

Abstract
Genetic susceptibility to type 1 diabetes (T1D) has been a subject of intensive study for nearly four decades. This article will present the history of these studies, beginning with observations of the Human Leukocyte Antigen (HLA) association in the 1970s, through the advent of DNA-based genotyping methodologies, through recent large, international collaborations and genome-wide association studies. More than 40 genetic loci have been associated with T1D in multiple studies; however, the HLA region, with its multiple genes and extreme polymorphism at those loci, remains by far the greatest contributor to the genetic susceptibility to T1D. Even after decades of study, the complete story has yet to unfold, and exact mechanisms by which HLA and other associated loci confer T1D susceptibility remain elusive.
More than 40 genetic loci have been associated with type 1 diabetes (T1D). Human leukocyte antigen (HLA) is, by far, the strongest predictor of T1D risk.
Type 1 diabetes (T1D) is a complex, multigenic disease. The first reports of genetic association to T1D were for the human leukocyte antigen (HLA) region (Singal and Blajchman 1973; Cudworth and Woodrow 1974; Nerup et al. 1974). In the nearly four decades since this discovery, researchers have searched not only to determine which alleles of which HLA-encoding genes are responsible for the T1D association but also for which other genetic loci, in addition to HLA, contribute to T1D risk, with dozens of loci reported to be associated with T1D (Bluestone et al. 2010; Pociot et al. 2010; Steck and Rewers 2011). The largest of these studies, completed in 2010, was the type 1 Diabetes Genetics Consortium (T1DGC) (Rich et al. 2006). The T1DGC is an international collaboration, conceived to create a repository of sufficient size and diversity to identify all of the genetic loci that contribute to T1D risk, through which >14,000 samples were collected and genotyped. Of all of the T1D associated genes and gene regions revealed by all of the studies, however, the HLA association remains the strongest by far, with reported odds ratios (ORs) ranging from 0.02 to >11 for specific DR-DQ haplotypes (Erlich et al. 2008). After HLA, the strongest T1D genetic association comes from polymorphism in the promoter region of the insulin gene (OR = 2.38) (Pociot et al. 2010). Only two other loci, PTPN22 and IL2RA, have ORs greater than 1.5; most are in the range of 1.1–1.3, which underscores the importance of the HLA region compared with other loci (Pociot et al. 2010). All studies of T1D genetic susceptibility must take HLA into account to interpret association data for any other candidate loci. For this reason, the focus of this article will be primarily on HLA-encoding genes, although other susceptibility loci will be described.
HLA STRUCTURE, FUNCTION, AND NOMENCLATURE
Structure
The HLA region maps to chromosome 6p21.31. The classical HLA loci are encoded in a region of DNA approximately 4 Mb, with the class II loci at the centromeric end of the region and the class I loci at the telomeric end. The region contains >200 identified genes, over half of which are predicted to be expressed. A schematic representation of the HLA region, with T1D-relevant genes indicated, is shown in Figure 1. Only some of the HLA region genes are involved in the immune response; in particular, the genes that encode the classical HLA class I (A, B, and C) and class II (DR, DQ, and DP) antigens. Genes encoding classical HLA class I and class II antigens flank a chromosomal region that is sometimes referred to as the “class III region,” which contains some immunologically relevant genes (e.g., tumor necrosis factor [TNFA]) but no classical HLA genes. Products of loci encoding the six classical class I (A, B, and C) and class II (DR, DQ, and DP) antigens are structurally similar, cell-surface proteins that bind antigenic peptides and present them to T cells. DR-encoding genes differ from those encoding DQ and DP in two important ways. First, the DRA1 gene, which encodes the α chain of the DR molecule, is essentially monomorphic and does not require genotyping. Second, the DRB1 gene is present on all chromosomes, but additional DRB genes are present on specific haplotypes. Some of the additional DRB genes, e.g., DRB2, are pseudogenes; however, three of these (DRB3, DRB4, and DRB5) encode functional polypeptide chains that can pair with the DRA1 gene product to create a functional antigen. The role of these additional DR antigens in disease susceptibility is not yet understood. Molecules resembling the classical class I antigens are encoded in the HLA region, including HLA-E, HLA-G, and the HFE gene, at the telomeric end of the HLA region, and MIC-A, in the class III region. These molecules, although structurally similar to classical class I antigens, do not have the capacity to bind and present peptides.
Figure 1.

Schematic representation of selected genes in the HLA region. Classical HLA genes are shown in black. Other T1D-associated genes are shown with patterned boxes. Gray boxes show genes not reported to be T1D-associated. Approximate boundaries of class I, class III, and class II regions are indicated by brackets.
The extracellular portions of the HLA proteins are comprised of four domains, shown schematically in Figure 2A. In the case of class II, two of the immunoglobulin-like domains are provided by each of the products of the genes encoding the α and β chains of the heterodimer. For class I, three of the extracellular domains are encoded by the HLA gene, and the fourth is provided by binding of the class I molecule to β-2 microglobulin. The outer two domains form a groove into which endogenous (for class I) or exogenous (for class II) peptides can bind and be presented to T cells. Nearly all of the polymorphism in the HLA genes resides in the regions encoding the amino acid residues that form the peptide-binding groove. Hence, the polymorphic residues affect the shape of the groove and, therefore, determine the repertoire of peptides that can bind to a given allele. Although a number of other cell-surface molecules, such as CD4, CD8, CD28, B7, and CTLA-4, are involved in mounting an immune response, the “trimolecular complex” created by the binding of the T-cell receptor (TCR) binding to the HLA/peptide complex (Fig. 2B) determines the specificity of the immune response.
Figure 2.

(A) Schematic representation of HLA class II and class I molecules. Protein domains are depicted as gray circles, plasma membrane is depicted by black ball and stick motifs, transmembrane regions are depicted as lines going through the plasma membrane, and the peptide antigen is depicted as a black diamond. (B) Schematic representation of the trimolecular complex. HLA molecule and peptide are depicted as in A; T-cell receptor is depicted as a gray Y-shape that contacts both the HLA molecule and the peptide antigen.
