

Abstract
BACKGROUND AND PURPOSE
Classic H1 histamine receptor (H1R) antagonists are non-selective for H1R and known to produce drowsiness. Modern antihistamines are more selective for H1R, and are ‘non-drowsy’ presumably due to reduced permeability through the blood-brain barrier. To characterize both histaminergic sleep regulation and the central actions of antihistamines, in the present study we analysed the effect of classic and modern antihistamines on rats' sleep using continuous i.c.v. infusions.
EXPERIMENTAL APPROACH
Effects of classic (d-chlorpheniramine; d-CPA) and second-generation (cetirizine) antihistamines on sleep were compared after i.p. injections or continuous i.c.v. infusions into rats. Fluorescent cetirizine/DBD-pz was synthesized to trace the approximate distribution of cerebral cetirizine. Furthermore, the effects of H1R antagonists on cultured preoptic neurons were examined using calcium imaging.
KEY RESULTS
d-CPA 4 mg·kg−1 i.p. increased non-rapid eye movement (REM) sleep whereas 10–40 mg·kg−1d-CPA decreased non-REM sleep at dark onset time. Nocturnal i.c.v. infusions of d-CPA (10 µmol·100 µL−1·10 h−1) increased drowsiness but not non-REM sleep, whereas the same i.c.v. infusions of cetirizine significantly increased non-REM sleep, abolished REM sleep, and decreased wakefulness for more than 10 h. The medial preoptic area contained the greatest fluorescent labelling after i.c.v. cetirizine/DBD-pz infusions. Histamine-induced Ca2+ increases in medial preoptic neurons were blocked by d-CPA or cetirizine, whereas d-CPA, but not cetirizine, increased Ca2+ irrespective of antihistaminergic activity at ≥100 µM.
CONCLUSION AND IMPLICATIONS
The excitatory action of d-CPA may explain the seemingly inconsistent actions of d-CPA on sleep. Cerebral H1R inhibition by cetirizine induces synchronization of cerebral activity and prolonged, continuous slow-wave sleep.
Keywords: continuous micro-infusion, intracellular calcium, preoptic sleep centres, second generation antihistamines, sleep encephalogram, yellow cameleon, third ventricle
Introduction
Classic antihistamines, such as d-chlorpheniramine (d-CPA), mepyramine and diphenhydramine, produce drowsiness in humans (Risberg et al., 1975; Sunshine et al., 1978; Bassano and Caille, 1979) and experimental animals (Jewett, 1968; Nisticòet al., 1980; Wauquier et al., 1981; Monti et al., 1986; Saitou et al., 1999; Tokunaga et al., 2007). Based on this, diphenhydramine is currently sold as a sleeping pill (e.g. Drewell® and Neoday®) in the Japanese market. The sedative effects of antagonists of the H1 histamine receptor (H1R; Alexander et al., 2011) appear to depend on their transport across the blood-brain barrier (Mahar Doan et al., 2004), because more modern H1R antagonists such as cetirizine, fexofenadine and mizolastine, which were designed with reduced permeability through the blood-brain barrier (Gupta et al., 2006), do not produce drowsiness (Nicholson, 1983; Seidel et al., 1987; Depoortere et al., 1995; review by Slater et al., 1999). As modern H1R antagonists are less permeable to the brain than classic H1R antagonists, their ability to induce sleep is still poorly understood.
In the brain, histaminergic neuronal cell bodies are located in the tuberomammillary nucleus (TMN) of the posterior hypothalamus (Inagaki et al., 1988; Lin et al., 1988; review by Onodera et al., 1994). TMN neurons are known to modulate arousal levels and have widespread projections to diverse brain areas such as neocortical and thalamic neurons (McCormick, 1992; Reiner and Kamondi, 1994; Chu et al., 2004), cholinergic neurons in the basal forebrain (Khateb et al., 1995; Ramesh et al., 2004) and the mesopontine tegmentum (MPT) (Khateb et al., 1990; Crochet and Sakai, 1999). Also, hypothalamic histamine release is positively correlated with locomotor activity in rats, suggesting a link with arousal levels (Mochizuki et al., 1992; Prast et al., 1992). Histological studies have shown that broad hypothalamic areas including the medial and lateral preoptic areas of the anterior hypothalamus (POAH) contain histaminergic fibres (Inagaki et al., 1988) and localization via an isotope-labelled mepyramine derivative reveals significant expression of H1R in these areas (Bouthenet et al., 1988). Furthermore, a recent electrophysiological study demonstrated that many POAH neurons fire action potentials at a rate that can be directly related to wakefulness or wakefulness/rapid eye movement (REM) sleep episodes (Takahashi et al., 2009). This neuronal activity may provide feedback to control non-REM sleep, because a reduction in firing rates in these neurons precedes the occurrence of non-REM sleep (Takahashi et al., 2009). Consistent with this, inhibition of histamine synthesis by micro-injections of the histidine decarboxylase inhibitor α-fluoromethylhistidine into the POAH significantly decreases wakefulness and increases non-REM and REM sleep in cats (Lin et al., 1994). Also, micro-injections of mepyramine into the POAH increase non-REM sleep (Lin et al., 1994), thus H1Rs in the POAH may regulate sleep relative to arousal-coupling activity.
The involvement of histaminergic systems in arousal has been further confirmed using gene-knockout mice lacking histidine decarboxylase, which exhibit shorter sleep latency after various behavioural stimuli (Parmentier et al., 2002). Also, gene-knockout mice lacking H1R exhibit fewer brief waking bouts and do not respond to orexin-A or H3R-mediated increases in wakefulness (Huang et al., 2001; 2006;). These data support a recent model describing the sleep-control network, where sleep-active ventrolateral preoptic nucleus (VLPO) neurons send inhibitory projections to the TMN, by which arousal-inducing histaminergic activity is reduced (Sherin et al., 1996; Gallopin et al., 2000; review by Huang et al., 2007).
Although the above model is widely accepted, classic antihistamines are also known to be less selective for H1R than second generation antihistamines; they bind to muscarinic receptors and thus have anticholinergic actions (Sherrod et al., 1947; Reuse, 1948; Kubo et al., 1987; Orzechowski et al., 2005). Also, classic antihistamines at relatively high concentrations have receptor-independent actions, such as inhibition of dopamine, noradrenaline or 5-HT transporters (Bergman, 1990; Karamanakos et al., 2004; Tanda et al., 2008), and various K+ channels (Taglialatela et al., 2000; Sato et al., 2005), direct activation of G-protein-mediated intracellular Ca2+ mobilization (Burde et al., 1996); all of these potentially affect sleep and wakefulness. Continuous i.c.v. infusion techniques to evaluate steady-state functions of hypnotic reagents or endogenous sleep substances directly accessible to the brain have been successfully applied to sleep assays in rats (Inoué, 1993; Ikeda et al., 1995; 2001; 2005;). Thus, to clarify the net effect of cerebral H1R inhibition on sleep, in the present study we compared the effects of d-CPA and cetirizine on sleep in rats using conventional i.p. injections and continuous i.c.v. infusion.
Methods
Animals
Adult male Sprague-Dawley rats (300–450 g), 60–70 days old, purchased from Ninox Labo Supply Inc., Ishikawa, Japan) were maintained on a 12:12 light : dark cycle at a constant ambient temperature (25 ± 1°C) and housed with our inbred colony at the Gofuku campus of the University of Toyama. Food and water were available ad libitum. All animal care and experimental procedures complied with international guidelines and were approved by the Animal Care and Use Committee at the University of Toyama.
Sleep recordings
Animals were anaesthetized with an i.p. injection of sodium pentobarbital (50 mg·kg−1 body weight) and placed in a stereotaxic apparatus. For EEG recording, rats were implanted with four gold-plated screw-electrodes (Unique Medical Co., Tokyo, Japan) placed through the skull on the frontal and occipital cortex, according to standard methods (Ikeda et al., 2001; 2005;). Briefly, three recording electrodes were placed over the right frontal cortex (mm) 1.8-, 4.2- and 7.4 rostral and 1.8 lateral to bregma, and one reference electrode was placed over the left frontal cortex 4.2 rostral and 1.8 lateral to bregma. To achieve continuous third ventricular infusion, a stainless steel cannula (0.35 mm inner diameter) was inserted into the third ventricle, 2.3 lateral and 0.9 caudal to bregma at a 15° angle. In addition, two stainless steel hook-electrodes were inserted into the cervical portion of the trapezius muscle for EMG recording. All electrodes and the cannulae were permanently affixed to the skull using dental acrylic resin. During and at the end of surgery, a total of 40 000 U of penicillin G potassium (Meiji Pharmaceutical Company, Tokyo, Japan) was injected s.c. and locally applied to the incision. Further details of the surgery have been described previously (Ikeda et al., 2001; 2005;).
One week was allowed for recovery from surgery, then rats were placed in individual sleep-recording cages in a sound-proof and electromagnetically shielded chamber. The lead wires of the EEG and EMG electrodes were connected to a multiple channel amplifier (MEG-6116; Nihon Kohden, Tokyo, Japan) via a feed-through slip ring (CAY-675; Airflyte Electronics Company, Bayonne, NJ, USA) fixed above the cage. The EEG and EMG signals were relayed to a computer via an analogue-to-digital converter (Ad 16-16U; Contec, Osaka, Japan) at a sampling rate of 128 Hz/channel and stored on a hard diskette. Sleep-wake stages were determined semi-automatically based on the amplitudes of the EEG and EMG and Fast-Fourier-Transform analysis of the EEG spectrum using SleepSign ver 2.0 software (Kissei Comtec, Nagano, Japan). After recording, the sleep-wake stages were replayed on the monitor and visually re-evaluated by two experienced investigators to correct improper scoring of sleep due to artefactual EEG/EMG patterns during active wakefulness.
Drug administration during sleep recordings
For i.p. injections of H1R antagonists, rats were habituated in recording chambers for daily i.p. injections of sterilized saline (0.2 mL) immediately before dark onset (19h 30min–19h55min). Subsequently (+)-chlorpheniramine maleate (d-CPA; F.W. 461.81) (−)-chlorpheniramine maleate (l-CPA; both purchased from Wako Pure Chemical Industries, Ltd. Tokyo), or cetirizine dihydrochloride (F.W. 390.86; CAS 83881-52-1, Sigma/RBI, St Louis, MO, USA) dissolved in 0.2 mL saline was injected instead of saline. Animals received multiple i.p. injections of d-CPA in the dose–response experiments. For example, an initial d-CPA injection of less than 1 mg·kg−1 was administered and then, after at least 1 week, a second d-CPA injection of greater than 4 mg·kg−1 was administered. The EEG and EMG recordings were initiated 1 day before the drug injection, and continued through to 1 day after the drug injection. A total of 24 rats were used for the i.p. injection studies.
