

Abstract
The mechanisms underlying the ability of nonsteroidal anti-inflammatory drugs (NSAIDs) to cause ulceration in the stomach and proximal duodenum are well understood, and this injury can largely be prevented through suppression of gastric acid secretion (mainly with proton pump inhibitors). In contrast, the pathogenesis of small intestinal injury induced by NSAIDs is less well understood, involving more complex mechanisms than those in the stomach and proximal duodenum. There is clear evidence for important contributions to NSAID enteropathy of enteric bacteria, bile and enterohepatic recirculation of the NSAID. There is no evidence that suppression of gastric acid secretion will reduce the incidence or severity of NSAID enteropathy. Indeed, clinical data suggest little, if any, benefit. Animal studies suggest a significant exacerbation of NSAID enteropathy when proton pump inhibitors are co-administered with the NSAID. This worsening of damage appears to be linked to changes in the number and types of bacteria in the small intestine during proton pump inhibitor therapy. The distinct mechanisms of NSAID-induced injury in the stomach/proximal duodenum versus the more distal small intestine likely dictate distinct strategies for prevention.
Keywords: gastrointestinal pharmacology, anti-inflammatory drugs, inflammation, peptic ulceration, eicosanoids, nitric oxide, analgesic drugs
Introduction
The first clear demonstration that a nonsteroidal anti-inflammatory drug (NSAID) could trigger bleeding in the stomach was provided by the gastroscopy study of Douthwaite and Lintott (1938). Since that time, the gastrointestinal (GI) damaging effects of NSAIDs have been very well characterized, and recognized as the major limitation to the use of this class of drugs for treating inflammatory conditions (Wallace, 2008). Despite many efforts to develop ‘GI-sparing’ NSAIDs, the bleeding and ulceration caused by these drugs remains a major clinical concern (McCarthy, 2009; Scarpignato and Hunt, 2010). For example, in a prospective analysis of over 18 000 patients in which adverse drug reactions (ADRs) were the cause of admission to hospital in Britain, NSAIDs were the most common class of drug associated with ADRs (30%) and NSAID-associated GI bleeding and ulceration accounted for 61% of ADR-related deaths (Pirmohamed et al., 2004).
Over the past few decades, a great deal has been learned about the pathogenesis of NSAID gastropathy. In particular, the critical role of suppression of mucosal PG synthesis in triggering mucosal ulceration has been clearly demonstrated (Whittle, 1981; Rainsford and Willis, 1982; Wallace et al., 2000). There have also been significant advances in the treatment and prevention of NSAID-induced damage to the stomach and duodenum. Histamine H2 receptor antagonists (H2RAs) and proton pump inhibitors (PPIs) (particularly the latter) have become mainstay prophylactic therapies. Several new drugs are in development that consist of both an NSAID and an inhibitor of gastric acid secretion (PPI or H2RA) in the same tablet, the goal being reduced gastric damage. One such drug, Vimovo, was launched recently (naproxen + esomeprazole).
It became clear from the studies of post-mortem samples by Bjarnason and colleagues (1993) that NSAID use is also associated with significant damage to the more distal regions of the small intestine (i.e. distal to the ligament of Treitz). Because this damage occurs in regions beyond the reach of typical endoscopic examinations, in contrast to the gastroduodenal damage, NSAID enteropathy has been under-examined or even ignored in most clinical studies. Also, simple and reliable surrogate markers for NSAID enteropathy are lacking. A number of recent video capsule endoscopy studies have demonstrated the high incidence (55–75%) of small intestinal damage in healthy volunteers taking NSAIDs plus a PPI over a 2 week period (Goldstein et al., 2005; Graham et al., 2005; Maiden et al., 2005; Fujimora et al., 2010). It is noteworthy that this high incidence of intestinal damage was observed in a group considered to be at low risk for NSAID-related GI damage, with a short period of treatment and with co-administration of a ‘gastro-protective’ drug.
NSAIDs reproducibly elicit enteropathy in rodents that is similar in appearance to that observed in humans, and these models have proven very useful for gaining a better understanding of the pathogenesis of this condition. There is no evidence to suggest that suppression of gastric acid secretion would have any positive impact with respect to the incidence of NSAID-induced enteropathy (Hunt et al., 2009). Indeed, some evidence suggests the opposite (Wallace et al., 2011). This raises questions about the common practice of co-prescribing a PPI together with an NSAID (or their combined use in a single pill).
In this review, the mechanisms through which NSAIDs produce damage to the upper GI tract (i.e. stomach and proximal duodenum) and to more distal regions of the intestine are reviewed. The distinct pathogenic mechanisms in these two regions likely necessitate different strategies for prevention of ulceration and bleeding.
Pathogenesis of gastroduodenal damage
The ability of different NSAIDs to cause gastroduodenal damage correlates very well with their ability to inhibit mucosal PG synthesis (Whittle, 1981; Rainsford and Willis, 1982). While most PGs produced by the healthy stomach are derived from COX-1, there is ample evidence that COX-2-derived PGs also play a key role in gastroduodenal mucosal defence (Wallace and Devchand, 2005), as well as in the repair of mucosal injury throughout the GI tract (Reuter et al., 1996; Mizuno et al., 1997; Ma et al., 2002). When the gastric mucosa is inflamed, COX-2 expression is markedly increased and the contribution of COX-2-derived eicosanoids to mucosal defence is much greater (Souza et al., 2003). NSAIDs that selectively inhibit either COX-1 or COX-2 cause less gastroduodenal damage than NSAIDs that inhibit both enzymes (Wallace et al., 2000). In clinical use, selective COX-2 inhibitors reduce, but do not eliminate, gastroduodenal damage (Laine et al., 2003a; Lanas et al., 2007). However, this beneficial effect is lost if these drugs are co-administered with aspirin, even at low doses (Laine et al., 2003b). Low-dose aspirin is often prescribed to patients taking selective COX-2 inhibitors in an attempt to reduce the incidence of serious cardiovascular events that have been associated with the use of the latter drugs (Kearney et al., 2006).
