1111/j.1471-4159.1987.tb03393.x. [DOI] [PubMed] [Google Scholar]
- Ogunrinu TA, Sontheimer H. Hypoxia increases the dependence of glioma cells on glutathione. J Biol Chem. 2010;285:37716–37724. doi: 10.1074/jbc.M110.161190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco R, Oliva H, Martinez-Navio JM, Climent N, Ciruela F, Gatell JM, et al. Glutamate released by dendritic cells as a novel modulator of T cell activation. J Immunol. 2006;177:6695–6704. doi: 10.4049/jimmunol.177.10.6695. [DOI] [PubMed] [Google Scholar]
- Pacheco R, Gallart T, Lluis C, Franco R. Role of glutamate on T-cell mediated immunity. J Neuroimmunol. 2007;185:9–19. doi: 10.1016/j.jneuroim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Palacin M, Nunes V, Jimenez-Vidal M, Font-Llitjos M, Gasol E, Pineda M, et al. The genetics of heteromeric amino acid transporters. Physiology. 2005;20:112–124. doi: 10.1152/physiol.00051.2004. [DOI] [PubMed] [Google Scholar]
- Patel SA, Warren BA, Rhoderick JF, Bridges RJ. Differentiation of substrate and non-substrate inhibitors of transport system x(c)(-): an obligate exchanger of L-glutamate and L-cystine. Neuropharmacology. 2004;46:273–284. doi: 10.1016/j.neuropharm.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Patel SA, Rajale T, O'Brien E, Burkhart DJ, Nelson JK, Twamley B, et al. Isoxazole analogues bind the System xc- transporter: structure-activity relationship and pharmacophore model. Bioorg Med Chem. 2010;18:202–213. doi: 10.1016/j.bmc.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Nat Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piani D, Fontana A. Involvement of the cystine transport system xc- in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol. 1994;152:3578–3585. [PubMed] [Google Scholar]
- Pin JP, Bockaert J, Recasen M. The Ca++/Cl- dependent L-[3H]glutamate binding: a new receptor or a particular transport process. FEBS Lett. 1984;175:31–36. doi: 10.1016/0014-5793(84)80563-3. [DOI] [PubMed] [Google Scholar]
- Prochaska HJ, De Long MJ, Talalay P. On the mechanisms of induction of cancer-protective enzymes: a unifying proposal. Proc Natl Acad Sci U S A. 1985;82:8232–8236. doi: 10.1073/pnas.82.23.8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Cahill JM, Liu J, Kuang X, Liu N, Scofield VL, et al. Activation of transcription factor Nrf-2 and its downstream targets in response to moloney murine leukemia virus ts1-induced thiol depletion and oxidative stress in astrocytes. J Virol. 2004;78:11926–11938. doi: 10.1128/JVI.78.21.11926-11938.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reig N, Chillaron J, Bartoccioni P, Fernandez E, Bendahan A, Zorzano A, et al. The light subunit of system b(o,+) is fully functional in the absence of the heavy subunit. EMBO J. 2002;21:4906–4914. doi: 10.1093/emboj/cdf500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein J, Patel S, Regan M, Haenggeli C, Huang Y, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Sagara J, Miura K, Bannai S. Maintenance of neuronal glutathione by glial cells. J Neurochem. 1993;61:1672–1676. doi: 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. Electrophilic response element-mediated induction of the cystine/gluamate exchange transporter gene expression. J Biol Chem. 2002;274:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- Sato H, Fujiwara K, Sagara J, Bannai S. Induction of cystine, transport activity in mouse peritoneal macrophages by bacterial lipopolysaccharide. Biochem J. 1995;310:547–551. doi: 10.1042/bj3100547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, et al. Distribution of the cystine/glutamate exchange transporter. system xc-, in the mouse brain. J Neurosci. 2002;22:8028–8033. doi: 10.1523/JNEUROSCI.22-18-08028.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophy Res Comm. 2004;325:109–116. doi: 10.1016/j.bbrc.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Seib TM, Patel S, Bridges RJ. Regulation of the System x(-) (C) cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia. 2011 doi: 10.1002/glia.21176. DOI: 10.1002/glia.21176 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. Structure and mechanism of a Na+-independent amino acid transporter. Science. 2009;325:1010–1014. doi: 10.1126/science.1176088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih A, Erb H, Sun X, Toda S, Kalivas P, Murphy T. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006a;41:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Fernandes HB, Choi FY, Kozoriz MG, Liu Y, Li P, et al. Policing the police: astrocytes modulate microglial activation. J Neurosci. 2006b;26:3887–3888. doi: 10.1523/JNEUROSCI.0936-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, et al. Characterization of novel L-threo-β-benzyloxyaspartate derivatives, potent blockers of glutamate transporters. Mol Pharmacol. 2004;65:1008–1015. doi: 10.1124/mol.65.4.1008. [DOI] [PubMed] [Google Scholar]
- Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J Neurochem. 2008;105:287–295. doi: 10.1111/j.1471-4159.2008.05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer SR, Xue LA, Klenz EM, Talalay P. The potency of inducers of NAD(P)H:(quinone-acceptor) oxidoreductase parallels their efficiency as substrates for glutathione transferases. Structural and electronic correlations. Biochem J. 1991;273:711–717. doi: 10.1042/bj2730711. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Tamba M, Bannai S, Sato H. Induction of cystine/glutamate transporter in bacterial lipopolysaccharide induced endotoxemia in mice. J Inflamm (Lond) 2007;4:20. doi: 10.1186/1476-9255-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Lin J, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001;7:1010–1015. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- Talalay P. Mechanisms of induction of enzymes that protect against chemical carcinogenesis. Adv Enzyme Regul. 1989;28:237–250. doi: 10.1016/0065-2571(89)90074-5. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med. 2009;11:e17. doi: 10.1017/S1462399409001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. CATs and HATs : the SLC7 family of amino acid transporters. Eur J Physiol. 2003;447:532–542. doi: 10.1007/s00424-003-1086-z. [DOI] [PubMed] [Google Scholar]
- Wang XF, Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem. 2000;74:1434–1442. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Characterization of L-glutamic acid transport by glioma cells in culture, evidence for sodium-independent, chloride-dependent high affinity uptake. J Neurosci. 1984;4:2237–2246. doi: 10.1523/JNEUROSCI.04-09-02237.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr O, Takahashi M, Attwell D. Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J Physiol. 1999;514.3:783–793. doi: 10.1111/j.1469-7793.1999.783ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren BA, Patel SA, Nunn PB, Bridges RJ. The Lathyrus excitotoxin B-N-oxalyl-L-α, β-diaminopropionic acid is a substrate of the L-cystine/L-glutamate exchanger system xc. Toxicol App Pharmacol. 2004;200:83–92. doi: 10.1016/j.taap.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Watabe M, Aoyama K, Nakaki T. Regulation of glutathione synthesis via interaction between glutamate transport-associated protein 3-18 (GTRAP3-18) and excitatory amino acid carrier-1 (EAAC1) at plasma membrane. Mol Pharmacol. 2007;72:1103–1110. doi: 10.1124/mol.107.039461. [DOI] [PubMed] [Google Scholar]
- Waxman E, Lynch D. N-methyl-D-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11:37–49. doi: 10.1177/1073858404269012. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Shen H, Baker DA, Kalivas PW. Inhibition of non-vesicular glutamate release by group III metabotropic glutamate receptors in the nucleus accumbens. J Neurochem. 2003;87:1204–1212. doi: 10.1046/j.1471-4159.2003.02093.x. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl – dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–4391. [PubMed] [Google Scholar]
- Ye Z, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci. 1999;19:10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Li Y, Chen RS, He X, Yang L, Li W. Overexpression of xCT induces up-regulation of 14-3-3beta in Kaposi's sarcoma. Biosci Rep. 2010;30:277–283. doi: 10.1042/BSR20090163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. ASCT-1 is a neutral amino acid exchanger with chloride channel activity. J Biol Chem. 1996a;271:27991–27994. doi: 10.1074/jbc.271.45.27991. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Interaction of L-cysteine with a human excitatory amino acid transporter. J Physiol. 1996b;493:419–423. doi: 10.1113/jphysiol.1996.sp021393. [DOI] [PMC free article] [PubMed] [Google Scholar]