For i.c.v. infusions of H1R antagonists, the rats implanted with an i.c.v. cannula received a continuous i.c.v. infusion of sterilized saline (10 µL·h−1) for a week before the EEG/EMG recordings. The i.c.v. cannula was connected through the slip ring to an infusion pump via polyethylene tubing and Teflon connecting tubing (0.5 mm inner diameter). This apparatus guaranteed unrestrained movement of the rats during the study. After the rats had acclimatized to the i.c.v. saline infusion, d-CPA or cetirizine dissolved in saline (0.2 or 10 µmol in 100 µL saline) was infused for 10 h from dark onset (20h00min). After drug infusion, the saline infusion was continued for 2 days. The EEG and EMG recordings were initiated 1 day before the drug infusion. A total of 24 rats was implanted with EEG/EMG electrodes and an i.c.v. infusion cannula, and 20 of the 24 rats were successfully used for the i.c.v. infusion studies described above. Studies were terminated after three days of recording.
Synthesis and characterization of a fluorescent labelled cetirizine
To synthesize fluorescent labelled cetirizine, 4-(N,N-dimethylaminosulphonyl)-7-piperazinobenzofurazan (DBD-pz; λex = 440 nm; λem = 569 nm; Tokyo Chemical Industry, Co., Tokyo, Japan) was conjugated to the carboxyl acid terminal of cetirizine (Figure 1A) as follows. Firstly, N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (6.1 mg, 0.064 mmol) was added to a stirred anhydrous dichloromethane (1 mL) of cetirizine dihydrochloride (10.1 mg, 0.032 mmol; compound 1), DBD-pz (15.0 mg, 0.032 mmol; compound 2) and N,N-dimethylaminopyridine (catalytic amount). The resulting mixture was stirred at room temperature under argon atmosphere for 2 h. Subsequently, the reaction mixture was diluted with dichloromethane, washed with saturated sodium bicarbonate buffer, and then dried over magnesium sulphate. The solvent was evaporated to leave a residue, which was subjected to silica gel column chromatography (eluent: acetone-MeOH-Et3N) to afford the amide compound 3 (19.6 mg, 90%) as yellow solids. Finally, the solids were dissolved in dichloromethane, and anhydrous HCl (1 M solution in Et2O) was added dropwise with stirring. The precipitated HCl salts were collected using filtration and dried at ambient temperature.
Figure 1.
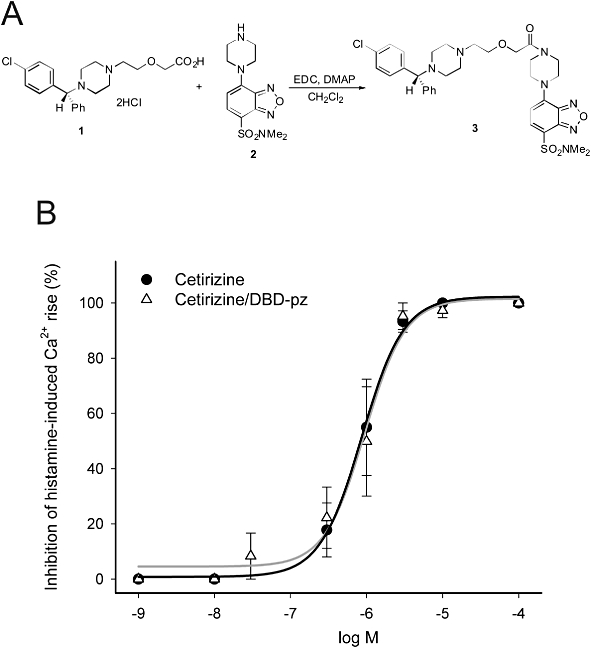
(A) Chemical structure of cetirizine (1), DBD-pz (2) and their conjugated forms (3). EDC, N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide; DMPA, N,N-dimethylaminopyridine. (B) Dose–response curve of inhibition of histamine (30 µM, 1 min)-induced Ca2+ increase by cetirizine and cetirizine/DBD-pz in HeLa cells.
Compound 3 (cetirizine/DBD-pz hydrochloride) was characterized using NMR as follows; Mp 158–160°C; 1H-NMR (500 MHz, DMSO-d6): δ 10.02–9.90 (1H, br), 7.87 (1H, d, J = 8.1 Hz), 7.52–7.39 (6H, m), 7.34 (2H, t, J = 7.5 Hz), 7.28–7.21 (1H, m), 6.61 (1H, d, J = 8.1 Hz), 4.60–4.51 (1H, br), 4.35 (2H, s), 3.96 (2H, brs), 3.91 (2H, brs), 3.81 (2H, brs), 3.70 (2H, brs), 3.61–3.45 (6H, br), 3.33 (2H, brs), 3.29–3.15 (2H, br), 2.90–2.79 (2H, br), 2.72 (6H, s); MS (EI): m/z 681 (M+); HRMS (EI) calculated for C33H40ClN7O5S: 681.2500, found: 681.2463.
The antihistamine activity of the conjugated forms of cetirizine remained intact. The affinity of cetirizine/DBD-pz for H1Rs was analysed using fura-2-based conventional Ca2+-imaging in HeLa cells, which intrinsically express H1Rs. The IC50 value of the inhibition of histamine (30 µM, 1 min)-induced Ca2+ rise was 951 nM for cetirizine/DBD-pz, which is equivalent to the IC50 value of cetirizine (867 nM; Figure 1B).
Histological analysis
To estimate cerebral cetirizine distribution at a time when non-REM sleep was enhanced, cetirizine/DBD-pz (4 µmol·40 µL−1·4 h−1 at dark onset) was infused into the third ventricle (n= 3) at the end of the i.c.v. infusion experiments. Because cetirizine/DBD-pz at this concentration is not soluble in physiological saline, cetirizine/DBD-pz was dissolved in dimethyl sulphoxide (5% of final volume) and then diluted with saline. For the background estimation, a mixture of cetirizine/DBD-pz (4 µmol·40 µL−1) and cetirizine (4 µmol·40 µL−1) was also infused as above following the cetirizine (8 µmol·40 µL−1·4 h−1) infusion (n= 3). Subsequently, rats were deeply anaesthetized with sodium pentobarbital (100 mg·kg−1 body weight) and perfused transcardially with PBS, pH 7.4, and 4% phosphate-buffered paraformaldehyde. Brains were removed, fixed further in 4% phosphate-buffered paraformaldehyde overnight at 4°C and immersed in PBS containing 30% sucrose for 12 h at 4°C. Using a cryostat at −20°C, 50 µm coronal sections were prepared, rinsed three times with PBS, and mounted on glass slides. These samples were imaged using a cooled charge-coupled-device (CCD) camera (Ds-5mc, Nikon, Tokyo, Japan) mounted on a fluorescence stereomicroscope (SMZ-1500; Nikon). The background fluorescence was pre-averaged and subtracted using Photoshop ver. 6.0 software (Adobe Systems, San Jose, CA, USA). The field fluorescence intensity (FI) of an 8-bit depth (i.e. 128-graded) bitmap image was calculated for sleep-related brain areas.
Ca2+ imaging in HeLa cells
HeLa cells plated on 35 mm dishes were incubated for 40 min in culture medium containing 3 µM fura-2 AM (Molecular Probes, Eugene, OR, USA) in a CO2 incubator at 36.5 ± 0.5°C and 5% CO2. Cells were rinsed three times with buffered salt solution consisting of (in mM): 128 NaCl, 5 KCl, 2.7 CaCl2, 1.2 MgCl2, 1 Na2HPO4, 10 glucose and 10 HEPES/NaOH (pH 7.3). Then, the culture dish was transferred to a temperature-controlled chamber on the microscope (TC344B; Warner Instruments, Hamden, CT, USA). Fluorescence images were obtained using an upright microscope (Axioplan 2; Carl Zeiss) with a water immersion objective (Achroplan × 40 NA0.75, Carl Zeiss). The wavelength of the excitation UV light (340 nm or 380 nm pulse; 100 ms) was switched using a filter wheel (Lambda 10-2; Sutter Instruments, Novato, CA, USA). The UV light was generated by a full-spectrum 175W Xenon bulb (Lambda LS; Sutter), conducted to the microscope through a liquid light guide and reflected using a dichroic mirror (FT 395 nm; Carl Zeiss). The pair of fluorescence images was processed using a band-pass filter (BP 485–515 nm; Carl Zeiss) and captured using a multiple format cooled CCD camera (CoolSnap-FS; Photometrics, Tokyo) at 6 s intervals. The filter wheel and the CCD camera were controlled using digital imaging software (MetaFluor ver. 6.0; Japan Molecular Devices, Tokyo). The background fluorescence was also subtracted using the software. During recording, HeLa cells were perfused with buffered salt solution at a flow rate of 2.5 mL·min−1. The histamine (30 µM) was bath-applied two times for 1 min each with a 20 min gap. H1R antagonists (cetirizine or cetirizine/DBD-pz) were perfused for 4 min across the second histamine application.
Ca2+ imaging in hypothalamic slices
Hypothalamic slice cultures were prepared from newborn Sprague-Dawley rats from our inbred colony. Series of coronal hypothalamic slices (380–400 µm) were prepared using a vibrating-blade microtome in ice-cold high-Mg2+ artificial cerebrospinal fluid (ACSF) containing (mM): 138.6 NaCl, 3.35 KCl, 21 NaHCO3, 0.6 NaH2PO4, 9.9 d-glucose, 0.5 CaCl2 and 3 MgCl2, bubbled with 95% O2/5% CO2, placed in a 0.40 µm filter cup (Millicell-CM, Millipore, Bedford, MA, USA), and incubated with 50% Eagles basal medium (Invitrogen, Carlsbad, CA, USA), 25% Earle's balanced salt solution (Invitrogen) and 25% heat-inactivated horse serum, supplemented with 5 mg·mL−1 glucose and 1:100 Glutamax (Invitrogen) in a CO2 incubator at 35.5 ± 0.5°C and 5% CO2.
To visualize the shape and activity of neurons, slice cultures were transfected with yellow cameleon (YC2.1) with a neuron-specific enolase promoter using a Helios gene-gun kit (Bio-Rad Laboratories, Hercules, CA, USA) 7–9 days after in vitro cultures had been established (Ikeda et al., 2003, 2005). The Ca2+ responses in 1–4 neuron(s) were observed immediately using an upright microscope with a water immersion objective as above. Slices were perfused with ACSF containing 2 mM CaCl2 and 1 mM MgCl2 that was bubbled with 95% O2 and 5% CO2 for at least 30 min prior to the experiments. Tetrodotoxin (TTX; 0.5 µM; Wako Pure Chemicals) perfusion was initiated 15 min prior to and continued for the duration of imaging. Cameleon-expressing neurons were exposed to 440 ± 5 nm light using a high power xenon lamp with a band-pass filter (440NBD10, Omega optical) at 6 s intervals. The resultant fluorescence image was separated using a dichroic mirror (455DRLP; Omega optical) and directed into a CCD camera (CoolSnap-Ez; Photometrics) through double-view optics (Hamamatsu Photonics, Hamamatsu, Japan), in which one image was split into bilateral images using internal reflection mirrors and processed using two dichroic mirrors (515 DRLP; Omega optical) and band-pass filters (480DF30 and 535DF25 filters). The monochromator and the CCD camera were controlled using digital imaging software (MetaFluor ver 6.2, Molecular Devices).