Why does suppression of mucosal PG synthesis contribute to mucosal injury? PGs have been found to enhance or stimulate many aspects of mucosal defence (Wallace, 2008). For example, PGs stimulate bicarbonate and mucus secretion by the epithelium, which contributes to the resistance of these cells to damage induced by luminal acid and pepsin, as well as promoting repair of damaged epithelium. The major PGs synthesized by the gastroduodenal mucosa (PGE2 and PGI2) are potent vasodilators, increasing mucosal blood flow when epithelial barrier function is compromised. The increased blood flow helps to neutralize back-diffusing acid and to dilute and remove any potentially toxic substances that have entered the subepithelial space. Maintenance of mucosal blood flow during times when the epithelium is damaged is particularly important in terms of facilitating the rapid repair of the damaged tissue, before it progresses deeper into the mucosa. PGs play a particularly important role in regulating mucosal blood flow at those times (Wallace and McKnight, 1990). PGs have also been shown to play a key role in the promotion of ulcer healing. These effects are likely related primarily to suppression of COX-1-derived PG synthesis (indeed, this has been demonstrated with respect to the decrease in gastric blood flow that occurs following NSAID administration) (Wallace et al., 2000). Suppression of PG synthesis also leads to leukocyte (mainly neutrophil) adherence to the vascular endothelium in the GI microcirculation, and this has been shown to result in endothelial damage (Asako et al., 1992a,b; Wallace, 1993; McCafferty et al., 1995). This event appears to be one of the earliest steps in the pathogenesis of mucosal injury (Wallace et al., 1990), and is a consequence of inhibition of COX-2 activity (Wallace et al., 2000). Moreover, prevention of this event results in prevention of mucosal damage (Wallace et al., 1990, 1991, 1993). The increased leukocyte-endothelial adhesion is related to up-regulation of both endothelial (e.g. inter-cellular adhesion molecule-1, P-selectin) and leukocyte (CD11/18) adhesion molecule expression (Andrews et al., 1994; Fiorucci et al., 2005), which appears to occur in part because of increased release of leukotriene B4 (Asako et al., 1992a) and TNF-α (Santucci et al., 1994; Appleyard et al., 1996).
While suppression of gastroduodenal PG synthesis is an important element of the pathogenesis of mucosal injury, such injury can be prevented through delivery of exogenous PGs (Robert et al., 1979) or of other mediators of mucosal defence, most notably NO (MacNaughton et al., 1989; Wallace et al., 1994) or hydrogen sulphide (Fiorucci et al., 2005; Wallace et al., 2007; 2010;). Both NO and hydrogen sulphide are potent inhibitors of leukocyte adherence to the vascular endothelium (Kubes et al., 1991; Zanardo et al., 2006), which may be one of the key reasons that they can reduce or prevent NSAID-induced gastroduodenal damage. They are also vasodilators, and can therefore prevent the decrease in gastric blood flow that is usually seen following administration of an NSAID.
Selective COX-2 inhibitors cause less upper GI injury/bleeding than non-selective COX inhibitors in part because they do not inhibit platelet aggregation. However, when co-administered with anti-thrombotic doses of aspirin, the benefit of a selective COX-2 inhibitor over a conventional NSAID in terms of upper GI ulceration and bleeding is lost (Laine et al., 2003b). While this may be partly because of the suppression of platelet aggregation by the aspirin, there is another reason for this interesting interaction between aspirin and selective COX-2 inhibitors. Aspirin irreversibly acetylates COX-1 and COX-2, preventing the conversion of arachidonic acid to PGH2. However, acetylated COX-2 can still metabolize arachidonic acid to 15(R)-hydroxyepitetraenoic acid, which in turn can be converted by 5-lipoxygenase to 15(R)-epi-lipoxin A4. 15(R)-epi-lipoxin A4 and its epimer, lipoxin A4, are potent anti-inflammatory substances, suppressing neutrophil adherence to the endothelium (Claria and Serhan, 1995; Serhan and Oliw, 2001; Serhan et al., 2007). Moreover, lipoxin A4 exhibits potent protective effects in the stomach (Fiorucci et al., 2002; Souza et al., 2003). When a COX-2 inhibitor is co-administered with aspirin, the conversion of arachidonic acid to 15(R)-hydroxyepitetraenoic acid (and therefore the production of the gastro-protective 15(R)-epi-lipoxin A4) is blocked. The result is more severe gastric damage than is seen with aspirin alone or with the selective COX-2 inhibitor alone (Fiorucci et al., 2002; 2003; Souza et al., 2003).
While there is considerable evidence that suppression of mucosal PG synthesis is central to the mechanism of action of NSAIDs in damaging the gastroduodenal mucosa (Wallace, 2008), there remains debate with respect to the contribution of topical irritant effects of these drugs to mucosal injury (Somasundaram et al., 2000; Lichtenberger, 2001). Topical exposure of the mucosa to NSAIDs (particularly acidic ones) can result in formation of erosions, but this mucosal injury does not necessarily progress to clinically significant ulcers and bleeding in the absence of concurrent suppression of mucosal PG synthesis. Indeed, there is evidence that topical exposure of the gastroduodenal mucosa to an NSAID is not necessary for ulcer formation. Parenteral administration of NSAIDs or oral administration of NSAID prodrugs cause clinically significant ulcer formation at rates similar to orally administered NSAIDs (Estes et al., 1993; Henry et al., 1993; Wallace and McKnight, 1993).
The effectiveness of H2RAs and PPIs in reducing the incidence of NSAID gastropathy strongly suggests a key role of acid in the pathogenesis of this injury. When mucosal defence is weakened, through suppression of mucosal PG synthesis, the tissue is less able to resist the damaging effects of acid. In areas of superficial mucosal injury, a microenvironment of relatively high pH, formed from mucus and coagulation proteins (the ‘mucoid cap’), can be maintained which is conducive to restitution of the epithelium (Wallace and McKnight, 1990). However, if mucosal blood flow is significantly reduced (such as through administration of an NSAID or a vasoconstrictor), the pH within the mucoid cap rapidly falls as acid penetrates into the mucosa, causing further damage and bleeding (Wallace and McKnight, 1990). The latter effect is attributable in part to the inability of platelets to aggregate at a pH of less than 4 (Green et al., 1978).