HLA Class II Antigens
The three HLA class II antigens include DR, DQ, and DP. HLA class II molecules are heterodimeric, consisting of α and β chains, encoded by separate genetic loci, i.e., the products of the DPA1 and DPB1 genes encode the DPα and DPβ polypeptides that combine to create the DP heterodimeric protein. The DRA1 gene is essentially monomorphic, so DR genotyping is performed only on the DRB1 gene, which encodes the β chain of the antigen and, occasionally, on the other DRB genes that are present only on certain haplotypes. All HLA regions have a copy of DRB1, and some DRB1 haplotypes also include other DRB genes, including DRB3, DRB4, and DRB5. Whether or not these additional genes confer, T1D risk is unclear at present and is just beginning to be studied. The DQA1, DQB1, DPA1, and DPB1 genes are all polymorphic. For DQ and DP, the genetic diversity produced by formation of heterodimers of α and βchains encoded in trans, along with formation of heterodimers encoded in cis, greatly increases the functional diversity of the DQ and DP molecules. In other words, a cell with two different DQA1 alleles and two different DQB1 alleles can be capable of producing four different DQ proteins. The ability to form DQ heterodimers encoded in trans is a leading hypothesis for why the T1D risk conferred by the DR3/DR4 genotype is greater than the additive risk for the two haplotypes (Fig. 3). An alternative hypothesis can be drawn from analysis of the T1DGC genome-wide association study (GWAS) single-nucleotide polymorphism (SNP) data in HLA-stratified subgroups, in which the strongest signals in DRB1*04:01-positive individuals were in protein networks involved in antigen procession and presentation, whereas the strongest signals in DRB1*03:01-positive individuals were in pathways involved with stress response and inflammation (Brorsson et al. 2010).
Figure 3.

Trans-encoded heterodimers. DR3 and DR4 haplotypes from the highly predisposing DR3/DR4 genotype are shown; putative high-T1D-risk heterodimers encoded in trans are circled.
HLA Class I Antigens
HLA class I antigens, known as A, B, and C, are encoded as a single chain that forms a complex with the essentially nonpolymorphic molecule β-2-microglobulin (Fig. 2A). All three of the class I loci are extremely polymorphic (see Table 1), and the majority of the polymorphisms can be found in the exons encoding the α1 and α2 domains of the mature protein (encoded by exons 2 and 3), which form the peptide-binding groove (Fig. 2A).
Table 1.
Extent of polymorphism at the classical HLA loci
| Locus | Number of alleles |
|---|---|
| A | 1601 |
| B | 2125 |
| C | 1102 |
| DRA | 7 |
| DRB | 1027 |
| DRB1 | 928 |
| DRB2 | 1 |
| DRB3 | 57 |
| DRB4 | 15 |
| DRB5 | 19 |
| DRB6 | 3 |
| DRB7 | 2 |
| DRB8 | 1 |
| DRB9 | 1 |
| DQA1 | 44 |
| DQB1 | 153 |
| DPA1 | 32 |
| DPB1 | 149 |
| Total | 6240 |
Reported number of alleles for each of the classical HLA loci are shown. DRB alleles are shown in total as well as by locus (in italics). Data are taken from the HLA informatics group website (http://www.ebi.ac.uk/imgt/hla/stats.html) and are current as of July 1, 2011.
HLA Function
The function of the HLA/peptide complex in directing the immune response suggests a role for HLA in both the immune response to environmental pathogens and in autoimmune disease. HLA was originally identified through its role in transplant rejection (Thorsby 1974). The importance of HLA typing in disease association studies was noted as early as 1975 (Oh and MacLean 1975). HLA has been implicated in the etiology of >100 diseases, including, but not limited to, complex autoimmune diseases, such as type 1 diabetes, rheumatoid arthritis, and multiple sclerosis; cancers, such as Hodgkin’s disease; infectious diseases, such as malaria and AIDS; and other diseases, such as narcolepsy (Lechler and Warrens 1999).
HLA Nomenclature
The HLA region is the most polymorphic observed in the human genome, with >6543 unique allele sequences reported as of July 2011 (http://www.ebi.ac.uk/imgt/hla/stats.html) (Table 1). The extreme polymorphism of the HLA-encoding loci creates challenges in maintaining a consistent nomenclature among laboratories and studies. Development of new and better genotyping technologies has led to a dramatic increase in the number of reported HLA alleles, which, in turn, has led to an evolution of the nomenclature. Before the advent of molecular genotyping technology, HLA variation was determined with cell-based assays using antisera derived from multiparous women. These antisera needed to be carefully characterized, were difficult to standardize among laboratories, and recognized only a fraction of the variation that is now known to exist for the HLA-encoding loci. Nomenclature for serologic HLA typing was simple, consisting of a letter to describe the locus and a number to specify the allele (e.g., A1 or DR4). As serologic genotyping technology became more refined, and as more variation was discovered in HLA, some of the original designations were subdivided, e.g., the “DR2” group was subdivided into “DR15” and “DR16.”
The emergence of DNA-based genotyping led to the creation of a nomenclature system that included the locus name, followed by an asterisk, followed by a numerical designation for the allele, for example, DRB1*0401. For most loci, the first two digits of the numerical designation refer to the serologic specificity of a particular allele. DRB1*0401, for example, reacts with antisera that are classified in the DR4 group. DRB1*0405 would react with the same DR4 antisera, but DRB1*0101 or DRB1*0301 would not. DR antigens were originally categorized into 10 groups, DR1 through DR10, by serologic typing. When DNA-based nomenclature was adopted, designations reflected the state of serologic typing at the time. Consequently, alleles that react with DR2-specific antisera were not named DRB1*02xx but, rather, DRB1*15xx or DRB1*16xx.