Statistical analysis
All data are presented as means with SEM. A two-tailed unpaired t-test was used for pair-wise comparisons. One-way anova followed by Duncan's multiple range test was used for the statistical comparisons among multiple groups. One-way repeated-measures anova was used to analyse differences in hourly sleep-wake periods before and after drug infusions. A four-parameter Hill function was used for the estimation of the dose–response curve for d-CPA and cetirizine analogues. A 95% confidence level was considered to be significant.
Results
Effects of peripheral administration of H1 blockers on sleep
The effects of peripheral administration of d-CPA on sleep were analysed using a conventional i.p. injection strategy with a detailed dose–response curve. Rats injected with 4 mg·kg−1d-CPA at the onset of dark exhibited a transient increase in non-REM sleep (148% of saline-injected controls; F6,45= 17.7, P < 0.05 by Duncan's multiple range test following one-way anova) and a decrease in REM sleep (34% of saline-injected controls; F6,45= 11.4, P < 0.01 by Duncan's multiple range test following one-way anova) during the 3 h following the injection (Figures 2 and 3). The peak non-REM sleep increase and decrease in wakefulness were observed 2 h after the injection, whereas the reduction in REM sleep was observed immediately (≤1 h) after the injection (Figure 2). There was no consistent increase or decrease in sleep or wakefulness during the subsequent dark period (data not shown). The effects of d-CPA on non-REM sleep were dose-dependent up to 10 mg·kg−1, with an EC50 of 0.68 mg·kg−1 (Figure 3A). The inhibitory effects of d-CPA on REM sleep were also dose- dependent, with an IC50 of 1.09 mg·kg−1 (excluding data points greater than 10 mg·kg−1) and 2.68 mg·kg−1 (including data points greater than 10 mg·kg−1; Figure 3B). On the other hand, higher doses of d-CPA (10 mg·kg−1 or 40 mg·kg−1) strongly decreased non-REM sleep (F6,45= 17.7, P < 0.01 by Duncan's multiple range test following one-way anova, Figure 3A) and produced complete insomnia for an hour after injection (Figure 2). Similarly, 40 mg·kg−1l-CPA reduced non-REM sleep (F2,29= 14.6, P < 0.01 by Duncan's multiple range test following one-way anova, Figure 3A) and REM sleep (F2,29= 7.8, P < 0.01 by Duncan's multiple range test following one-way anova, Figure 3B).
Figure 2.
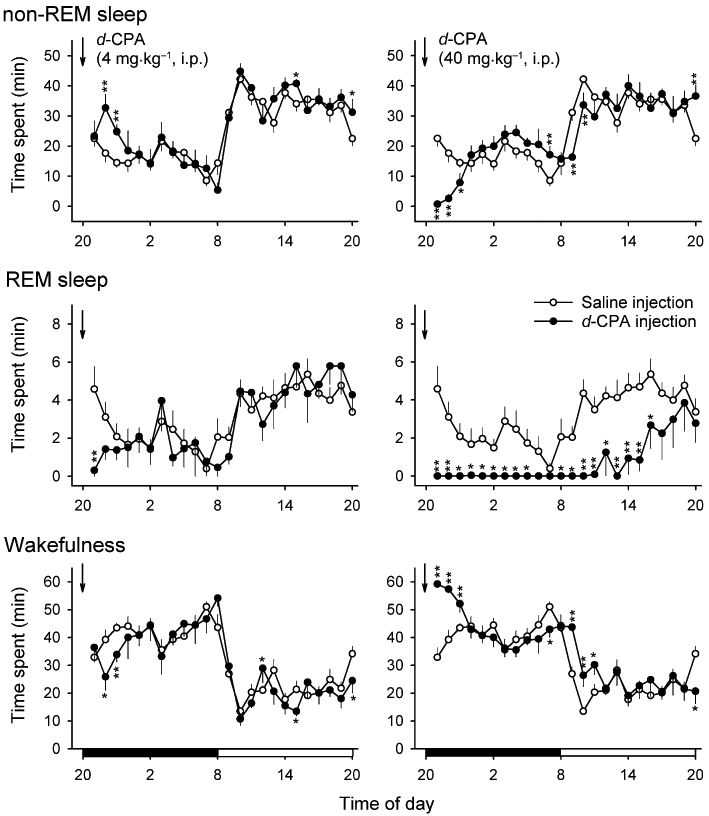
Effects of i.p. injection of d-CPA on daily sleep-wake activity in rats. Graphs show the amount of time spent in non-REM sleep, REM sleep, and wakefulness during 1 day after d-CPA or saline control (n= 24 pooled) injections (arrows). Black and open bars on the bottom row indicate dark and light periods, respectively. Note that there was a transient increase in non-REM sleep and decreases in REM sleep and wakefulness after 4 mg·kg−1d-CPA injections (n= 6) whereas non-REM sleep and REM sleep were both significantly reduced after 40 mg·kg−1d-CPA injections (n= 6). *P < 0.05, **P < 0.01.
Figure 3.
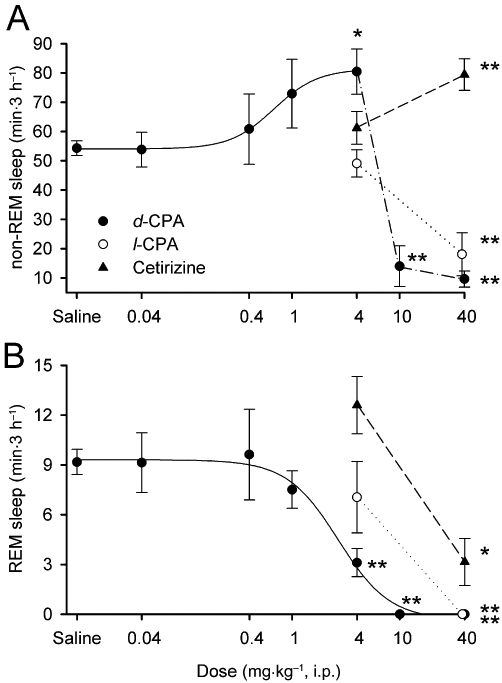
Dose–response curves for the effect of d-CPA on non-REM sleep (A) and REM sleep (B). The 3 h cumulative amount of sleep after i.p. injections of d-CPA were plotted. Non-REM sleep increased and REM sleep decreased in a dose-dependent manner with doses of d-CPA between 0.04–4 mg·kg−1. Higher doses of d-CPA (10 or 40 mg·kg−1) significantly inhibited non-REM and REM sleep. The less-active enantiomer l-CPA similarly reduced non-REM and REM sleep at 40 mg·kg−1. Cetirizine failed to modulate sleep at 4 mg·kg−1 but increased non-REM sleep and decreased REM sleep at 40 mg·kg−1. (n= 4–6 for each drug concentration; n= 24 for saline-injected control). *P < 0.05, **P < 0.01 compared with saline-injected controls.
The effects of i.p. injections of cetirizine on sleep were also tested at 4 mg·kg−1 and 40 mg·kg−1. Lower doses (4 mg·kg−1) of cetirizine failed to produce significant effects on non-REM and REM sleep when compared with saline-injected controls (Figure 3A and B). On the other hand, cetirizine 40 mg·kg−1 significantly increased non-REM sleep (146% of saline-injected controls; F2,29= 7.4, P < 0.01 by Duncan's multiple range test following one-way anova; Figure 3A) and decreased REM sleep (34% of saline-injected controls; F2,29= 4.9, P < 0.05 by Duncan's multiple range test following one-way anova; Figure 2B).
Effects of continuous third ventricular infusion of H1 blockers on sleep
The effect of cerebral H1R blockade on sleep-wake cycles was further examined using continuous, 10 h i.c.v. infusions of d-CPA (Figure 4) or cetirizine (Figure 5) into the vicinity of the POAH in unrestrained rats at two different doses (0.2 or 10 µmol·100 µL−1·10 h−1). While 10 µmol d-CPA failed to evoke a consistent increase or decrease in the amount of non-REM sleep (Figures 4 and 6), infusion of 10 µmol d-CPA resulted in a long-term reduction in REM sleep during the dark period of drug infusion (F4,35= 13.7, P < 0.05 by Duncan's multiple range test following one-way anova) and during the subsequent light period (F4,35= 37.1, P < 0.01 by Duncan's multiple range test following one-way anova; Figures 4 and 7).
Figure 4.
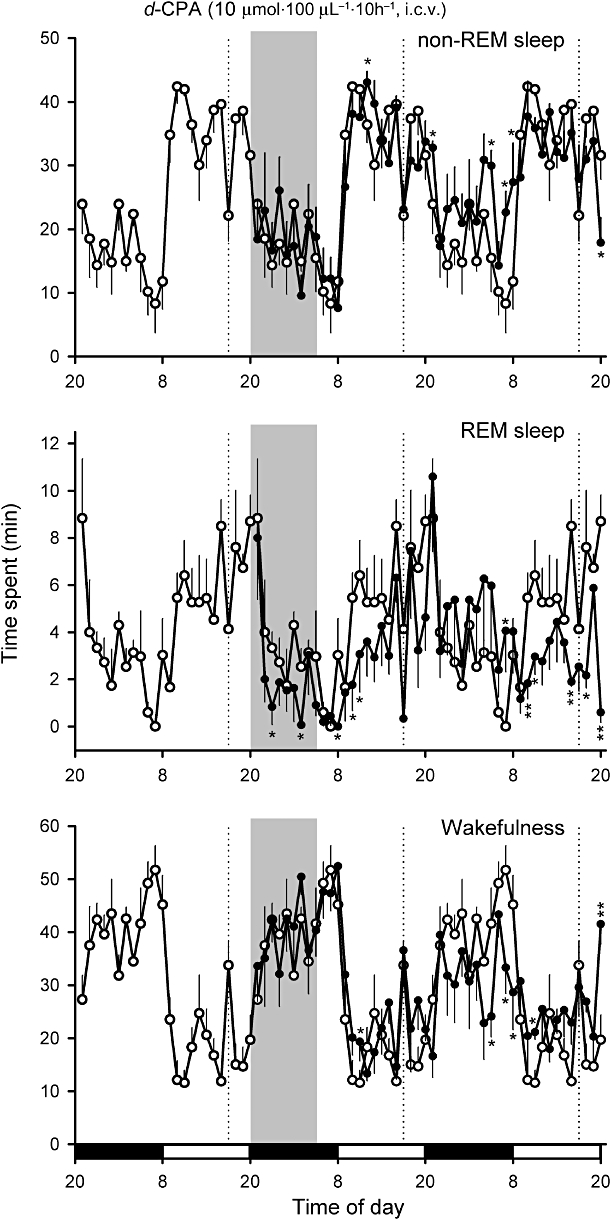
Effects of continuous i.c.v. infusion of 10 µmol·100 µL−1·10 h−1d-CPA (as indicated by the shaded bar) on non-REM sleep and REM sleep in rats. Continuous vehicle infusion (10 µL·h−1) was initiated 1 week before d-CPA infusion and continued through the end of the experiment. Black and open bars on the bottom indicate dark and light periods respectively. The amount of sleep under control conditions before the d-CPA infusion was superimposed on the plots during the d-CPA infusion day and recovery day (open circles). *P < 0.05, **P < 0.01 (n= 5). Dashed lines denote the replacement of infusion tubing, which caused temporary reductions in sleep.