Pathogenesis of small intestinal injury
The pathogenesis of NSAID enteropathy is distinct from that of NSAID gastropathy (Wallace, 2008). It is also more challenging to investigate in a controlled manner because the manifestation of the damage takes place over a much longer period of time than the lesions that develop in the stomach. Inhibition of PG synthesis by NSAIDs renders the intestinal mucosa more susceptible to injury and less able to undergo repair (Reuter et al., 1997; Tanaka et al., 2002). As in the stomach (Davies et al., 1997b), inhibition of COX-1 activity leads to a rapid, compensatory increase in expression of COX-2, and suppression of both enzymes leads to exacerbation of tissue injury (Tanaka et al., 2002). Unlike the stomach, however, there does not appear to be a primary role of COX inhibition in the mechanism of NSAID-induced enteropathy (Reuter et al., 1997). Thus, intestinal PG synthesis can be markedly suppressed, but ulceration and bleeding does not necessarily occur, and when it does occur, it is not temporally synchronized with the suppression of intestinal PG synthesis (Whittle, 1981; Reuter et al., 1997). Likewise, increases in intestinal epithelial permeability occur within 12 h of administration of an NSAID to a rat, but this does not necessarily predict the development of intestinal ulcers (Reuter et al., 1997). The extent to which the increase in permeability is due to the topical irritant effects of the NSAID versus the suppression of PG synthesis is not clear.
A critical feature of some NSAIDs that appears to be essential for induction of significant intestinal ulceration is their re-absorption in the ileum and subsequent secretion back into the duodenum via the enterohepatic circulation (Figure 1). NSAIDs that do not undergo enterohepatic recirculation do not cause significant intestinal damage in animal models (Kent et al., 1969; Reuter et al., 1997). Ligating the bile duct to prevent enterohepatic recirculation of an NSAID prevents intestinal damage (Kent et al., 1969). Of course, ligating the bile duct also prevents the entry of bile into the small intestine, and this may also contribute to the observed prevention of NSAID-induced damage. NSAIDs can themselves cause damage to intestinal epithelial cells (Somasundaram et al., 2000; Zhou et al., 2010), and these damaging effects are enhanced when the NSAIDs are combined with bile (Zhou et al., 2010). Uncoupling of oxidative phosphorylation has been suggested as an underlying mechanism for NSAID-induced epithelial damage (Somasundaram et al., 2000), as has disruption of the lipid bilayer of epithelial cells (Zhou et al., 2010). Whereas leukocyte adherence to the vascular endothelium is a critical event in the pathogenesis of NSAID gastropathy, this does not appear to be the case in the intestine, although infiltrating neutrophils likely contribute to tissue injury once the process of ulceration has started (Antoon and Perry, 1997; Konaka et al., 1999). There is evidence for a contribution of TNF-α to NSAID enteropathy (Santucci et al., 1994; Appleyard et al., 1996), but its effects are independent of induction of leukocyte adherence to the vascular endothelium (Appleyard et al., 1996).
Figure 1.
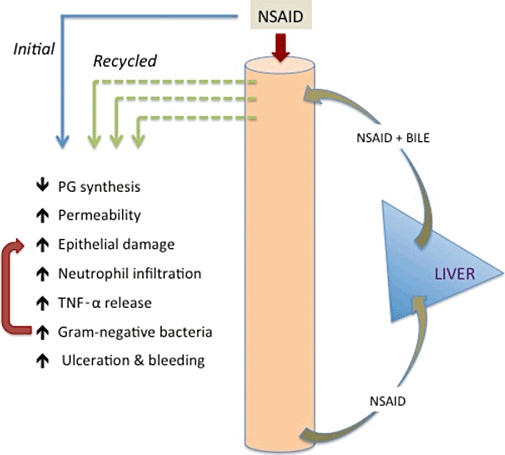
Pathogenesis of NSAID enteropathy. NSAIDs that undergo enterohepatic recirculation exhibit much greater capacity to induce small intestinal injury than those that are not absorbed and delivered into the upper small intestine in bile. Inhibition of PG synthesis and increased intestinal permeability can be observed with all NSAIDs, but only those that undergo enterohepatic recirculation will cause significant ulceration. It is likely that NSAIDs, particularly when combined with bile, can directly damage epithelial cells in the intestine. Neutrophil infiltration and release of TNF-α contribute to injury, but the increase in gram-negative bacteria in the small intestine is particularly important for the generation of ulcers. Thus, broad-spectrum antibiotics can prevent experimental NSAID enteropathy, and germ-free rodents do not develop intestinal ulcers when given NSAIDs.
Administration of NSAIDs to rodents results in profound changes in the numbers and types of enteric bacteria, and this appears to contribute significantly to the development of small intestinal ulcers (Figure 1). In particular, the numbers of gram-negative bacteria increase substantially (Reuter et al., 1997; Hagiwara et al., 2004; Wallace et al., 2011). Treatment with broad-spectrum antibiotics can reduce the severity of NSAID enteropathy (Kent et al., 1969; Konaka et al., 1999). Germ-free rats and mice do not develop NSAID enteropathy (Robert and Asano, 1977; Uejima et al., 1996). When germ-free mice were colonized with E. coli or Eubacterium limosum they became susceptible to NSAID enteropathy, but when colonized with Bifidobacter adolescentis or Lactobacillus acidophilus (both considered as ‘probiotics’) they did not (Uejima et al., 1996). A role for gram-negative bacteria in the development of NSAID enteropathy is further supported by the observation that mice genetically lacking toll-like receptor 4 (the ‘receptor’ for bacterial endotoxin) did not develop intestinal ulceration when administered an NSAID (Watanabe et al., 2008). Importantly, the changes in intestinal bacteria are only seen with NSAIDs that undergo enterohepatic recirculation (Reuter et al., 1997). This may be related to the repeated injury to the epithelium that is induced as the NSAID is recirculated through the intestine. It is also possible that the ability of some bacteria to deconjugate bile may be an important factor in promoting intestinal injury (Shindo et al., 1998).
In contrast to the stomach, there is no evidence that gastric acid plays an important role in the pathogenesis of NSAID-induced damage distal to the ligament of Treitz (Hunt et al., 2009). As discussed in more details below, chronic suppression of gastric acid secretion has been shown to result in overgrowth of bacteria in the small intestine, which could increase the severity of NSAID enteropathy (Wallace et al., 2011).
Prevention strategies: gastroduodenal injury
Current
Most patients being treated chronically with NSAIDs would also be co-prescribed a PPI, or in some cases, an H2RA. There is extensive evidence that these agents can significantly reduce the incidence and severity of NSAID-induced gastroduodenal damage, as well as accelerating the healing of such damage (Taha et al., 1996; Yeomans et al., 1998; Scheiman et al., 2006; Scarpignato and Hunt, 2010). It is likely that the use of PPIs to protect the stomach and duodenum from NSAID-induced damage will increase now that many PPIs are off-patent and there are a number of combination tablets of an NSAID and a PPI (or H2RA) coming to market.