DPB1 is an exception to the serology-based nomenclature. Because most DPB1 alleles were discovered by DNA-based genotyping, their allele designations were assigned sequentially (e.g., DPB1*2301, DPB1*2401, DPB1*2501, etc.), rather than by serologic reactivity, such that no inferences can be made about the antigen itself simply from the allele designation. Silent polymorphisms add additional complexity to HLA nomenclature; these were originally designated as one additional digit, e.g., A*68011, but later changed to an additional two digits, e.g., A*680101. Polymorphic sequences in introns are also sometimes designated as an additional two digits, and low or null expression of an allele can be designated with a letter. Thus, HLA allele names can be as complex as A*03010102N, which, depending on the level of resolution of the genotyping and the preference of the investigator, could be referred to as A3, A*03, A*0301, A*030101, A*03010102, or A*03010102N. This variation in reported nomenclature complicates comparison or merging of data sets generated in different laboratories.
In 2010, the nomenclature system was again changed, this time to add a colon delimiter between the sections of an allele designation, such that each section is no longer limited to two digits, e.g., A*01:01:01. This change was implemented, in part, to clarify reporting for serologic groups with <99 alleles. For example, the B*15 allele discovered after B*15:99 can now be reported as B*15:100. Its designation in the previous system was B*9501 to avoid confusion with B*1510. Tools are available to convert nomenclature from one system to another, but reporting of HLA genotyping data is still a mixed bag, with some papers utilizing the new nomenclature, other papers using the old, and others still using old, serologic nomenclature.
The primary amino acid sequence of an HLA protein is specified by the first two fields of its designation, for example, A*01:01 and A*01:02 encode slightly different polypeptides, but A*01:01:01 and A*01:01:02 differ only by a silent polymorphism and are translated into exactly the same polypeptide. Because the protein encoded by the gene, rather than the gene itself, is (presumably) the source of susceptibility to or protection from disease, most disease association studies report HLA alleles using the first two fields of the designation to reduce ambiguity and facilitate statistical analysis.
HLA GENOTYPING METHODOLOGY
Dozens to thousands of alleles exist for each of the HLA genes (Table 1). The extent of this polymorphism necessitates a much more complex and costly process for genotyping than those commonly used for microsatellites or SNPs. HLA genotyping cost is driven in part by level of resolution. Paradoxically, increased resolution of HLA genotyping can decrease statistical power of HLA disease association analyses by increasing the number of categories and, therefore, reducing the numbers seen for each allele category. However, low-resolution genotyping necessarily bins HLA alleles together and can mask the effects of individual alleles. For example, in general, DRB1*04:xx alleles are highly predisposing for T1D. DRB1*04:03, however, is T1D protective. Low-resolution genotyping for DRB1*04, without allele-level resolution, could mask the protective effect of DRB1*04:03. In populations where DRB1*04:03 is at a high frequency, its protective effect could decrease the apparent predisposing effect of high-risk DRB1*04:xx alleles, such as DRB1*04:05. Table 2 shows the risk for various haplotypes including DRB1*04xx alleles seen in the T1DGC. Genotyping methodologies and their capacity for resolution are discussed below.
Table 2.
DRB1-DQA1-DQB1 haplotypes that reached statistical significance in the published T1DGC data set
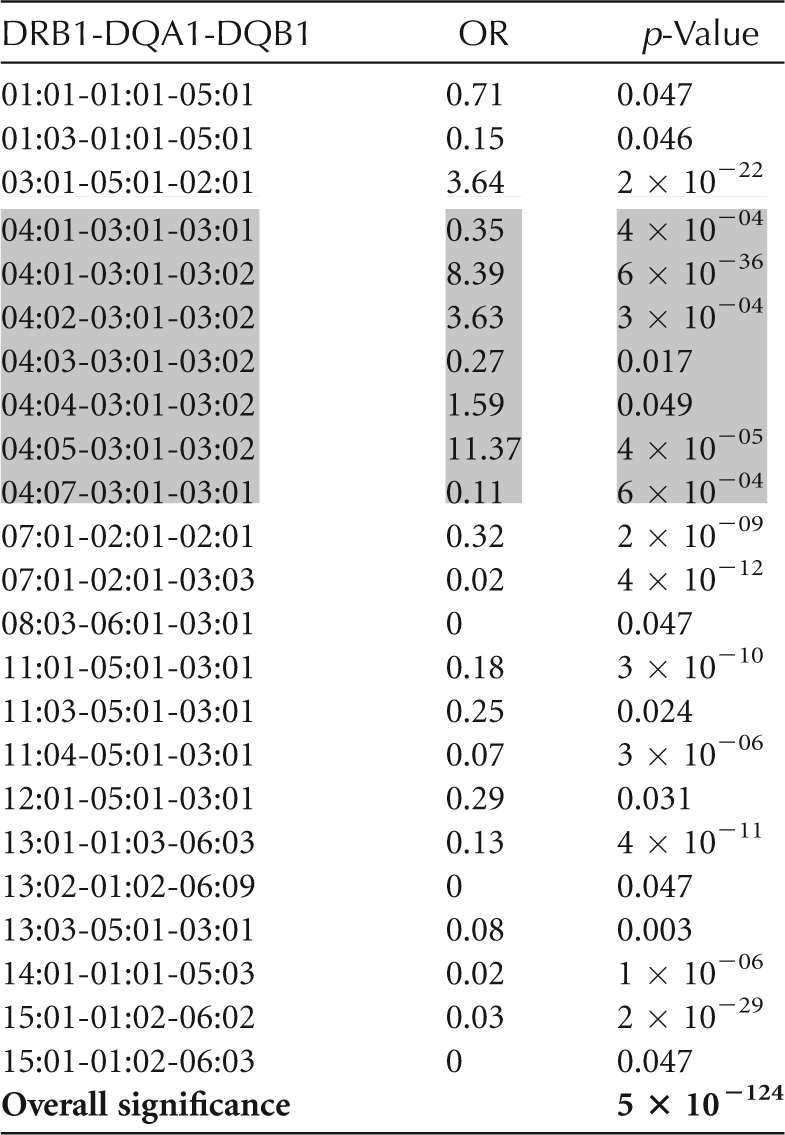
DR4 haplotypes are in shaded boxes. Additional data can be found in Erlich et al. (2008).