Figure 5.
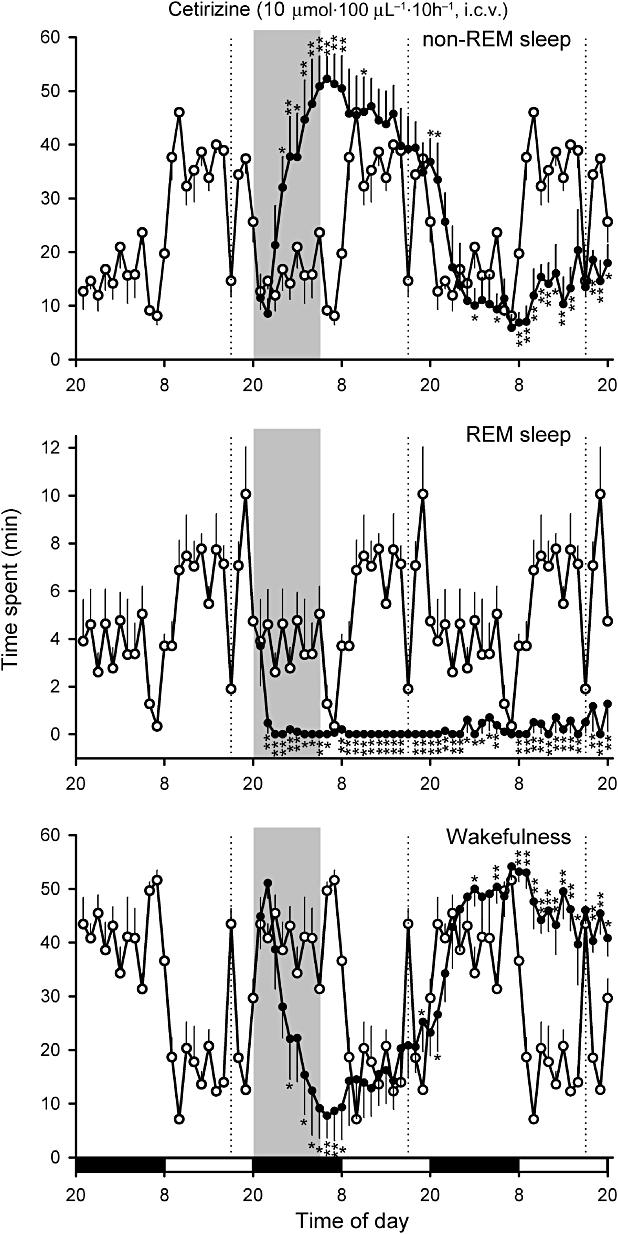
Effects of continuous i.c.v. infusion of 10 µmol·100 µL−1·10 h−1 cetirizine (as indicated by the shaded bar) on non-REM sleep and REM sleep in the rat. Black and open bars on the bottom indicate the dark and light periods respectively. The amount of sleep under control conditions before the cetirizine infusion was superimposed on the plots during the cetirizine infusion day and recovery day (open circles). *P < 0.05, **P < 0.01 (n= 5). Dashed lines denote the replacement of infusion tubing, which caused temporary reductions in sleep.
Figure 6.
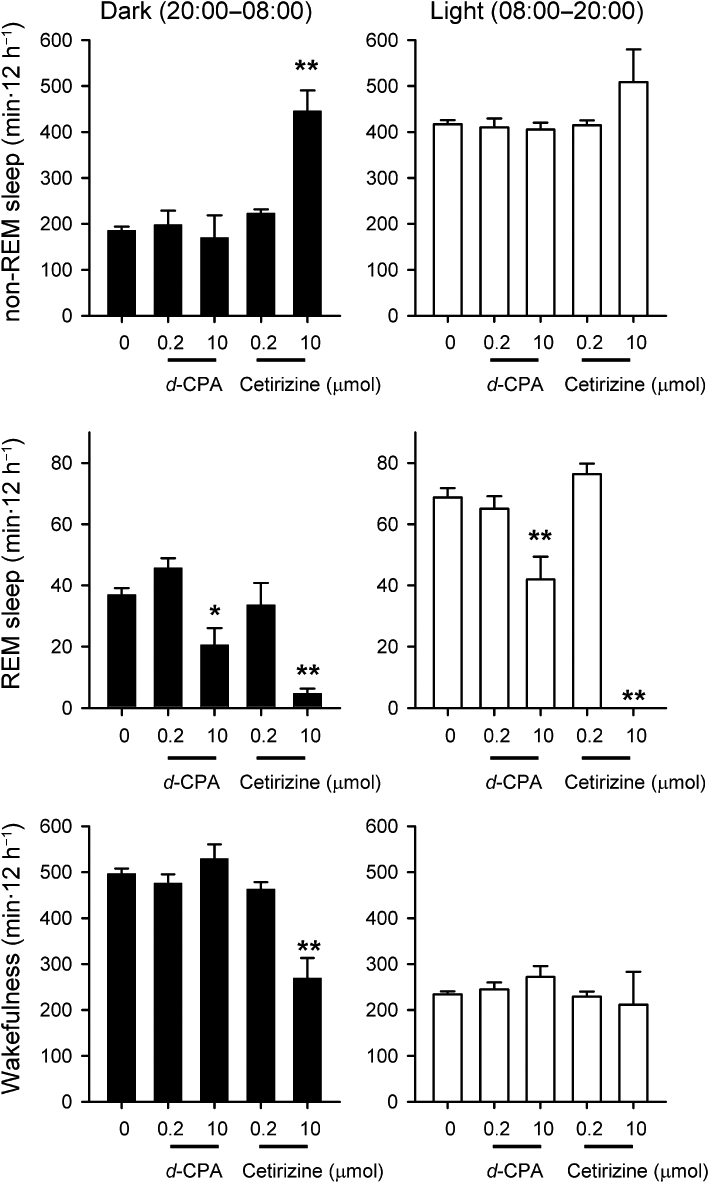
The 12 h cumulative amounts of sleep and wakefulness during and after i.c.v. infusion of d-CPA or cetirizine. Black and open bars denote mean amount of sleep during dark and light periods, respectively (n= 5 for each dose of drug; n= 20 for saline-infused controls). *P < 0.05, **P < 0.01 compared with the corresponding saline infusion group.
Figure 7.
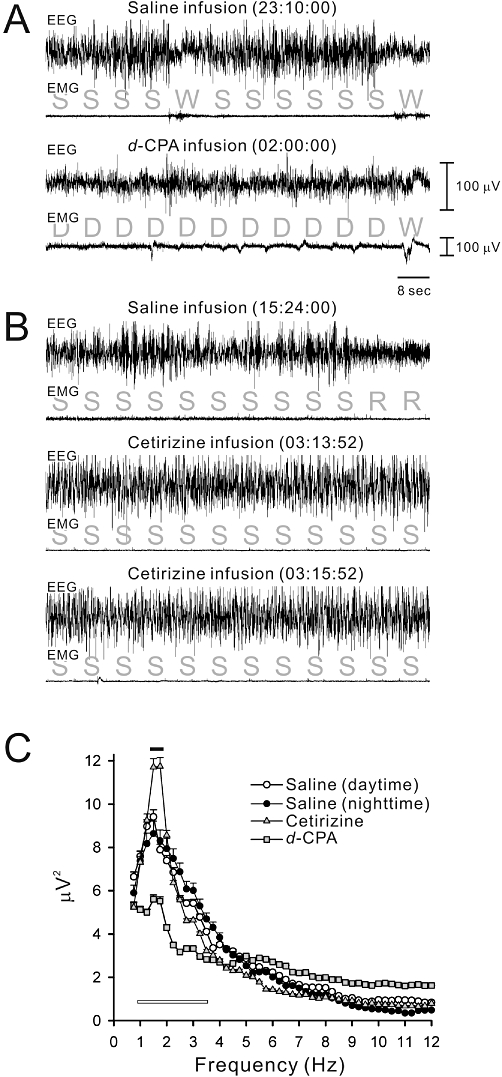
(A) An example of a sleep polygraph (combination of EEG and EMG) during the i.c.v. infusion of saline (upper) or 10 µmol d-CPA (lower) in a rat. (B) An example sleep polygraph during the infusion of saline (upper) or 10 µmol cetirizine (lower two panels are polygraphs from one continuous non-REM sleep episode) in a rat. Based on the sleep polygraph, sleep stages were determined in 8 s bins. S, slow-wave sleep (i.e. non-REM sleep); W, wakefulness; D, drowsiness; R, REM sleep. Drowsiness was composed of slow-wave-like EEG patterns with incomplete EMG relaxation, which was not categorized as non-REM sleep. Drowsiness was frequent during the d-CPA infusions. On the other hand, steady high-amplitude slow-wave EEG coinciding with EMG relaxation was dominant during cetirizine infusions. (C) Fast-Fourier-Transform (FFT) analysis of non-REM sleep EEG waves (i.e. slow waves) during saline-infusion during the light and dark periods or cetirizine-infusion during the dark. For comparison, the FFT spectrum of drowsy EEG waves during d-CPA infusion was plotted as grey squares. Means were calculated from 30 pre-averaged typical epochs in five animals. Frequencies less than 0.5 Hz were eliminated using a filter installed in the amplifiers. Note that the slow-wave spectrum at 1.5–1.75 Hz (black bar) was higher during cetirizine infusion (P < 0.01). The drowsy EEG spectrum during d-CPA infusion was of significantly lower amplitude at 1.0–3.5 Hz (open bar) compared with the slow-wave EEG spectrum during saline or cetirizine infusions (P < 0.01).