Chronic use of drugs that markedly suppress gastric acid secretion has been associated with a number of problems, including bacterial overgrowth (Williams and McColl, 2006; Lombardo et al., 2010). Use of PPIs has also been associated with a significant increase in the incidence of various infections, most notably Clostridium difficile (McCarthy, 2010). Absorption of calcium, iron, magnesium and vitamin B12 can be impaired, and there are several published reports of increased rates of osteoporosis-associated bone fractures in patients chronically treated with PPIs (Ito and Jensen, 2010). As discussed in more detail below, recent animal studies suggest that PPI-induced changes in small intestinal bacteria may contribute to a significant worsening of NSAID enteropathy (Wallace et al., 2011).
The PGE1 analogue, misoprostol, has been shown to be effective in reducing the incidence of gastroduodenal ulcers in NSAID users (Graham et al., 1993). A high incidence of adverse effects, most notably diarrhoea, has limited the use of this drug.
NSAID prodrugs have been developed, and continue to be developed, based on the notion that if the drug passes through the stomach without inhibiting PG synthesis, it will not cause ulceration. These prodrugs are typically metabolized in the liver to yield the active drug (which will inhibit COX activity). Clinical studies suggest that the incidence of significant ulcers and bleeding is not markedly reduced with NSAID prodrugs versus conventional NSAIDs (Henry et al., 1993), and this is reflected in the relatively small market share drugs such as sulindac have captured (Graham et al., 1985).
In development
A number of NSAID prodrugs and PPI-like drugs are in development, aimed at reducing the incidence of NSAID-induced gastroduodenal injury (see Scarpignato and Hunt 2010 and Wallace and Ferraz, 2010 for recent reviews). Another approach being investigated is the non-covalent linking of phosphatidylcholine to an NSAID, which in animal studies has been shown to markedly reduce the gastric damaging effects of the NSAID while not impairing the anti-inflammatory and analgesic effects (Lichtenberger et al., 2009). Two clinical studies on phosphatidylcholine-associated NSAIDs have been reported. One was a double-blind, 6 week, endoscopic study comparing ibuprofen to phosphatidylcholine-associated ibuprofen in patients with osteoarthritis. The two drugs exhibited similar anti-inflammatory efficacy, but there was no significant difference in GI safety (Lanza et al., 2008). The other study was an endoscopic examination of the gastroduodenal injury elicited by aspirin (325 mg) or an equimolar dose of phosphatidylcholine-associated aspirin, given once daily for 7 days. In this randomized, single-blind study, about half as many individuals in the phosphatidylcholine-associated aspirin group developed gastric damage as in the aspirin group, and the incidence of duodenal injury in the phosphatidylcholine-associated aspirin group was only about one-third that in the aspirin group (statistically significant in both the stomach and duodenum) (Cryer et al., 2011). Unfortunately, no measure of anti-thrombotic efficacy was included in this study, so there was no confirmation that phosphatidylcholine-associated aspirin exerted comparable effects on platelet function as was achieved with aspirin.
Nitric oxide-releasing NSAIDs have been extensively studied for several years, and one (NO-releasing naproxen) has been evaluated in advanced clinical trials. However, this drug has not yet achieved regulatory approval, because the safety advantages over the parent drug (naproxen) have not been sufficiently demonstrated. Also in development are NSAID derivatives that release hydrogen sulphide, which in animal studies do not produce gastric or intestinal damage despite producing comparable anti-inflammatory effects to the parent NSAIDs (Wallace, 2007; Wallace et al., 2007; 2010;).
Prevention strategies: intestinal injury
Current
There are presently no therapies specifically designed or approved for the prevention of NSAID-induced enteropathy. As mentioned above, PPIs or H2RAs are very commonly co-prescribed with NSAIDs, but this has only been shown to be effective at reducing the incidence of damage in the stomach and proximal duodenum. Indeed, the four video capsule endoscopy studies that were described above provided evidence that in a population at low risk for NSAID gastroenteropathy there was a high incidence (55–75%) of small intestinal damage despite co-administration of a PPI with the NSAID (Goldstein et al., 2005; Graham et al., 2005; Maiden et al., 2005; Fujimora et al., 2010). Recent studies in rodents suggest that PPIs actually exacerbate NSAID-induced enteropathy, rather than provide any beneficial effects. Rats treated with a PPI (omeprazole or lansoprazole) developed substantially more intestinal ulceration and bleeding when concurrently treated with an NSAID (naproxen or celecoxib) than the control group treated with vehicle plus the NSAID (Wallace et al., 2011). The PPI did not alter the plasma levels of the NSAID nor its biliary excretion, and did not cause mucosal inflammation or injury. However, treatment with the PPI did significantly alter the intestinal flora. Along with a substantial increase in gram-negative bacteria in the small intestine, there was a significant decrease in the numbers of Actinobacteria. Replenishment of Actinobacteria levels in the intestine through administration of selectively cultured jejunal contents (from healthy rats) restored resistance to NSAID-induced intestinal injury. These results suggested that changes in the intestinal flora were responsible for the PPI-induced increase in susceptibility to small intestinal injury. This was further supported by studies using germ-free mice. Jejunal contents from rats treated with vehicle or a PPI were transferred (orally) into two groups of germ-free mice. When subsequently treated with an NSAID, the mice with flora from PPI-treated rats developed significantly more small intestinal damage than the mice with flora from vehicle-treated rats.
As in the case of NSAID gastropathy, misoprostol is not widely used for prevention of NSAID enteropathy. There is some limited evidence suggesting that PGs would exert benefit in this indication. Bjarnason et al. (1989) demonstrated a significant reduction of NSAID-induced intestinal permeability with misoprostol, but whether or not a reduction of changes in permeability translates into reduction of clinically significant injury is unclear. Fujimori et al. (2009) reported benefit of treatment with misoprostol in a small pilot study in which intestinal damage was assessed by video capsule endoscopy.
In development
Many of the drugs that are in development with an aim of causing less gastroduodenal damage have not yet been evaluated for safety in the more distal small intestine (e.g. new PPIs and combination NSAID-PPI tablets, phosphatidylcholine-associated NSAIDs). NO-releasing NSAIDs have been shown to be better tolerated in the small intestine in animal studies (Reuter et al., 1994; Davies et al., 1997a), and in a clinical trial, to cause significant less of an increase in small intestinal permeability than the parent drug (naproxen) (Hawkey et al., 2003). Hydrogen sulphide-releasing NSAIDs have been shown to cause negligible damage in the small intestine of rats (Wallace et al., 2010), but have not yet been evaluated in humans.