For HLA genes, the intron-exon structure of the gene corresponds to the structure of the encoded protein (Fig. 2A). For both class I and class II genes, exon 1 encodes the signal sequence. For class II genes, exon 2 encodes the domain that is furthest from the cell membrane and participates in the formation of the peptide-binding groove (α1 for the α chain and β1 for the β chain). The domain encoded by exon 3 (α2 for the α chain and β2 for the β chain) does not make contact with the peptide, and exon 4 encodes the transmembrane region. For class I, exons 2 and 3 encode the two immunoglobulin-like domains (α1 and α2) that form the peptide-binding groove, exon 4 encodes the α3 domain, which does not contact the peptide, and exon 5 encodes the transmembrane region. Minimally, complete genotyping to determine the primary sequence for all of the peptide-binding pockets for the classical HLA loci in a given individual requires genotyping the following: DRB1 exon 2 (and exon 2 for DRB3, DRB4, and DRB5 when present), DQA1 exon 2, DQB1 exon 2, DPA1 exon 2, A exons 2 and 3, B exons 2 and 3, and C exons 2 and 3. To further increase resolution, genotyping of DQB1 exon 3 and exon 4 for the class I loci may be included.
The earliest methods of determining HLA genotypes involved cell-based assays that used serum from multiparous women. The number of categories was small, reflecting the major antigenic epitopes on the HLA. The extreme diversity of the HLA genes was only recognized with the advent of DNA-based genotyping technology, which led to an exponential increase in the number of recognized alleles. DNA-based genotyping methods fall into three general categories, sequence-specific priming (SSP), sequence-specific oligonucleotide (SSO) probe, and sequence-based typing (SBT).
SSP involves multiple polymerase chain reaction (PCR) amplifications of genomic DNA with primer pairs designed to produce a product only if a given polymorphism is present in the sample. Results are visualized as the presence or absence of a product by gel electrophoresis. Genotypes consistent with the data are deduced by comparing data to expected patterns for all genotype combinations possible from known alleles. In most cases, multiple genotypes are consistent with the data. The resolution of this technique is dependent on the number of initial amplifications; thus, the higher the desired resolution, the more template, amplification reagents, and gels are required. Polymorphisms that are not represented in the test kit, including novel polymorphisms, will be missed by this technique.
SSO involves a single amplification of the locus to be tested, followed by hybridization of the PCR product with a set of oligonucleotide probes corresponding to known polymorphisms. Initial SSO technology involved immobilization of the PCR product, followed by multiple rounds of hybridization with individual labeled probes. Later versions of SSO technology use a single hybridization of the labeled PCR product to a set of oligonucleotide probes immobilized to a solid support, such as a nylon membrane or bead. Advantages of this technology include the single amplification and hybridization to test each locus, allowing genotyping to be performed with a smaller amount of template than what is required for SSP. The level of resolution of the assay is dependent on the number of oligonucleotide probes included. Like SSP, SSO only queries known polymorphisms, so novel polymorphisms will be missed.
SBT produces higher resolution than either SSP or SSO, because every position of the target sequence is included, rather than just selected polymorphic motifs. SBT also has the advantage of requiring a small amount of template compared with SSP. Until recently, however, the cost of SBT was significantly higher than that of SSP or SSO. Even with SBT, the two alleles in a genotype could not always be distinguished easily.
The most recent development in HLA genotyping technology uses next-generation sequencing (NGS) technology in which hundreds to thousands of sequence reads of polymorphic exons are generated from single molecules. With sequence read lengths long enough to span an exon in an HLA locus, the two sequences originating from two chromosomes in a sample can be determined individually. Thus, the only remaining ambiguity comes from assembling exons in loci with more than one polymorphic exon, or from polymorphisms that lie outside the tested exons. HLA genotyping data generated by NGS provides the highest resolution presently available.
Multiple technologies and varying resolution levels make combining data from different studies quite challenging. The same allele can be given two different designations, depending on the level of resolution used to genotype it. As resolution improves and costs of new technologies decrease, consistency among studies is improving; however, caution must be used when interpreting genotyping data from different studies or different laboratories.
HLA ASSOCIATIONS WITH T1D
Sorting out HLA associations is complicated not only by the extremely large numbers of reported alleles at the HLA genetic loci but also by differences in allele frequencies and haplotypic combinations among populations, incomplete penetrance of the HLA susceptibility loci, and epistatic interactions of HLA with other susceptibility factors. Studies of HLA association with T1D have been ongoing for nearly 40 years. By far, the majority of studies of HLA association with T1D have used Caucasian subjects. Although great advances have been made, more is still to be learned, especially in non-Caucasian populations.
CLASS II: DR-DQ
The association of specific HLA alleles with T1D is remarkable in several respects: (1) for DRB1 and DQB1, the risk is determined by specific combinations of DRB1 and DQB1 alleles rather than by genotypes for individual loci; (2) both susceptible and protective strong, highly significant DR-DQ associations with T1D are seen, dependent on the particular DR-DQ haplotype; (3) multiple haplotypes are positively associated with T1D (susceptible), and many haplotypes are negatively associated (protective); and (4) specific genotypic combinations (most notably DRB1*03:01-DQA1*05:01-DQB1*02:01, DRB1*04:xx-DQA1*03:01-DQB1*03:02 heterozygotes, referred to as DR3/DR4) are associated with increased risk. In addition to this complex pattern of DR-DQ associations, which represent the major genetic determinant of T1D risk, other HLA loci, such as DPB1 and the class I loci (see below), contribute significantly to genetic risk for T1D. Based on data from the T1DGC, Table 2 shows a subset of the DRB1-DQA1-DQB1 haplotypes that have the strongest effect on T1D risk. DR4 haplotypes are shaded to illustrate the extent of risk heterogeneity in that subgroup (Erlich et al. 2008).