Infusion of 10 µmol cetirizine produced similar but stronger effects on REM sleep. REM sleep during the dark period of drug infusion (F4,35= 13.7, P < 0.01 by Duncan's multiple range test following one-way anova) and REM sleep during the subsequent light period (F4,35= 37.1, P < 0.01 by Duncan's multiple range test following one-way anova; Figures 5 and 6) were almost completely suppressed. Non-REM sleep during the dark period during cetirizine infusion was significantly increased above the amount of sleep during the light period (F4,35= 19.46, P < 0.01 in comparison with saline-infused control by Duncan's multiple range test following one-way anova; Figures 5 and 6). The increase in amount of non-REM sleep coincided with prolongation of the non-REM sleep episode and the maximum duration was 47.8 ± 18.6 min (n= 5) during cetirizine infusion. Infusion of d-CPA or cetirizine at 0.2 µmol had no effects on daily sleep-wake cycles (n= 5 for each group; Figure 6).
d-CPA 10 µmol evoked an unusual drowsy state (Figure 7A), which coincided with higher EMG tones than those during non-REM sleep (Figure 7A) and the predominant EEG spectrum in the slow-wave range (1–3.5 Hz) was a lower amplitude than that during normal non-REM sleep (Figure 7A and C). After review of the video of the rats' behaviour during the drowsy state, this state was judged to be a part of wakefulness rather than non-REM sleep. The EEG spectrum during cetirizine-induced non-REM sleep had a peak at 1.75 Hz with a power larger than that during the light and dark under control conditions or during the dark period with d-CPA infusion (F3,16= 75.28, P < 0.01 by Duncan's multiple range test following one-way anova; Figure 7B and C).
Cerebral cetirizine distribution following i.c.v. infusion
Cetirizine/DBD-pz (40 µmol·4 h−1) was infused into the third ventricle using the same experimental procedures for sleep recordings, and brain sections were prepared from rats killed at the approximate time of cetirizine-initiated non-REM sleep enhancement (24:00–24:30; Figure 8A and B). The tip of the infusion cannula was located in the third ventricle, −0.6 ± 0.4 mm from bregma on the midline in these animals. Cetirizine/DBD-pz fluorescence was observed most significantly in the medial preoptic area (FI = 33.6 ± 6.0), near the coronal plane of pipette placement. The sleep-active neuronal nucleus, the median preoptic nucleus (MnPO; FI = 29.7 ± 6.4), but not VLPO (FI = 0.7 ± 0.5; P < 0.01 compared with MnPO levels), was significantly labelled (Figure 8A and B). This difference in labelling was due to the limited lateral diffusion of cetirizine/DBD-pz from the third ventricle, as there was also no fluorescent signal detected in the substantia innominata. In contrast, cetirizine/DBD-pz diffused into the bilateral lateral ventricles, as evidenced by the fibrous staining patterns detected in the septum (Figure 8Aa; FI = 25.4 ± 4.9 for the lateral septum and 24.0 ± 4.1 for the medial septum). In addition, cetirizine/DBD-pz diffused caudally through the mesencephalic duct and permeated the periaqueductus parenchyma, including the ventrolateral periaqueductal grey matter (vlPAG; FI = 28.1 ± 6.3; Figure 8B). A signal was also detected in the dorsal raphe nucleus (FI = 6.2 ± 0.8), whereas it was almost absent in the dorsal end of the laterodorsal tegmental nuclei (LDTg; FI = 0.8 ± 0.2) 4 h after i.c.v. infusions. These findings indicate that there are cetirizine concentration gradients in the brain during continuous i.c.v. infusion.
Figure 8.
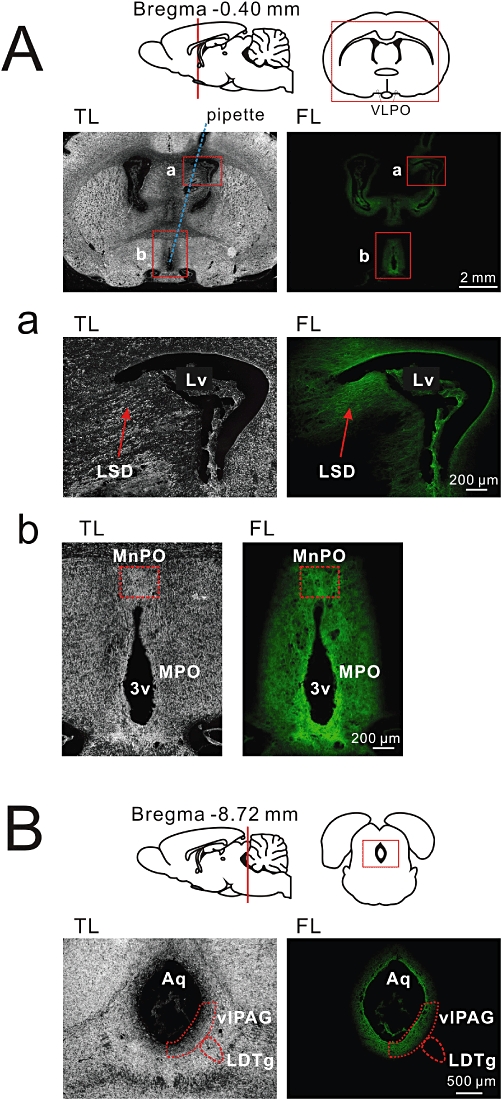
Approximate cetirizine distribution in the rat brain was estimated using i.c.v. infusion of fluorescent-conjugated cetirizine (cetirizine/DBD-pz). (A) An example of a coronal brain section c.a. 100 µm anterior to the infusion plane, which was fixed after a 4 h third-ventricular infusion of cetirizine/DBD-pz (4 µmol·40 µL−1·4 h−1). TL, transmitted light image; FL, background-subtracted fluorescence image. Approximate location of the cannula is indicated as a blue dotted line. Cetirizine/DBD-pz fluorescence was observed both near the lateral ventricle (Lv) and third ventricle (3v). Squares marked as (a) and (b) were enlarged below (Aa,b). (a) The dorsolateral septal nucleus (LSD) is observed as fibrous staining with cetirizine/DBD-pz. (b) The most significant signal was found in the medial preoptic area (MPO) including the median preoptic nucleus (MnPO). (B) Cetirizine/DBD-pz diffused into the mesencephalic duct and permeated the ventrolateral periaqueductal grey matter (vlPAG), but the signal was not detected in the dorsal end of the laterodorsal tegmental nuclei (LDTg).
Effects of H1R antagonists on cytosolic Ca2+ levels in POAH neurons
To analyse the effects of histamine and H1R antagonists on cultured POAH neurons, the yellow cameleon gene was randomly transfected into cultured POAH neurons (Figure 9). When added to circulating ACSF, histamine (100 µM for 45 s) evoked Ca2+ transients in 41% of medial POAH neurons (11 out of 27 neurons in eight slices). These histamine-induced cytosolic Ca2+ responses were completely abolished by d-CPA (10 µM) and recovered after washout of d-CPA (Figure 9A). Cetirizine (10 µM) also completely blocked histamine-induced cytosolic Ca2+ responses in POAH neurons (number of neurons = eight in three slices). Histamine-induced Ca2+ transients were also observed in periventricular neurons (four out of six neurons in two slices) but not in neurons located in the VLPO, although the number of recordings in this area was limited (four neurons in two slices; Figure 9B). To visualize the net histamine response without synaptic interactions, the same histamine stimulation was examined with perfusion of ACSF containing TTX (0.5 µM). The proportion of medial POAH neurons that responded to histamine (10 out of 26 neurons in six slices; 38%) was similar to that without TTX (41%; Figure 9B).
Figure 9.
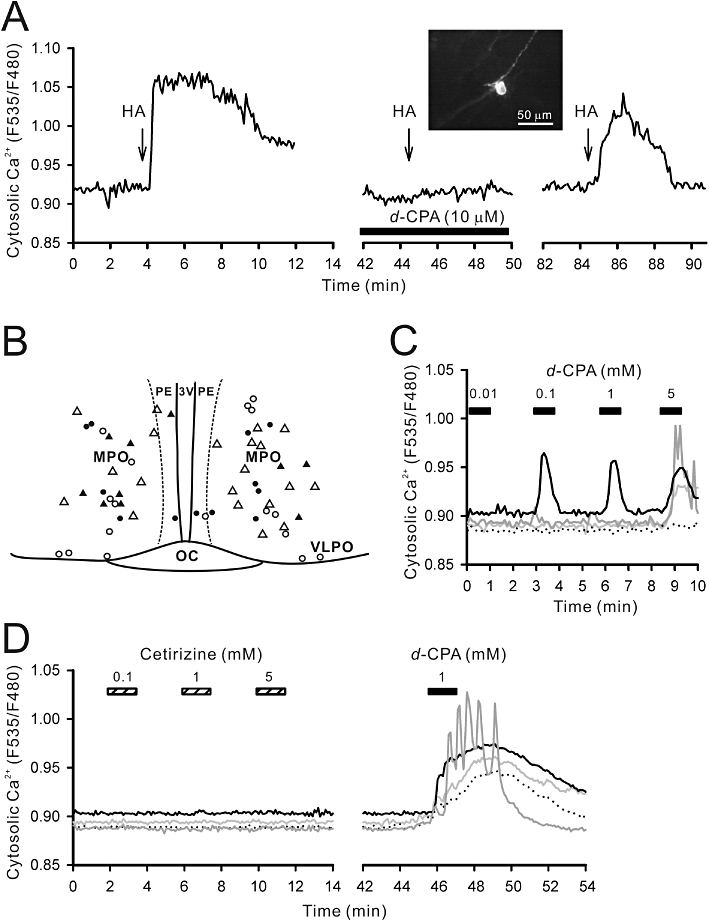
Calcium imaging of POAH slice cultures. A Ca2+ sensor (YC2.1) gene linked to a neuron-specific enolase promoter was transfected into slice cultures to visualize individual neuronal responses to histamine and H1R antagonists. (A) Typical histamine-induced cytosolic Ca2+ response in a medial POAH neuron. Arrows indicate a 30 µM histamine (HA) pulse. The perfusion of d-CPA (10 µM) completely blocked the histamine response. The inset is a fluorescence image of the recorded neuron. (B) Approximate location of the recorded neurons in the preoptic slice. Black and white circles indicate location of histamine-responsive and non-responsive neuronal cell bodies respectively. Black and white triangles indicate the location of histamine-responsive and non-responsive cell bodies respectively, in the presence of the Na+ channel blocker, tetrodotoxin (0.5 µM). Medial POAH (MPO) but not ventrolateral preoptic (VLPO) neurons responded to histamine. (C) d-CPA at higher concentrations increased Ca2+ in medial POAH neurons. Four neurons (as different grey scale or dotted traces) were simultaneously monitored in this experiment and 100 µM d-CPA evoked mobilization of Ca2+ in one neuron. Most medial POAH neurons responded to >5 mM d-CPA. (D) In a different set of experiments, cetirizine (0.1–5 mM) was perfused prior to d-CPA (1 mM) perfusion. Cetirizine did not evoke any Ca2+ responses.