Future directions
Gastric damage induced by NSAIDs can largely be managed through the use of inhibitors of acid secretion. With combination NSAID-PPI and NSAID-H2RA tablets becoming available, this usage will likely increase. Increasingly concerns about the long-term use of PPIs are emerging (increased risk of certain infections, malabsorption of certain vitamins and nutrients, etc.). The small intestinal damage caused by NSAIDs is more complex in terms of its pathogenesis. The prevalence and clinical relevance of this damage has been underestimated until recently, but this is changing with improvements to video capsule endoscopy and more widespread availability of this technology. The approaches taken to prevent NSAID-induced damage in the stomach and duodenum are unlikely to provide significant benefit in the small intestine. Indeed, there is substantial evidence from laboratory studies to suggest that chronic acid suppression markedly alters the small intestinal flora, and this can have detrimental consequences, including a marked worsening of NSAID-induced enteropathy.
Given the evidence for an important role of enteric bacteria (particularly gram negative) in the development of NSAID-induced intestinal ulceration, exploration of the potential of probiotics and prebiotics is warranted. Antibiotics are another option, but there is a strong possibility of development of resistance to the antibiotics (Lanas and Scarpignato, 2006), because patients would need to take these drugs over long periods of time.
Of course, the ideal solution to the significant toxicity of NSAIDs in the GI tract would be the development of anti-inflammatory drugs that do not damage the GI mucosa. The distinct underlying mechanisms for injury in the stomach/duodenum versus more distal small intestine make this a very challenging prospect, but there are some promising developments in this regard, based on studies of laboratory animals. Future studies of new and existing NSAIDs need to include a more rigorous evaluation of their injurious effects in the small intestine, rather than focusing almost entirely on gastroduodenal damage. This likely necessitates more widespread use of video capsule endoscopy, and the development of surrogate markers of intestinal injury and bleeding.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research.
Glossary
Abbreviations
- ADR
adverse drug reaction
- GI
gastrointestinal
- H2RA
histamine H2 receptor antagonist
- PPI
proton pump inhibitor
Conflict of interest
Dr Wallace is a founder of Antibe Therapeutics Inc., which is developing novel anti-inflammatory drugs.
References
- Andrews FJ, Malcontenti-Wilson C, O'Brien PE. Effect of nonsteroidal anti-inflammatory drugs on LFA-1 and ICAM-1 expression in gastric mucosa. Am J Physiol Gastrointest Liver Physiol. 1994;266:G657–G664. doi: 10.1152/ajpgi.1994.266.4.G657. [DOI] [PubMed] [Google Scholar]
- Antoon JS, Perry MA. Role of neutrophils and mast cells in acute indomethacin-induced small bowel injury in the rat. J Gastroenterol. 1997;32:747–757. doi: 10.1007/BF02936950. [DOI] [PubMed] [Google Scholar]
- Appleyard CB, McCafferty DM, Tigley AW, Swain MG, Wallace JL. Tumor necrosis factor mediation of NSAID-induced gastric damage: role of leukocyte adherence. Am J Physiol. 1996;270:G42–G48. doi: 10.1152/ajpgi.1996.270.1.G42. [DOI] [PubMed] [Google Scholar]
- Asako H, Kubes P, Wallace J, Gaginella T, Wolf RE, Granger DN. Indomethacin-induced leukocyte adhesion in mesenteric venules: role of lipoxygenase products. Am J Physiol Gastrointest Liver Physiol. 1992a;262:G903–G908. doi: 10.1152/ajpgi.1992.262.5.G903. [DOI] [PubMed] [Google Scholar]
- Asako H, Kubes P, Wallace J, Wolf RE, Granger DN. Modulation of leukocyte adhesion in rat mesenteric venules by aspirin and salicylate. Gastroenterology. 1992b;103:146–152. doi: 10.1016/0016-5085(92)91107-f. [DOI] [PubMed] [Google Scholar]
- Bjarnason I, Smethurst P, Fenn GC, Lee CE, Menzies IS, Levi AJ. Misoprostol reduces indomethacin induced changes in human small intestinal permeability. Dig Dis Sci. 1989;34:407–411. doi: 10.1007/BF01536263. [DOI] [PubMed] [Google Scholar]
- Bjarnason I, Hayllar J, MacPherson AJ, Russell AS. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology. 1993;104:1832–1847. doi: 10.1016/0016-5085(93)90667-2. [DOI] [PubMed] [Google Scholar]
- Claria J, Serhan CN. Aspirin triggers previously unrecognized bioactive eicosanoids in human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A. 1995;92:9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryer B, Bhatt DL, Lanza FL, Dong JF, Lichtenberger LM, Marathi UK. Low-dose aspirin-induced ulceration is attenuated by aspirin-phosphatidylcholine: a randomized clinical trial. Am J Gastroenterol. 2011;106:272–277. doi: 10.1038/ajg.2010.436. [DOI] [PubMed] [Google Scholar]
- Davies NM, Røseth AG, Appleyard CB, McKnight W, Del Soldato P, Calignano A, et al. NO-naproxen vs. naproxen: ulcerogenic, analgesic and anti-inflammatory effects. Aliment Pharmacol Ther. 1997a;11:69–79. doi: 10.1046/j.1365-2036.1997.115286000.x. [DOI] [PubMed] [Google Scholar]
- Davies NM, Sharkey KA, ASFAHA S, MacNaughton WK, Wallace JL. Aspirin induces a rapid up-regulation of cyclooxygenase-2 expression in the rat stomach. Aliment Pharmacol Ther. 1997b;11:1101–1108. doi: 10.1046/j.1365-2036.1997.00247.x. [DOI] [PubMed] [Google Scholar]
- Douthwaite AH, Lintott SAM. Gastroscopic observation of the effect of aspirin and certain other substances on the stomach. Lancet. 1938;2:1222–1225. [Google Scholar]