Although PCR-based HLA typing has greatly refined and increased our understanding of these HLA associations with T1D, many of the genetic associations noted above were initially observed and reported based on serological typing. Following the development of DR serology, the association of DR3 and DR4 with T1D was reported (Rodey et al. 1979; Solow et al. 1979). Some reports noted that an increased risk for DR3/DR4 heterozygotes relative to DR3/DR3 and DR4/DR4 homozygotes was observed (Deschamps et al. 1980). In addition, a serological epitope, TA10, was found to distinguish between high- and low-risk DR4 serotypes (Tait and Boyle 1986; Held et al. 1999).
PCR-based sequence analysis showed that the high- and low-risk DR4 haplotypes differed at the DQB1 locus; high-risk DQB1*0302 (TA10+) differed from low-risk DQB1*0301 (TA10−). DQB1*0301 and *0302 differ at four amino acid positions encoded by exon 2 sequences. At codon 57, DQB1*0302 encodes an Ala whereas DQB1*0301 encodes an Asp. Other pairs of DQB1 alleles that differed in their T1D association also differed at a codon for Ala, Ser, or Val versus Asp. Structural analysis indicated that the Asp at position 57, a residue in pocket P9 of the peptide-binding groove, contributes to a salt bridge that is absent in the presence of the other neutral amino acids at this position. A model was proposed in which Asp-57 confers protection, whereas the other amino acids at this position are associated with neutral or susceptible DQB1 alleles (Todd et al. 1987; Horn et al. 1988). In support of this model, sequence analysis of the A-β gene in the nonobese diabetic (NOD) mouse, an inbred strain widely used as a mouse model for T1D, showed the presence of His-Ser at positions 56 and 57, whereas the nonsusceptible parent strain, NON, has a Pro-Asp at these positions (Singer et al. 1998). Of note, however, the mouse NOD strain does not express the E-β gene, the homolog to DRB1. Expression of I-E in the NOD mouse was shown to prevent insulitis, even in the presence of Ser at position 57, suggesting that the identity of the amino acid residue at position 57 of I-A in the mouse, or DQB1 in the human, was not sufficient to fully explain disease susceptibility (Miyazaki et al. 1990). As noted below, polymorphisms at the DRB1 gene also influence T1D susceptibility (Erlich et al. 1991). Moreover, not all Asp-57-containing alleles are protective; some, such as the DRB1*04:05-DQB1*04:01 and DRB1*04:05-DQB1*04:02 haplotypes, common in Asia, confer susceptibility (Erlich et al. 2008). Notwithstanding these caveats and exceptions, the correlation of the amino acid residue at DQB1 position 57 with T1D risk is striking, if not absolute.
On the DRB1*04 haplotype, polymorphism at the DRB1 locus also influences T1D risk. This is illustrated by the different T1D risk associated with different DRB1 alleles linked to the same DQA1-DQB1 haplotype, DQA1*03:01-DQB1*03:02 (Table 2). The DRB1*04:05-DQA1*03:01-DQB1*03:02 haplotype has an OR = 11.37, whereas the DRB1*04:03-DQA1*03:01-DQB1*03:02 haplotype has an OR = 0.27. Table 2 shows the risk for all DR4 haplotypes reported to be significantly T1D associated in the T1DGC data set (Erlich et al. 2008).
One of the striking patterns of T1D association with DR-DQ haplotypes is the observation, reported in many different studies, that DRB1*03:01-DQA1*05:01-DQB1*02:01/DRB1*04:xx-DQA1*03:01-DQB1*03:02 heterozygotes have a higher risk than do homozygotes for either haplotype. Because the DQ molecule is a heterodimer, up to four DQ antigens can be expressed on a given cell; thus, trans-encoded DQ heterodimers, in particular, the one encoded by DQA1*0501 from the DRB1*03 haplotype and DQB1*0302 encoded by the DRB1*04 haplotype, which has never been seen encoded in cis, may help explain the high risk of the DR3/DR4 heterozygous genotype (Fig. 3).
CLASS II: DP
The DP molecule is encoded by the DPA1 and DPB1 genes. DPA1 has only a small number of alleles, and either DPA1*01:03 or DPA1*02:01 is found on most haplotypes. Most of the DP variation is provided by the DPB1 gene. Although not as strong as the effect of the established predisposing and protective DR-DQ haplotypes, variation at DPB1 also contributes to T1D risk (Noble et al. 2000; Cucca et al. 2001; Valdes et al. 2001; Cruz et al. 2004; Stuchlikova et al. 2006; Baschal et al. 2007; Varney et al. 2010). Demonstrating that an observed association in the HLA region is a true disease susceptibility effect, and not due simply to linkage disequilibrium (LD) with risk DR-DQ haplotyes, is a necessary step in establishing other genes in the HLA region as potential T1D risk loci. This can be achieved by a variety of methods. One method is to stratify data based on DR-DQ haplotypes and examine other loci within subsets. This method is useful for DR3 haplotypes, which are nearly invariable (DRB1*03:01-DQA1*05:01-DQB1*02:01) in all populations except Africans. Because DR3 is quite common in T1D patients, assessing modulation of DR3 risk by other HLA region loci (e.g., HLA DPB1, HLA class I, TNFA) is relatively straightforward (Robinson et al. 1993; Noble et al. 1996, 2006, 2008; Aly et al. 2006). For other, more variable haplotypes, stratification leads to small numbers per category and very little statistical power. An alternate approach adjusts for LD by estimating the expected frequency of a given HLA region allele based on the observed LD to DR-DQ haplotypes among controls in the data set. Then the observed frequency among patients is compared with the LD-based expected frequency, assuming the null hypothesis of no association. This approach has been previously described (Valdes et al. 2005). This approach, as well as others, showed that DPB1*0301 and *0202 showed a positive association with T1D and DPB1*0402 showed a negative association (Noble et al. 2000; Cucca et al. 2001; Cruz et al. 2004; Varney et al. 2010).
Other observed associations in the HLA region, such as the class I and class III regions discussed below, also require an adjustment for the extensive LD between alleles at the target loci and the established DR-DQ risk alleles and haplotypes in this region.