To further analyse possible non-histaminergic effects of CPA compounds on sleep regulatory circuits, we also applied millimolar concentrations of d-CPA to cultured POAH neurons (Figure 9C). In about 42% (five out of 12 neurons in seven slices) of medial POAH neurons, there was an increase in cytosolic Ca2+ in response to d-CPA at 100 µM. Cytosolic Ca2+ was increased in half the neurons in response to d-CPA at 1 mM (eight out of 16 neurons in eight slices) and most neurons responsed to d-CPA 5 mM (20 out of 21 neurons in 14 slices). Because the slice culture used did not contain histaminergic neurons and their axon fragments are completely lost during the culturing process, the d-CPA-induced increase in cytosolic Ca2+ is not explained by antihistaminergic activity. Indeed, cetirizine 100 µM–5 mM did not induce Ca2+ mobilization in cultured POAH neurons (13 out of 13 neurons in five slices; Figure 9D).
Discussion
The affinities of d-CPA (pKi= 8.2) and cetirizine (pKi= 8.6) for H1R were previously reported in CHO cells over-expressing human H1 receptors (Gillard et al., 2002), but their pharmacokinetics and specificity for producing the desired actions in vivo remained to be analysed. In the present study the dose-dependent effects of d-CPA and cetirizine were compared on REM sleep, non-REM sleep and wakefulness in rats. d-CPA facilitated non-REM sleep and suppressed REM sleep within a narrow dose window (≤4 mg·kg−1, i.p.). Inconsistent decreases in non-REM sleep and total REM sleep suppression (i.e. arousal effects) were observed with higher doses of d-CPA (10 or 40 mg·kg−1, i.p.). The present results also demonstrated that (i) l-CPA, which has less than 10% H1R affinity of d-CPA (Tanda et al., 2008), similarly suppressed sleep at 40 mg·kg−1, i.p., and (ii) continuous i.c.v. infusion of d-CPA (10 µmol·100 µL−1·10 h−1) induced unusual drowsiness rather than non-REM sleep. These sleep suppressive effects of d-CPA at high concentrations were consistent with the psychomotor stimulant actions reported for CPA compounds (Bergman, 1990; Tanda et al., 2008). d-CPA but not cetirizine mobilized intracellular Ca2+ in cultured POAH neurons irrespective of antihistaminergic activity at ≥100 µM; therefore, the bimodal effects of d-CPA on sleep can be explained by this non-specific pharmacological action. However, the increase in non-REM sleep and the reduction in REM sleep with i.p. injections of d-CPA at 4 mg·kg−1 may be the result of blocking cerebral H1R, because similar but more prolonged effects were observed with higher i.p. doses of cetirizine (40 mg·kg−1, i.p.) or with a continuous i.c.v. infusion of cetirizine (10 µmol·100 µL−1·10 h−1). Taken together, these results indicate that blocking cerebral H1Rs induces prolonged, continual non-REM sleep in rats.
Classic H1R blocker actions on sleep
Monti et al. (1986) examined the effects of a single i.c.v. injection of the H1R agonist 2-thiazolylethylamine (2-TEA) into the lateral ventricle of rats, and observed a dose-dependent (0.5–2 µmol dissolved in 5 µL saline) and transient (<1 h) increase in wakefulness. The 2-TEA effect was blocked by i.p. injections of mepyramine (1–2 mg·kg−1), thus it was hypothesized that the arousal-inducing effects of 2-TEA are mediated via H1R. Lin et al. (1994) further examined the effects of bilateral micro-injections of mepyramine (120 µg·1 µL−1; 420 mM) in the POAH of cats and observed an increase in non-REM sleep and prolongation of REM sleep latency (i.e. delayed onset of REM sleep after injections). These results strongly suggest that direct H1R antagonist actions in the POAH facilitate non-REM sleep, whereas it should be noted that most classic H1R antagonists cause non-histaminergic effects when applied at high concentrations or in large volumes. For example, mepyramine greater than 30 µM is known to reduce a potassium current (M-current) in dissociated cortical neurons (Sato et al., 2005). In addition, l-CPA as well as d-CPA injections at 5.6 mg·kg−1 induce the release of dopamine in the nucleus accumbens of rats, suggesting that higher doses of these CPA compounds have non-histaminergic effects (Tanda et al., 2008). For this reason, in the present study we re-evaluated the net central actions of H1R antagonists on sleep-wake activity.
In the present study we demonstrated that 10 h infusions of 10 µmol d-CPA (c.a. 6 mg·kg−1, i.c.v. per night) had little effect on non-REM sleep, but instead, induced an unusual state of drowsiness. According to our previous study with a synthetic peroxide reagent (tert-butyl hydroperoxide), applied using the same i.c.v. infusion technique, the concentrations of drugs in the brain parenchyma nearby the infusion pipette were approximately 1000–10 000 times less than that in the infusion pipette (Ikeda et al., 2005). Although the permeability of drugs from the third ventricle to the brain parenchyma may depend on their chemical formula, 10 µmol·100 µL−1·10 h−1d-CPA (i.e. 100 mM in the infusion pipette) probably diffuses into the brain parenchyma at levels similar to those observed in the previous tert-butyl hydroperoxide studies (i.e. 10–100 µM), because d-CPA is highly membrane permeable. This is supported by the present results showing that i.c.v. infusion of 0.2 µmol·100 µL−1·10 h−1d-CPA or cetirizine (i.e. 2 mM in the infusion pipette) failed to modulate sleep, probably because the levels diffused into the brain parenchyma were too low. We also demonstrated that d-CPA at 100 µM or higher induced a histamine-independent mobilization of cytosolic Ca2+ in cultured POAH neurons. Therefore, it is likely that non-specific and excitatory actions of high concentrations of d-CPA may influence many neuronal circuits in the vicinity of the i.c.v. infusion pipette, which would mask its inhibitory actions on H1R.
The results of the present study confirmed that i.p. injections of d-CPA increase non-REM sleep, although the effective dose window was quite limited (4 mg·kg−1). Taken together with the fact that most classic H1 antagonists produce non-histaminergic effects at high concentrations, we suggest careful use of classic H1 antagonists as sleeping pills.
Specific H1R blocker actions on sleep
Several second-generation H1R antagonists, astemizole, loratadine and terfenadine have been shown to facilitate non-REM sleep and/or reduce REM sleep when administered peripherally (Wauquier et al., 1981; Marzanatti et al., 1989; Depoortere et al., 1995). To our knowledge, however, no reports clearly demonstrate how specific H1R antagonists generate sleepiness if they are directly and continuously administered in the brain.
Cetirizine is one of the most specific H1R antagonists and exhibits few side effects. For example, cetirizine does not inhibit M-currents in cortical neurons up to 300 µM (Sato et al., 2005). In the present study, cetirizine up to 5 mM did not mobilize cytosolic Ca2+ in cultured POAH neurons, whereas continuous i.c.v. infusion of cetirizine increased non-REM sleep and suppressed REM sleep. Thus, we suggest that blocking cerebral H1Rs induces prolonged, continuous slow-wave sleep. A reduction in REM sleep has been frequently observed following the peripheral administration of classic H1R antagonists (Jewett, 1968; Risberg et al., 1975; Wauquier et al., 1981; Nicholson et al., 1985; Adam and Oswald, 1986; Lin et al., 1988; 1994; Marzanatti et al., 1989; review by White and Rumbold, 1988), but this effect has been usually regarded as a non-specific action of the classic H1R antagonists. Here, we propose that specific H1R inhibition in the brain causes significant suppression of REM sleep and facilitation of non-REM sleep.
Another important aspect of the results of the present study is the long-term modulation in the amount of sleep with the continuous i.c.v. infusion of cetirizine. Numerous reports have demonstrated a transient increase in sleep or a reduction in sleep latency with the administration of H1R antagonists in humans (Risberg et al., 1975; Sunshine et al., 1978; Bassano and Caille, 1979; Boyle et al., 2006) or in animal models (Jewett, 1968; Nisticòet al., 1980; Wauquier et al., 1981; Monti et al., 1986; Saitou et al., 1999; Tokunaga et al., 2007). Consistent with these findings, our 24 h sleep-EEG recordings following i.p. injection of d-CPA demonstrated a transient increase in non-REM sleep (an increase of less than 30 min during the 3 h after injections). Nevertheless, the possibility exists that the transient hypnotic activity of classic H1R antagonists is due to clearance of the drugs from sleep centres or due to competitive non-specific excitatory actions. In the present study we examined the effect of a continuous 10 h infusion of cetirizine in the vicinity of sleep-regulatory centres, and observed a sustained increase in non-REM sleep and the abolition of REM sleep. The long-lasting rebound wakefulness following cetirizine-induced non-REM sleep demonstrates that continuous cetirizine infusion influenced not only transient switching between sleep states, but also homeostatic control of the total amount of sleep. It is also possible that cetirizine metabolites in the brain modulate sleep-wake activities. These aspects are important for the future development of somnogenic agents based on specific H1R antagonism.
Neuronal circuits underlying the effects of H1R inhibition on sleep
To determine the site(s) at which cetirizine acts to produce non-REM sleep, we synthesized a fluorescent conjugate of cetirizine (cetirizine/DBD-pz), and analysed the its distribution in the brain following 4 h i.c.v. infusions. Cetirizine/DBD-pz and cetirizine had equivalent IC50s; therefore, a comparison between the sleep-inducing effects of cetirizine/DBD-pz and post mortem visualization of fluorescent signals in the individual rats is a valid experiment. However, as cetirizine/DBD-pz was dissolved in 5% dimethyl sulphoxide, which is cytotoxic and affects basal sleep levels, a continuous i.c.v. infusion experiment was not feasible. Therefore, we infused cetirizine/DBD-pz at the end of the i.c.v. infusion experiment and roughly estimated the drug distribution in the brain during the time cetirizine promoted non-REM sleep.