- Estes LL, Fuhs DW, Heaton AH, Butwinick CS. Gastric ulcer perforation, associated with the use of injectable ketorolac. Ann Pharmacother. 1993;27:42–43. doi: 10.1177/106002809302700111. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, de Lima OM, Mencarelli A, Palazzetti B, Distrutti E, McKnight W, et al. Cyclooxygenase-2-derived lipoxin A4 increases gastric resistance to aspirin-induced damage. Gastroenterology. 2002;123:1598–1606. doi: 10.1053/gast.2002.36558. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Santucci L, Wallace JL, Sardina M, Fransioli R, Romano M, et al. Interaction of COX-2 inhibitor with aspirin and NO-aspirin in the human gastric mucosa: evidence for a protective role of nitric oxide. Proc Natl Acad Sci U S A. 2003;100:10937–10941. doi: 10.1073/pnas.1933204100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S, Antonelli E, Distrutti E, Rizzo G, Mencarelli A, Orlandi S, et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129:1210–1224. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- Fujimora S, Gudis K, Takahashi Y, Seo T, Yamada Y, Ehara A, et al. Distribution of small intestinal mucosal injuries as a result of NSAID administration. Eur J Clin Invest. 2010;40:504–510. doi: 10.1111/j.1365-2362.2010.02290.x. [DOI] [PubMed] [Google Scholar]
- Fujimori S, Seo T, Ehara A, Kobayashi T, Mitsui K, Yonezawa M, et al. Prevention of nonsteroidal anti-inflammatory drug-induced small-intestinal injury by prostaglandin: a pilot randomized controlled trial evaluated by capsule endoscopy. Gastrointest Endosc. 2009;69:1339–1346. doi: 10.1016/j.gie.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Eisen GM, Lewis B, Gralnek IM, Zlotnick S, Fort JG. Video capsule endoscopy to prospectively assess small bowel injury with celecoxib, naproxen plus omeprazole, and placebo. Clin Gastroenterol Hepatol. 2005;3:133–141. doi: 10.1016/s1542-3565(04)00619-6. [DOI] [PubMed] [Google Scholar]
- Graham DY, Smith JL, Holmes GI, Davies RO. Nonsteroidal anti-inflammatory effect of sulindac sulfoxide and sulfide on gastric mucosa. Clin Pharmacol Ther. 1985;38:65–70. doi: 10.1038/clpt.1985.136. [DOI] [PubMed] [Google Scholar]
- Graham DY, White RH, Moreland LW, Schubert TT, KATZ R, Jaszewski R, et al. Duodenal and gastric ulcer prevention with misoprostol in arthritis patients taking NSAIDs. Misoprostol Study Group. Ann Intern Med. 1993;119:257–262. doi: 10.7326/0003-4819-119-4-199308150-00001. [DOI] [PubMed] [Google Scholar]
- Graham DY, Opekun AR, Willingham FF, Qureshi WA. Visible small-intestinal mucosal injury in chronic NSAID users. Clin Gastroenterol Hepatol. 2005;3:55–59. doi: 10.1016/s1542-3565(04)00603-2. [DOI] [PubMed] [Google Scholar]
- Green FW, Kaplan MM, Curtis LE, Levine PH. Effect of acid and pepsin on blood coagulation and platelet aggregation. A possible contributor prolonged gastroduodenal mucosal hemorrhage. Gastroenterology. 1978;74:38–43. [PubMed] [Google Scholar]
- Hagiwara M, Kataoka K, Arimochi H, Kuwahara T, Ohnishi Y. Role of unbalanced growth of Gram-negative bacteria in ileal ulcer formation in rats treated with a nonsteroidal anti-inflammatory drug. J Med Invest. 2004;51:43–51. doi: 10.2152/jmi.51.43. [DOI] [PubMed] [Google Scholar]
- Hawkey CJ, Jones JI, Atherton CT, Skelly MM, Bebb JR, Fagerholm U, et al. Gastrointestinal safety of AZD3582, a cyclooxygenase inhibiting nitric oxide donator: proof of concept study in humans. Gut. 2003;52:1537–1542. doi: 10.1136/gut.52.11.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry D, Dobson A, Turner C. Variability in the risk of major gastrointestinal complications from nonaspirin nonsteroidal anti-inflammatory drugs. Gastroenterology. 1993;10:1078–1088. doi: 10.1016/0016-5085(93)90952-9. [DOI] [PubMed] [Google Scholar]
- Hunt RH, Lanas A, Stichtenoth DO, Scarpignato C. Myths and facts in the use of anti-inflammatory drugs. Ann Med. 2009;8:1–16. doi: 10.1080/07853890902887295. [DOI] [PubMed] [Google Scholar]
- Ito T, Jensen RT. Association of long-term proton pump inhibitor therapy with bone fractures and effects on absorption of calcium, vitamin B12, iron, and magnesium. Curr Gastroenterol Rep. 2010;12:448–457. doi: 10.1007/s11894-010-0141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332:1302–1308. doi: 10.1136/bmj.332.7553.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent TH, Cardelli RM, Stamler FW. Small intestinal ulcers and intestinal flora in rats given indomethacin. Am J Pathol. 1969;54:237–245. [PMC free article] [PubMed] [Google Scholar]
- Konaka A, Kato S, Tanaka A, Kunikata T, Korolkiewicz R, Takeuchi K. Roles of enterobacteria, nitric oxide and neutrophil in pathogenesis of indomethacin-induced small intestinal lesions in rats. Pharmacol Res. 1999;40:517–524. doi: 10.1006/phrs.1999.0550. [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine L, Connors LG, Reicin A, Hawkey CJ, Burgos-Vargas R, Schnitzer TJ, et al. Serious lower gastrointestinal adverse clinical events with non-selective NSAID or coxib use. Gastroenterology. 2003a;124:288–292. doi: 10.1053/gast.2003.50054. [DOI] [PubMed] [Google Scholar]
- Laine L, Maller ES, Yu C, Quan H, Simon T. Ulcer formation with low-dose enteric-coated aspirin and the effect of COX-2-selective inhibition: a double-blind trial. Gastroenterology. 2003b;127:395–402. doi: 10.1053/j.gastro.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Lanas A, Scarpignato C. Microbial flora in NSAID-induced intestinal damage: a role for antibiotics? Digestion. 2006;73(Suppl. 1):136–150. doi: 10.1159/000089789. [DOI] [PubMed] [Google Scholar]
- Lanas A, Baron JA, Sandler RS, Horgan K, Bolognese J, Oxenius B, et al. Peptic ulcer and bleeding events associated with rofecoxib in a 3-year colorectal adenoma chemoprevention trial. Gastroenterology. 2007;132:490–497. doi: 10.1053/j.gastro.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Lanza FL, Marathi UK, Anand BS, Lichtenberger LM. Clinical trial: comparison of ibuprofen-phosphatidycholine and ibuprofen on the gastrointestinal safety and analgesic efficacy in osteoarthritis patients. Aliment Pharmacol Ther. 2008;28:431–442. doi: 10.1111/j.1365-2036.2008.03765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenberger LM. Where is the evidence that cyclooxygenase inhibition is the primary cause of nonsteroidal anti-inflammatory drug (NSAID)-induced gastrointestinal injury? Topical injury revisited. Biochem Pharmacol. 2001;61:631–637. doi: 10.1016/s0006-2952(00)00576-1. [DOI] [PubMed] [Google Scholar]