HLA Class I
Initial reports of HLA association with T1D pointed to the class I loci B8 and B18. (Singal and Blajchman 1973; Cudworth and Woodrow 1974; Nerup et al. 1974). Later, these associations were attributed to LD of the class I alleles with predisposing HLA class II loci (Rodey et al. 1979; Solow et al. 1979). The primary HLA associations with T1D are now well established to be with class II genes, specifically those encoding the DR and DQ molecules, as described above. However, alleles at the class I loci A and B loci have also been shown to affect T1D susceptibility independently from class II; in particular, class I alleles appear to play a role in age of onset of T1D (Noble et al. 2002, 2010; Tait et al. 2003; Valdes et al. 2005; Nejentsev et al. 2007; Howson et al. 2009b). T1D is believed to result from an initial triggering event, followed by gradual autoimmune destruction of the pancreatic β cells, until the point that the residual β cells are insufficient to meet the insulin demands of the body, leading to hyperglycemia and disease diagnosis. Because the trigger is unknown, the autoimmune process is largely undetected until the point of diagnosis. The rate of the autoimmune destruction is not known, and may vary among individuals. The end point of the autoimmune process is pancreatic β cell destruction by cytotoxic T cells (Tc), and the CD8 molecules on Tc cells act as coreceptors for TCR binding to class I antigens on target cells. Thus, the notion that the class I allele with its bound peptide affects the rate of the autoimmune destruction is quite plausible.
Some of the early reports of class I association were with A*24 alleles. In a Japanese study, patients with the A*24 allele were shown to have little or no residual β-cell function compared with other T1D patients, consistent with the idea that the A*24 allele facilitates more rapid and complete destruction of β cells (Nakanishi et al. 1993, 1995). In patients with the highest-risk DR3/DR4 genotype, those that also carry the A*24:02 allele have a significantly lower age of onset than those who do not have A*24 (Noble et al. 2002). DNA-based genotyping methods have allowed examination of individual alleles in the A*24 serogroup. Although most A*24 alleles do show a predisposing effect, a study of T1D in a Filipino population showed that A*24:07, which differs from A*24:02 by the substitution of Gln for His at position 70, is not predisposing for T1D (Bugawan et al. 2002).
B*39 alleles appear to be the most highly predisposing class I alleles, with the B*39:06 allele most commonly associated with T1D (Ilonen et al. 1992; Nejentsev et al. 1997; Reijonen et al. 1997; Valdes et al. 2005; Noble et al. 2010). B*3906 increases T1D risk for moderate susceptibility haplotypes, such as DR1, DR8, and even DR16 (a type of DR2) haplotypes (Valdes et al. 2005; Baschal et al. 2011).
HLA-C appears to have little, if any, effect on T1D susceptibility. Observed HLA-C T1D associations appear to result from LD of HLA-C with predisposing or protective DR-DQ or HLA-B alleles (Valdes et al. 2005; Noble et al. 2010).
NON-HLA T1D SUSCEPTIBILITY LOCI IN THE HLA REGION
The region between the class II and class I genes is commonly referred to as “class III,” although it contains no loci encoding classical HLA loci. The class III region does include several immunologically relevant genes, which include TNFA and the complement C4-encoding genes C4A and C4B. The gene encoding the class I-like molecule MIC-A is also located between the classical HLA class II and class I genes, although MIC-A is sometimes considered to be in the class I, rather than the class III, region. Candidate gene studies have been reported on all of these loci and are described below. In addition, analysis of GWAS data from the T1DGC revealed T1D associations in the class III region that are not attributable to LD with classical HLA-encoding loci (Valdes et al. 2010).
The TNFA gene has been extensively studied for T1D association, especially for the −238 and −308 promoter SNPs, with conflicting reports. Some studies, particularly early studies, reported data that were not adjusted to reflect LD with DR-DQ; thus, strong disease associations were observed, e.g., Perez et al. (2004). Other studies, in which LD was taken into account, did not report significant association (Ilonen et al. 1992; Nishimura et al. 2003; Noble et al. 2006).
The genes-encoding complement C4 (C4A and C4B) vary in copy number, and this variation is associated with systemic lupus erythematosus (SLE) (Yang et al. 2007). A number of studies report T1D association for C4A null patients and for the C4B “short” allele (Rich et al. 1985; Caplen et al. 1990; Marcelli-Barge et al. 1990; Jenhani et al. 1992; Lhotta et al. 1996).
The “MHC class I-related” gene, MIC-A, produces a cell-surface antigen that resembles classical HLA class I molecules. MIC-A appears to be induced in response to stress in the intestinal epithelium and is recognized by γδ intestinal epithelial cells (IELs) (Groh et al. 1998, 2002). Several studies have implicated the allele MIC-A5 in T1D susceptibility (Gambelunghe et al. 2000; Bilbao et al. 2002; Gupta et al. 2003).
NON-HLA T1D SUSCEPTIBILITY LOCI OUTSIDE THE HLA REGION
After HLA, the next-strongest genetic association with T1D is seen for the insulin gene. This association was initially described as a difference in size for three classes of alleles of a region flanking the insulin gene (Bell et al. 1984; Permutt et al. 1984). The insulin promoter region was found to have a variable number of tandem repeats (VNTR) that could be genotyped to study disease association, with the shorter alleles (class I) predisposing for T1D and the longer alleles (class III) showing a negative (protective) association (Rotwein et al. 1986). Differences in insulin expression levels from these alleles are postulated to account for the susceptibility, perhaps by modulating thymic expression of insulin and affecting T-cell education (Pugliese 2005). VNTR typing of the insulin gene has largely been supplanted by genotyping of a tagging SNP, −23HphI (Meigs et al. 2005). This SNP has the second-highest OR (2.38) for T1D in the T1DGC GWAS studies (Pociot et al. 2010).
The PTPN22 gene encodes a protein tyrosine phosphatase important in down-regulation of the immune response. The association of PTPN22 was first described by Bottini in 2004 and quickly replicated in other studies (Bottini et al. 2004; Onengut-Gumuscu et al. 2004; Smyth et al. 2004; Ladner et al. 2005; Qu et al. 2005; Zheng and She 2005). This gain-of-function mutation affects binding of the PTPN22 gene product to Csk and is associated with multiple autoimmune diseases, including rheumatoid arthritis, Grave’s disease, juvenile idiopathic arthritis, SLE, and vitiligo (Begovich et al. 2004; Velaga et al. 2004; Hinks et al. 2005; Bottini et al. 2006).