Despite the limitations of the experiment described above, the POAH, especially its medial portion, is one of the most plausible sites where H1R antagonists evoke non-REM sleep, as this area contained the greatest concentration of cetirizine/DBD-pz labelling. This is consistent with former studies demonstrating significant labelling of the medial preoptic area by a [125I] iodo-labelled mepyramine derivative (Bouthenet et al., 1988). Also consistent with this are electrophysiological data showing that the majority of thermo-sensitive preoptic neurons, which are known to be located predominantly in the medial preoptic area, facilitate action potential firing in response to histamine in rat brain slices, and mepyramine inhibits most of this response (Tsai et al., 1989). Furthermore, Lundius et al. (2010) recently demonstrated that H1Rs are expressed on medial POAH neurons that lack GAD, whereas H3 histamine receptors are expressed on medial POAH GAD-positive neurons. The MnPO is also known to contain sleep-active neurons (Gong et al., 2000), and the majority of sleep-active MnPO neurons are GAD positive (Gong et al., 2004). Therefore, it is possible that cetirizine diffusing into this area may directly inhibit GAD-negative MnPO neurons and indirectly activate sleep-active neurons. However, our data failed to indicate direct cetirizine/DBD-pz binding on VLPO cells concurrent with non-REM sleep induction. This does not necessarily mean that the VLPO is not involved in the cetirizine-induced non-REM sleep enhancement, because neurons sending their axons to the VLPO are located in the medial and median preoptic area or in the lateral septal nucleus (Chou et al., 2002), all of which were found to be labelled significantly with cetirizine/DBD-pz. Thus, it is reasonable to assume that i.c.v. cetirizine indirectly activates VLPO via broad neuronal networks.
TMN neurons send ascending histaminergic fibres (e.g. to the POAH and cerebral cortex) and also descending histaminergic fibres to the MPT, including the LDTg, locus coeruleus, locus coeruleus-α, and peri-locus coeruleus-α, known to be critical centres for switching in and out of REM sleep (Lin et al., 1996). As micro-injections of mepyramine (5 µg·0.25 µL−1; 70 mM) into the MPT promote non-REM sleep but fail to modulate REM sleep, it has been suggested that there is no direct involvement of descending histaminergic fibres in the regulation of REM sleep (Lin et al., 1996). In the present study, cetirizine/DBD-pz binding was observed only in the periaqueductal grey matter and not in the LDTg at the time that i.c.v. cetirizine abolished REM sleep. Therefore, it is likely that cetirizine-induced inhibition of REM sleep is not caused by a direct block of H1Rs within the MPT, as implicated by former micro-injection studies (Lin et al., 1996). It has also been shown that micro-injection of histamine in cat vlPAG significantly reduces REM sleep and enhances non-REM sleep, whereas micro-injections of 5-HT, noradrenaline, adrenaline or dopamine in this area had no effects (Crochet et al., 2006), and that the vlPAG functions as an interface of REM-off signalling (Lu et al., 2006). Therefore, a direct activation of REM-off circuits in the vlPAG is most consistent with previous data and currently proposed mechanisms, although the involvement of H1Rs in the periaqueductal grey remains to be analysed.
In conclusion, in the present study it was demonstrated that i.p. injections of d-CPA (4 mg·kg−1) and continuous i.c.v. infusion of cetirizine (10 µmol·100 µL−1·10 h−1) both facilitated non-REM sleep, with i.c.v. cetirizine being more potent. These results provide pharmacological evidence supporting a model of non-REM sleep and arousal involving histaminergic controls. In addition, significant REM sleep suppression by H1R antagonists suggests that cerebral H1Rs function to synchronize cerebral activity resulting in prolonged, continuous slow-wave sleep. The finding that cetirizine induced long-lasting non-REM sleep may provide a target for the development of somnogenic reagents based on this compound, although the risk of coincidental REM sleep suppression must be carefully evaluated.
Acknowledgments
We thank Dr Osamu Hayaishi (Osaka Bioscience Institute) for critical comments. This work was supported in part by grant-in-aids for scientific research (21659056 and 22300108) by the Ministry of Education, Culture, Sports, Science, and Technology Japan, to M. I.
Glossary
Abbreviations
- 2-TEA
2-thiazolylethylamine
- ACSF
artificial cerebrospinal fluid
- CCD
charge-coupled device
- CPA
chlorpheniramine
- DBD-pz
4-(N,N-dimethylaminosulphonyl)-7-piperazinobenzofurazan
- FI
fluorescence intensity
- H1R
H1 histamine receptor
- LDTg
laterodorsal tegmental nucleus
- LSD
dorsolateral septal nucleus
- MnPO
median preoptic nucleus
- MPT
mesopontine tegmentum
- POAH
preoptic areas of the anterior hypothalamus
- REM
rapid eye movement
- TMN
tuberomammillary nucleus
- TTX
tetrodotoxin
- vlPAG
ventrolateral periaqueductal grey matter
- VLPO
ventrolateral preoptic nucleus
Conflicts of interest
None to declare.
References
- Adam K, Oswald I. The hypnotic effects of antihistamine: promethazine. Br J Clin Pharmacol. 1986;22:715–717. doi: 10.1111/j.1365-2125.1986.tb02962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassano JL, Caille EJ. Effects of two antihistaminic compounds (mequitazine, dexchlorpheniramine) on sleep. Sleep distortion by antihistaminics. Waking Sleeping. 1979;3:57–61. [PubMed] [Google Scholar]
- Bergman J. Psychomotor stimulant effects of the stereoisomers of chlorpheniramine. Psychopharmacology (Berl) 1990;100:132–134. doi: 10.1007/BF02245804. [DOI] [PubMed] [Google Scholar]
- Bouthenet ML, Ruat M, Sales N, Garbarg M, Schwartz JC. A detailed mapping of histamine H1-receptors in guinea-pig central nervous system established by autoradiography with [125I] iodobolpyramine. Neuroscience. 1988;26:553–600. doi: 10.1016/0306-4522(88)90167-4. [DOI] [PubMed] [Google Scholar]
- Boyle J, Eriksson M, Stanley N, Fujita T, Kumagi Y. Allergy medication in Japanese volunteers: treatment effect of single doses on nocturnal sleep architecture and next day residual effects. Curr Med Res Opin. 2006;22:1343–1351. doi: 10.1185/030079906X112660. [DOI] [PubMed] [Google Scholar]
- Burde R, Dippel E, Seifert R. Receptor-independent G protein activation may account for the stimulatory effects of first-generation H1-receptor antagonists in HL-60 cells, basophils, and mast cells. Biochem Pharmacol. 1996;51:125–131. doi: 10.1016/0006-2952(95)02123-x. [DOI] [PubMed] [Google Scholar]
- Chou TC, Bjorkum AA, Gaus SE, Lu J, Scammell TE, Saper CB. Afferents to the ventrolateral preoptic nucleus. J Neurosci. 2002;22:977–990. doi: 10.1523/JNEUROSCI.22-03-00977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu M, Huang ZL, Qu WM, Eguchi N, Yao MH, Urade Y. Extracellular histamine level in the frontal cortex is positively correlated with the amount of wakefulness in rats. Neurosci Res. 2004;49:417–420. doi: 10.1016/j.neures.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Crochet S, Sakai K. Effects of microdialysis application of monoamines on the EEG and behavioural states in the cat mesopontine tegmentum. Eur J Neurosci. 1999;11:3738–3752. doi: 10.1046/j.1460-9568.1999.00760.x. [DOI] [PubMed] [Google Scholar]
- Crochet S, Onoe H, Sakai K. A potent non-monoaminergic paradoxical sleep inhibitory system: a reverse microdialysis and single-unit recording study. Eur J Neurosci. 2006;24:1404–1412. doi: 10.1111/j.1460-9568.2006.04995.x. [DOI] [PubMed] [Google Scholar]
- Depoortere H, Decobert M, Granger P, Françon D. Mizolastine, a novel selective histamine H1 receptor antagonist: lack of sedative potential on the EEG in the rodent. Neuropsychobiology. 1995;32:214–221. doi: 10.1159/000119238. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Fort P, Eggermann E, Cauli B, Luppi PH, Rossier J, et al. Identification of sleep-promoting neurons in vitro. Nature. 2000;40:992–995. doi: 10.1038/35010109. [DOI] [PubMed] [Google Scholar]
- Gillard M, van der Perren C, Moguilevsky N, Massingham R, Chatelain P. Binding characteristics of cetirizine and levocetirizine to human H1 histamine receptors: contribution of Lys191 and Thr194. Mol Pharmacol. 2002;61:391–399. doi: 10.1124/mol.61.2.391. [DOI] [PubMed] [Google Scholar]
- Gong H, Szymusiak R, King J, Steininger T, McGinty D. Sleep-related c-Fos protein expression in the preoptic hypothalamus: effects of ambient warming. Am J Physiol. 2000;279:R2079–R2088. doi: 10.1152/ajpregu.2000.279.6.R2079. [DOI] [PubMed] [Google Scholar]
- Gong H, McGinty D, Guzman-Marin R, Chew KT, Stewart D, Szymusiak R. Activation of c-fos in GABAergic neurones in the preoptic area during sleep and in response to sleep deprivation. J Physiol. 2004;556:935–946. doi: 10.1113/jphysiol.2003.056622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Chatelain P, Massingham R, Jonsson EN, Hammarlund-Udenaes M. Brain distribution of cetirizine enantiomers: comparison of three different tissue-to-plasma partition coefficients: K(p), K(p,u), and K(p,uu) Drug Metab Dispos. 2006;34:318–323. doi: 10.1124/dmd.105.007211. [DOI] [PubMed] [Google Scholar]
- Huang ZL, Qu WM, Li WD, Mochizuki T, Eguchi N, Watanabe T, et al. Arousal effect of orexin A depends on activation of the histaminergic system. Proc Natl Acad Sci U S A. 2001;98:9965–9970. doi: 10.1073/pnas.181330998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZL, Mochizuki T, Qu WM, Hong ZY, Watanabe T, Urade Y, et al. Altered sleep-wake characteristics and lack of arousal response to H3 receptor antagonist in histamine H1 receptor knockout mice. Proc Natl Acad Sci U S A. 2006;103:4687–4692. doi: 10.1073/pnas.0600451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZL, Urade Y, Hayaishi O. Prostaglandins and adenosine in the regulation of sleep and wakefulness. Curr Opin Pharmacol. 2007;7:33–38. doi: 10.1016/j.coph.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Honda K, Inoué S. Circadian time-dependent modulation of sleep and brain temperature (Tbr) by methylcobalamine and resultant prolongation of Tbr freerunning period in rats. Biol Rhythm Res. 1995;26:521–531. [Google Scholar]