- Lichtenberger LM, Barron M, Marathi U. Association of phosphatidylcholine and NSAIDs as a novel strategy to reduce gastrointestinal toxicity. Drugs Today (Barc) 2009;45:877–890. doi: 10.1358/dot.2009.45.12.1441075. [DOI] [PubMed] [Google Scholar]
- Lombardo L, Foti M, Ruggia O, Chiecchio A. Increased incidence of small intestinal bacterial overgrowth during proton pump inhibitor therapy. Clin Gastroenterol Hepatol. 2010;8:504–508. doi: 10.1016/j.cgh.2009.12.022. [DOI] [PubMed] [Google Scholar]
- Ma L, del Soldato P, Wallace JL. Divergent effects of new cyclooxygenase inhibitors on gastric ulcer healing: shifting the angiogenic balance. Proc Natl Acad Sci U S A. 2002;99:13243–13247. doi: 10.1073/pnas.202392199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNaughton WK, Cirino G, Wallace JL. Endothelium-derived relaxing factor (nitric oxide) has protective actions in the stomach. Life Sci. 1989;45:1869–1876. doi: 10.1016/0024-3205(89)90540-7. [DOI] [PubMed] [Google Scholar]
- Maiden L, Thjodleifsson B, Theodors A, Gonzalez J, Bjarnason I. A quantitative analysis of NSAID-induced small bowel pathology by capsule endoscopy. Gastroenterology. 2005;128:1172–1178. doi: 10.1053/j.gastro.2005.03.020. [DOI] [PubMed] [Google Scholar]
- McCafferty DM, Granger DN, Wallace JL. Indomethacin-induced gastric injury and leukocyte adherence in arthritic versus healthy rats. Gastroenterology. 1995;109:1173–1180. doi: 10.1016/0016-5085(95)90576-6. [DOI] [PubMed] [Google Scholar]
- McCarthy DM. GI bleeding: problems that persist. Gastrointest Endosc. 2009;70:225–228. doi: 10.1016/j.gie.2008.12.247. [DOI] [PubMed] [Google Scholar]
- McCarthy DM. Adverse effects of proton pump inhibitor drugs: clues and conclusions. Curr Opin Gastroenterol. 2010;26:624–631. doi: 10.1097/MOG.0b013e32833ea9d9. [DOI] [PubMed] [Google Scholar]
- Mizuno H, Sakamoto C, Matsuda K, Wada K, Uchida T, Noguchi H, et al. Induction of cyclooxygenase 2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice. Gastroenterology. 1997;112:387–397. doi: 10.1053/gast.1997.v112.pm9024292. [DOI] [PubMed] [Google Scholar]
- Pirmohamed M, James S, Meakin S, Green C, Scott AK, Walley TJ, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. Br Med J. 2004;329:15–19. doi: 10.1136/bmj.329.7456.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainsford KD, Willis C. Relationship of gastric mucosal damage induced in pigs by antiinflammatory drugs to their effects on prostaglandin production. Dig Dis Sci. 1982;27:624–635. doi: 10.1007/BF01297219. [DOI] [PubMed] [Google Scholar]
- Reuter BK, Cirino G, Wallace JL. Markedly reduced intestinal toxicity of a diclofenac derivative. Life Sci. 1994;55:PL1–PL8. doi: 10.1016/0024-3205(94)90083-3. [DOI] [PubMed] [Google Scholar]
- Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL. Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J Clin Invest. 1996;98:2076–2085. doi: 10.1172/JCI119013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter BK, Davies NM, Wallace JL. Nonsteroidal anti-inflammatory drug enteropathy in rats: role of permeability, bacteria, and enterohepatic circulation. Gastroenterology. 1997;112:109–117. doi: 10.1016/s0016-5085(97)70225-7. [DOI] [PubMed] [Google Scholar]
- Robert A, Asano T. Resistance of germ free rats to indomethacin-induced lesions. Prostaglandins. 1977;14:331–341. doi: 10.1016/0090-6980(77)90178-2. [DOI] [PubMed] [Google Scholar]
- Robert A, Nezamis JE, Lancaster C, Hanchar AJ. Cytoprotection by prostaglandins in rats. Prevention of gastric necrosis produced by alcohol, HCl, NaOH, hypertonic NaCl, and thermal injury. Gastroenterology. 1979;77:433–443. [PubMed] [Google Scholar]
- Santucci L, Fiorucci S, Giansanti M, Brunori PM, Di Matteo FM, Morelli A. Pentoxifylline prevents indomethacin induced acute gastric mucosal damage in rats: role of tumour necrosis factor alpha. Gut. 1994;35:909–915. doi: 10.1136/gut.35.7.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpignato C, Hunt RH. Nonsteroidal anti-inflammatory drug-related injury to the gastrointestinal tract: clinical picture, pathogenesis, and prevention. Gastroenterol Clin North Am. 2010;39:433–464. doi: 10.1016/j.gtc.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Scheiman JM, Yeomans ND, Talley NJ, VAKIL N, Chan FK, Tulassay Z, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101:701–710. doi: 10.1111/j.1572-0241.2006.00499.x. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Oliw E. Unorthodox routes to prostanoid formation: new twists in cyclooxygenase-initiated pathways. J Clin Invest. 2001;107:1481–1489. doi: 10.1172/JCI13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo K, Machida M, Fukumura M, Koide K, Yamazaki R. Omeprazole induces altered bile acid metabolism. Gut. 1998;42:266–271. doi: 10.1136/gut.42.2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram S, Sigthorsson G, Simpson RJ, Watts J, Jacob M, Tavares IA, et al. Uncoupling of intestinal mitochondrial oxidative phosphorylation and inhibition of cyclooxygenase are required for the development of NSAID-enteropathy in the rat. Aliment Pharmacol Ther. 2000;14:639–650. doi: 10.1046/j.1365-2036.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- Souza MHLP, Menezes de Lima O, Zamuner SR, Fiorucci S, Wallace JL. Gastritis increases resistance to aspirin-induced mucosal injury via COX-2-mediated lipoxin synthesis. Am J Physiol Gastrointest Liver Physiol. 2003;285:G54–G61. doi: 10.1152/ajpgi.00525.2002. [DOI] [PubMed] [Google Scholar]