GENOME-WIDE ASSOCIATION STUDIES
All of the associations discussed above were based on the candidate gene approach, the analysis of genetic variation in genes with an immunological function, or an islet-related function that might be plausibly connected to autoimmunity and/or insulin-related dysfunction. The development of high-throughput SNP genotyping technologies allowed “hypothesis-free” GWAS with hundreds of thousands of different SNPs. Although hypothesis-free, in the sense of not relying on specific candidate genes, the GWAS approach assumed that the panel of SNPs on the GWAS chips, whose variant allele frequency was generally greater than 5%, could “tag” potentially causal genomic polymorphisms, owing to LD. Given the very large number of SNPs used in the genotyping analysis, achieving genome-wide statistical significance for an individual SNP association required a very large number of cases and controls and, consequently, most of these GWAS have been performed by international collaborations and consortia, such as the T1DGC.
Several very large GWAS analyses for T1D have been reported recently, with the largest body of data generated on the T1DGC collection of samples (Barrett et al. 2009; Concannon et al. 2009; Qu et al. 2010). Data from these studies can be found at www.t1dbase.org. The most significant associations (represented by the highest peaks in the “Manhattan plots” with the lowest p-values) were in regions previously identified by linkage studies and candidate gene studies, namely, the HLA region and insulin. Of critical importance, however, was the identification of >40 different genetic regions with highly significant association (Pociot et al. 2010). Although the ORs conferred by these non-HLA-associated regions were quite modest, generally ranging from 1.1–1.3, the identification of these regions, each with a plausible candidate gene, provided significant insight into the pathological process that can lead to T1D. Not unexpectedly, most of these genes identified by GWAS had immunological functions, whereas some had metabolic functions (Pociot et al. 2010).
This powerful approach of using a dense panel of SNPs was recently applied to the analysis of the entire HLA region (∼4 Mb). This region is highly gene-rich, with many of the genes having known immunological functions. The hypothesis-free strategy could address the fundamental question of whether variation at the classical HLA loci was, in fact, the major risk determinant in the HLA region and potentially identify any other significantly associated gene regions. The results from this approach showed that the major peak of association was with SNPs in the DR-DQ region and, following adjustment for DR-DQ in the conditional logistic regression model, the next-highest peak was in the HLA-B region (Howson et al. 2009a). Following adjustment for HLA-B, the next-highest peaks were for SNPs in the HLA-A and DP regions.
GWAS studies based on SNP chips query common variants in the genome. With the development of high-throughput “next-generation DNA sequencing” technologies, researchers are now beginning to examine the effects of rare variants, with the idea that multiple low-penetrance rare variants in a locus may combine to create a phenotype. In other words, a single, common, causative polymorphism may not exist for a given susceptibility locus, but a cluster of rare variants may indicate a susceptibility locus. In addition, the diploid nature of the human genome is largely overlooked by traditional GWAS studies, in which the phase of SNPs on a chromosome cannot be determined (Tewhey et al. 2011). If, for example, two inactivating mutations are found in a single gene, knowing whether or not they reside on the same chromosome leads to different interpretations of the data. Both mutations on the same copy of the gene would indicate one dysfunctional and one functional allele, whereas a mutation on each chromosome would indicate two dysfunctional alleles. New sequencing technologies are addressing this issue by producing long sequence reads from single molecules, or by physical separation of chromosomes before sequencing. The results of studies of rare variants and phase-specific sequencing will represent yet another step forward in our understanding of the complexity of genetic susceptibility to T1D.
CONCLUSIONS
More than 35 years have passed since the first genetic associations with T1D were reported. After decades of research and thousands of reports, HLA remains, by far, the strongest predictor of T1D risk. However, the complexity of the genetics of T1D is far greater than might have been predicted by the early reports. “HLA” does not refer to a single genetic locus, but, rather, to a region of the genome that includes genes encoding three classical HLA class II and three classical class I antigens as well as a number of other genes whose products may also influence susceptibility. Polymorphisms in genes outside the HLA region, most notably the insulin and PTPN22 genes, also influence T1D susceptibility but to a far lesser extent than the classical HLA loci. The importance of considering HLA context in the analysis of genetic association data from non-HLA loci cannot be overstated.
The genes known to affect T1D susceptibility can be grouped into three general categories: immune function, insulin expression, and β-cell function. Most of the T1D susceptibility loci encode products that function in the immune response. After HLA, the strongest susceptibility locus is in the insulin gene itself, in which promoter polymorphisms affect insulin expression levels. Other candidate loci are involved in β-cell function.
Although much is known about the effects of certain HLA alleles on disease risk, the extreme polymorphism of the HLA loci, with new alleles still being discovered, and the LD in the HLA region, complicates association analyses. In addition, the exact biological mechanism of HLA-conferred susceptibility remains elusive. The highest genetic risk for T1D in Caucasians is conferred by the heterozygous genotype DR3/DR4, in which the DR4 is not DRB1*04:03, and the DR4 haplotype contains the DQB1*03:02 allele. This may be owing to the set of DQ heterodimers that are encoded in trans in the genotype. Or it may be because of factors that are not yet understood. In any case, the risk conferred by the DR3/DR4 genotype is so high that other susceptibility loci appear to have little or no effect. Separating non-DR3/DR4 individuals from the DR3/DR4 high-risk group has been useful in revealing other susceptibility loci.
Genetic susceptibility to T1D that is not attributable to classical HLA, INS, and PTPN22 genes appears to be conferred by many loci, each of which has a small (OR 1.1–1.3) effect. Loci identified to date have been identified through LD with common SNPs (minor allele frequency = 5% or greater). New sequencing technologies will allow identification of rare variants that may, in combination, reveal additional T1D susceptibility loci, and phase-specific sequencing data will allow better assessment of the biological basis of observed genetic associations.