- Ikeda M, Sagara M, Sekino Y, Shirao T, Honda K, Yoshioka T, et al. The sulphydryl reagent, N-ethylmaleimide, disrupts sleep and blocks A1 adenosine receptor-mediated inhibition of intracellular calcium signaling in the in vitro ventromedial preoptic nucleus. Neuroscience. 2001;106:733–743. doi: 10.1016/s0306-4522(01)00290-1. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Sugiyama T, Wallace CS, Gompf HS, Yoshioka T, Miyawaki A, et al. Circadian dynamics of cytosolic and nuclear Ca2+ in single suprachiasmatic nucleus neurons. Neuron. 2003;38:253–263. doi: 10.1016/s0896-6273(03)00164-8. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Ikeda-Sagara M, Okada T, Clement P, Urade Y, Nagai T, et al. Brain oxidation is an initial process in sleep induction. Neuroscience. 2005;130:1029–1040. doi: 10.1016/j.neuroscience.2004.09.057. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Yamatodani A, Ando-Yamamoto M, Tohyama M, Watanabe T, Wada H. Organization of histaminergic fibers in the rat brain. J Comp Neurol. 1988;273:283–300. doi: 10.1002/cne.902730302. [DOI] [PubMed] [Google Scholar]
- Inoué S. Sleep-promoting substance (SPS) and physiological sleep regulation. Zool Sci. 1993;10:557–576. [Google Scholar]
- Jewett RE. Effects of promethazine on sleep stages in the cat. Exp Neurol. 1968;21:368–382. doi: 10.1016/0014-4886(68)90048-4. [DOI] [PubMed] [Google Scholar]
- Karamanakos PN, Pappas P, Marselos M. Involvement of the brain serotonergic system in the locomotor stimulant effects of chlorpheniramine in Wistar rats: implication of postsynaptic 5-HT1A receptors. Behav Brain Res. 2004;148:199–208. doi: 10.1016/s0166-4328(03)00193-1. [DOI] [PubMed] [Google Scholar]
- Khateb A, Serafin M, Mühlethaler M. Histamine excites pedunculopontine neurones in guinea pig brainstem slices. Neurosci Lett. 1990;112:257–262. doi: 10.1016/0304-3940(90)90213-s. [DOI] [PubMed] [Google Scholar]
- Khateb A, Fort P, Pegna A, Jones BE, Mühlethaler M. Cholinergic nucleus basalis neurons are excited by histamine in vitro. Neuroscience. 1995;69:495–506. doi: 10.1016/0306-4522(95)00264-j. [DOI] [PubMed] [Google Scholar]
- Kubo N, Shirakawa O, Kuno T, Tanaka C. Antimuscarinic effects of antihistamines: quantitative evaluation by receptor-binding assay. Jpn J Pharmacol. 1987;43:277–282. doi: 10.1254/jjp.43.277. [DOI] [PubMed] [Google Scholar]
- Lin JS, Sakai K, Jouvet M. Evidence for histaminergic arousal mechanisms in the hypothalamus of cat. Neuropharmacology. 1988;27:111–122. doi: 10.1016/0028-3908(88)90159-1. [DOI] [PubMed] [Google Scholar]
- Lin JS, Sakai K, Jouvet M. Hypothalamo-preoptic histaminergic projections in sleep-wake control in the cat. Eur J Neurosci. 1994;6:618–625. doi: 10.1111/j.1460-9568.1994.tb00306.x. [DOI] [PubMed] [Google Scholar]
- Lin JS, Hou Y, Sakai K, Jouvet M. Histaminergic descending inputs to the mesopontine tegmentum and their role in the control of cortical activation and wakefulness in the cat. J Neurosci. 1996;16:1523–1537. doi: 10.1523/JNEUROSCI.16-04-01523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Lundius EG, Sanchez-Alavez M, Ghochani Y, Klaus J, Tabarean IV. Histamine influences body temperature by acting at H1 and H3 receptors on distinct populations of preoptic neurons. J Neurosci. 2010;30:4369–4381. doi: 10.1523/JNEUROSCI.0378-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar Doan KM, Wring SA, Shampine LJ, Jordan KH, Bishop JP, Kratz J, et al. Steady-state brain concentrations of antihistamines in rats: interplay of membrane permeability, P-glycoprotein efflux and plasma protein binding. Pharmacology. 2004;72:92–98. doi: 10.1159/000079137. [DOI] [PubMed] [Google Scholar]
- Marzanatti M, Monopoli A, Trampus M, Ongini E. Effects of nonsedating histamine H1-antagonists on EEG activity and behavior in the cat. Pharmacol Biochem Behav. 1989;32:861–866. doi: 10.1016/0091-3057(89)90049-x. [DOI] [PubMed] [Google Scholar]
- McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex. J Clin Neurophysiol. 1992;9:212–223. doi: 10.1097/00004691-199204010-00004. [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Yamatodani A, Okakura K, Horii A, Inagaki N, Wada H. Circadian rhythm of histamine release from the hypothalamus of freely moving rats. Physiol Behav. 1992;51:391–394. doi: 10.1016/0031-9384(92)90157-w. [DOI] [PubMed] [Google Scholar]
- Monti JM, Pellejero T, Jantos H. Effects of H1- and H2-histamine receptor agonists and antagonists on sleep and wakefulness in the rat. J Neural Transm. 1986;66:1–11. doi: 10.1007/BF01262953. [DOI] [PubMed] [Google Scholar]
- Nicholson AN. Antihistamines and sedation. Lancet. 1983;2:211–213. doi: 10.1016/s0140-6736(83)90185-x. [DOI] [PubMed] [Google Scholar]
- Nicholson AN, Pascoe PA, Stone BM. Histaminergic systems and sleep. Studies in man with H1 and H2 antagonists. Neuropharmacology. 1985;24:245–250. doi: 10.1016/0028-3908(85)90081-4. [DOI] [PubMed] [Google Scholar]
- Nisticò G, Rotiroti D, De Sarro A, Naccari F, Stephenson JD. Central effects of histamine and H1 and H2 receptors agonists and antagonists after intraventricular infusion in fowls. Res Commun Chem Pathol Pharmacol. 1980;27:431–450. [PubMed] [Google Scholar]
- Onodera K, Yamatodani A, Watanabe T, Wada H. Neuropharmacology of the histaminergic neuron system in the brain and its relationship with behavioral disorders. Prog Neurobiol. 1994;42:685–702. doi: 10.1016/0301-0082(94)90017-5. [DOI] [PubMed] [Google Scholar]
- Orzechowski RF, Currie DS, Valancius CA. Comparative anticholinergic activities of 10 histamine H1 receptor antagonists in two functional models. Eur J Pharmacol. 2005;506:257–264. doi: 10.1016/j.ejphar.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Parmentier R, Ohtsu H, Djebbara-Hannas Z, Valatx JL, Watanabe T, Lin JS. Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J Neurosci. 2002;22:7695–7711. doi: 10.1523/JNEUROSCI.22-17-07695.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prast H, Dietl H, Philippu A. Pulsatile release of histamine in the hypothalamus of conscious rats. J Auton Nerv Syst. 1992;9:105–110. doi: 10.1016/0165-1838(92)90050-q. [DOI] [PubMed] [Google Scholar]
- Ramesh V, Thakkar MM, Strecker RE, Basheer R, McCarley RW. Wakefulness-inducing effects of histamine in the basal forebrain of freely moving rats. Behav Brain Res. 2004;152:271–278. doi: 10.1016/j.bbr.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Reiner PB, Kamondi A. Mechanisms of antihistamine-induced sedation in the human brain: H1 receptor activation reduces a background leakage potassium current. Neuroscience. 1994;59:579–588. doi: 10.1016/0306-4522(94)90178-3. [DOI] [PubMed] [Google Scholar]
- Reuse JJ. Comparison of various histamine antagonists. Br J Pharmacol Chemother. 1948;3:174–180. doi: 10.1111/j.1476-5381.1948.tb00371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risberg AM, Risberg J, Ingvar DH. Effects of promethazine on nocturnal sleep in normal men. Psychopharmacology. 1975;43:279–284. doi: 10.1007/BF00429264. [DOI] [PubMed] [Google Scholar]
- Saitou K, Kaneko Y, Sugimoto Y, Chen Z, Kamei C. Slow wave sleep-inducing effects of first generation H1-antagonists. Biol Pharm Bull. 1999;22:1079–1082. doi: 10.1248/bpb.22.1079. [DOI] [PubMed] [Google Scholar]
- Sato I, Munakata M, Iinuma K. Histamine H1 antagonists block M-currents in dissociated rat cortical neurons. Brain Res. 2005;1057:81–87. doi: 10.1016/j.brainres.2005.07.040. [DOI] [PubMed] [Google Scholar]
- Seidel WF, Cohen S, Bliwise NG, Roth T, Dement WC. Cetirizine effects on objective measures of daytime sleepiness and performance. Ann Allergy. 1987;59:58–62. [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Sherrod TR, Loew ER, Schloemer HF. Pharmacological properties of antihistamine drugs, benadryl, pyribenzamine, and neoantergan. J Pharmacol Exp Ther. 1947;89:247–255. [PubMed] [Google Scholar]
- Slater JW, Zechnich AD, Haxby DG. Second-generation antihistamines: a comparative review. Drugs. 1999;57:31–47. doi: 10.2165/00003495-199957010-00004. [DOI] [PubMed] [Google Scholar]
- Sunshine A, Zighelboim I, Laska E. Hypnotic activity of diphenhydramine, methapyrilene, and placebo. J Clin Pharmacol. 1978;18:425–431. doi: 10.1002/j.1552-4604.1978.tb02459.x. [DOI] [PubMed] [Google Scholar]
- Taglialatela M, Timmerman H, Annunziato L. Cardiotoxic potential and CNS effects of first-generation antihistamines. Trends Pharmacol Sci. 2000;21:52–56. doi: 10.1016/s0165-6147(99)01437-6. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Lin JS, Sakai K. Characterization and mapping of sleep-waking specific neurons in the basal forebrain and preoptic hypothalamus in mice. Neuroscience. 2009;161:269–292. doi: 10.1016/j.neuroscience.2009.02.075. [DOI] [PubMed] [Google Scholar]
- Tanda G, Kopajtic TA, Katz JL. Cocaine-like neurochemical effects of antihistaminic medications. J Neurochem. 2008;106:147–157. doi: 10.1111/j.1471-4159.2008.05361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga S, Takeda Y, Shinomiya K, Hirase M, Kamei C. Effects of some H1-antagonists on the sleep-wake cycle in sleep-disturbed rats. J Pharmacol Sci. 2007;103:201–206. doi: 10.1254/jphs.fp0061173. [DOI] [PubMed] [Google Scholar]
- Tsai CL, Matsumura K, Nakayama T, Itowi N, Yamatodani A, Wada H. Effects of histamine on thermosensitive neurons in rat preoptic slice preparations. Neurosci Lett. 1989;102:297–302. doi: 10.1016/0304-3940(89)90095-5. [DOI] [PubMed] [Google Scholar]
- Wauquier A, Van den Broeck WA, Awouters F, Janssen PA. A comparison between astemizole and other antihistamines on sleep-wakefulness cycles in dogs. Neuropharmacology. 1981;20:853–859. doi: 10.1016/0028-3908(81)90078-2. [DOI] [PubMed] [Google Scholar]
- White JM, Rumbold GR. Behavioural effects of histamine and its antagonists: a review. Psychopharmacology (Berl) 1988;95:1–14. doi: 10.1007/BF00212757. [DOI] [PubMed] [Google Scholar]