- Taha AS, Hudson N, Hawkey CJ, Swannell AJ, Trye PN, Cottrell J, et al. Famotidine for the prevention of gastric and duodenal ulcers caused by nonsteroidal antiinflammatory drugs. N Engl J Med. 1996;334:1435–1439. doi: 10.1056/NEJM199605303342204. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Hase S, Miyazawa T, Takeuchi K. Up-regulation of cyclooxygenase-2 by inhibition of cyclooxygenase-1: a key to nonsteroidal anti-inflammatory drug-induced intestinal damage. J Pharmacol Exp Ther. 2002;300:754–761. doi: 10.1124/jpet.300.3.754. [DOI] [PubMed] [Google Scholar]
- Uejima M, Kinouchi T, Kataoka K, Hiraokam I, Ohnishi Y. Role of intestinal bacteria in ileal ulcer formation in rats treated with a nonsteroidal anti-inflammatory drug. Microbiol Immunol. 1996;40:553–560. doi: 10.1111/j.1348-0421.1996.tb01108.x. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Gastric ulceration: critical events at the neutrophil–endothelium interface. Can J Physiol Pharmacol. 1993;71:98–102. doi: 10.1139/y93-014. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol Sci. 2007;28:501–505. doi: 10.1016/j.tips.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn't the stomach digest itself? Physiol Rev. 2008;88:1547–1565. doi: 10.1152/physrev.00004.2008. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Devchand PR. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br J Pharmacol. 2005;145:275–282. doi: 10.1038/sj.bjp.0706201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Ferraz JG. New pharmacological therapies in gastrointestinal disease. Gastroenterol Clin North Am. 2010;39:709–720. doi: 10.1016/j.gtc.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Wallace JL, McKnight GW. The mucoid cap over superficial gastric damage in the rat. A high-pH microenvironment dissipated by nonsteroidal antiinflammatory drugs and endothelin. Gastroenterology. 1990;99:295–304. doi: 10.1016/0016-5085(90)91009-u. [DOI] [PubMed] [Google Scholar]
- Wallace JL, McKnight GW. Characterization of a simple animal model for nonsteroidal anti-inflammatory drug induced antral ulcer. Can J Physiol Pharmacol. 1993;71:447–452. doi: 10.1139/y93-066. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Keenan CM, Granger DN. Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Am J Physiol. 1990;259:G462–G467. doi: 10.1152/ajpgi.1990.259.3.G462. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Arfors K-E, McKnight GW. A monoclonal antibody against the CD18 leukocyte adhesion molecule prevents indomethacin-induced gastric damage in the rabbit. Gastroenterology. 1991;100:878–883. doi: 10.1016/0016-5085(91)90259-n. [DOI] [PubMed] [Google Scholar]
- Wallace JL, McKnight W, Miyasaka M, Tamatani T, Paulson J, Anderson DC, et al. Role of endothelial adhesion molecules in NSAID-induced gastric mucosal injury. Am J Physiol. 1993;265:C993–C998. doi: 10.1152/ajpgi.1993.265.5.G993. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Reuter B, Cicala C, McKnight W, Grisham MB, Cirino G. Novel nonsteroidal anti-inflammatory drug derivatives with markedly reduced ulcerogenic properties in the rat. Gastroenterology. 1994;107:173–179. doi: 10.1016/0016-5085(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Wallace JL, McKnight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement of inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;119:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Caliendo G, Santagada V, CIRINO G, Fiorucci S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulphide-releasing diclofenac derivative in the rat. Gastroenterology. 2007;132:261–271. doi: 10.1053/j.gastro.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Caliendo G, Santagada V, Cirino G. Markedly reduced toxicity of a hydrogen sulfide-releasing derivative of naproxen (ATB-346) Br J Pharmacol. 2010;159:1236–1246. doi: 10.1111/j.1476-5381.2009.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Denou E, Vong L, Syer S, McKnight W, Jury J, et al. Proton pump inhibitors and low-dose aspirin markedly exacerbate NSAID-induced small intestinal injury: link to dysbiosis? Gastroenterology. 2011;140:S-87. doi: 10.1053/j.gastro.2011.06.075. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Higuchi K, Kobata H, Tanigawa T, Shiba M, Tominaga K, et al. Non-steroidal anti-inflammatory drug-induced small intestinal damage is Toll-like receptor 4 dependent. Gut. 2008;57:181–187. doi: 10.1136/gut.2007.125963. [DOI] [PubMed] [Google Scholar]
- Whittle BJ. Temporal relationship between cyclooxygenase inhibition, as measured by prostacyclin biosynthesis, and the gastrointestinal damage induced by indomethacin in the rat. Gastroenterology. 1981;80:94–98. [PubMed] [Google Scholar]
- Williams C, McColl KE. Review article: proton pump inhibitors and bacterial overgrowth. Aliment Pharmacol Ther. 2006;23:3–10. doi: 10.1111/j.1365-2036.2006.02707.x. [DOI] [PubMed] [Google Scholar]
- Yeomans ND, Tulassay Z, Juhasz L, Racz I, Howard JM, van Rensburg CJ, et al. A comparison of omeprazole with ranitidine for ulcers associated with nonsteroidal anti-inflammatory drugs. Acid Suppression Trial: Ranitidine versus Omeprazole for NSAID-associated Ulcer Treatment (ASTRONAUT) Study Group. N Engl J Med. 1998;338:719–726. doi: 10.1056/NEJM199803123381104. [DOI] [PubMed] [Google Scholar]
- Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Dial EJ, Doyen R, Lichtenberger LM. Effect of indomethacin on bile acid-phospholipid interactions: implication for small intestinal injury induced by nonsteroidal anti-inflammatory drugs. Am J Physiol Gastrointest Liver Physiol. 2010;298:G722–G731. doi: 10.1152/ajpgi.00387.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]