Although much is known about the genetics of susceptibility to T1D, more data are needed to completely unravel this tangled web.
ACKNOWLEDGMENTS
The authors are grateful to Dr. Ana Maria Valdes for assistance with figure preparation and to Dr. Nancy Keller for assistance with literature searches.
Footnotes
Editors: Jeffrey A. Bluestone, Mark A. Atkinson, and Peter R. Arvan
Additional Perspectives on Type 1 Diabetes available at www.perspectivesinmedicine.org
REFERENCES
- Aly TA, Eller E, Ide A, Gowan K, Babu SR, Erlich HA, Rewers MJ, Eisenbarth GS, Fain PR 2006. Multi-SNP analysis of MHC region: Remarkable conservation of HLA-A1-B8-DR3 haplotype. Diabetes 55: 1265–1269 [DOI] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, et al. 2009. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 41: 703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baschal EE, Aly TA, Babu SR, Fernando MS, Yu L, Miao D, Barriga KJ, Norris JM, Noble JA, Erlich HA, et al. 2007. HLA-DPB1*0402 protects against type 1A diabetes autoimmunity in the highest risk DR3-DQB1*0201/DR4-DQB1*0302 DAISY population. Diabetes 56: 2405–2409 [DOI] [PubMed] [Google Scholar]
- Baschal EE, Baker PR, Eyring KR, Siebert JC, Jasinski JM, Eisenbarth GS 2011. The HLA-B*3906 allele imparts a high risk of diabetes only on specific HLA-DR/DQ haplotypes. Diabetologia 54: 1702–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, et al. 2004. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75: 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell GI, Horita S, Karam JH 1984. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 33: 176–183 [DOI] [PubMed] [Google Scholar]
- Bilbao JR, Martin-Pagola A, Calvo B, Perez de Nanclares G, Gepv N, Castano L 2002. Contribution of MIC-A polymorphism to type 1 diabetes mellitus in Basques. Ann NY Acad Sci 958: 321–324 [DOI] [PubMed] [Google Scholar]
- Bluestone JA, Herold K, Eisenbarth G 2010. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 464: 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, et al. 2004. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 36: 337–338 [DOI] [PubMed] [Google Scholar]
- Bottini N, Vang T, Cucca F, Mustelin T 2006. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol 18: 207–213 [DOI] [PubMed] [Google Scholar]
- Brorsson C, Tue Hansen N, Bergholdt R, Brunak S, Pociot F 2010. The type 1 diabetes - HLA susceptibility interactome—identification of HLA genotype-specific disease genes for type 1 diabetes. PloS One 5: e9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugawan TL, Klitz W, Alejandrino M, Ching J, Panelo A, Solfelix CM, Petrone A, Buzzetti R, Pozzilli P, Erlich HA 2002. The association of specific HLA class I and II alleles with type 1 diabetes among Filipinos. Tissue Antigens 59: 452–469 [DOI] [PubMed] [Google Scholar]
- Caplen NJ, Patel A, Millward A, Campbell RD, Ratanachaiyavong S, Wong FS, Demaine AG 1990. Complement C4 and heat shock protein 70 (HSP70) genotypes and type I diabetes mellitus. Immunogenetics 32: 427–430 [DOI] [PubMed] [Google Scholar]
- Concannon P, Rich SS, Nepom GT 2009. Genetics of type 1A diabetes. New Engl J Med 360: 1646–1654 [DOI] [PubMed] [Google Scholar]
- Cruz TD, Valdes AM, Santiago A, Frazer de Llado T, Raffel LJ, Zeidler A, Rotter JI, Erlich HA, Rewers M, Bugawan T, et al. 2004. DPB1 alleles are associated with type 1 diabetes susceptibility in multiple ethnic groups. Diabetes 53: 2158–2163 [DOI] [PubMed] [Google Scholar]
- Cucca F, Dudbridge F, Loddo M, Mulargia AP, Lampis R, Angius E, De Virgiliis S, Koeleman BP, Bain SC, Barnett AH, et al. 2001. The HLA-DPB1–associated component of the IDDM1 and its relationship to the major loci HLA-DQB1, -DQA1, and -DRB1. Diabetes 50: 1200–1205 [DOI] [PubMed] [Google Scholar]
- Cudworth AG, Woodrow JC 1974. Letter: HL-A antigens and diabetes mellitus. Lancet 2: 1153. [DOI] [PubMed] [Google Scholar]
- Deschamps I, Lestradet H, Bonaiti C, Schmid M, Busson M, Benajam A, Marcelli-Barge A, Hors J 1980. HLA genotype studies in juvenile insulin-dependent diabetes. Diabetologia 19: 189–193 [DOI] [PubMed] [Google Scholar]
- Erlich HA 1991. HLA class II sequences and genetic susceptibility to insulin dependent diabetes mellitus. Baillieres Clin Endoc 5: 395–411 [DOI] [PubMed] [Google Scholar]
- Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, et al. 2008. HLA DR-DQ haplotypes and genotypes and Type 1 diabetes risk: Analysis of the Type 1 Diabetes Genetics Consortium families. Diabetes 57: 1084–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambelunghe G, Ghaderi M, Cosentino A, Falorni A, Brunetti P, Sanjeevi CB 2000. Association of MHC class I chain-related A (MIC-A) gene polymorphism with type I diabetes. Diabetologia 43: 507–514 [DOI] [PubMed] [Google Scholar]
- Groh V, Steinle A, Bauer S, Spies T 1998. Recognition of stress-induced MHC molecules by intestinal epithelial γδ T cells. Science 279: 1737–1740 [DOI] [PubMed] [Google Scholar]
- Groh V, Wu J, Yee C, Spies T 2002. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419: 734–738 [DOI] [PubMed] [Google Scholar]
- Gupta M, Nikitina-Zake L, Zarghami M, Landin-Olsson M, Kockum I, Lernmark A, Sanjeevi CB 2003. Association between the transmembrane region polymorphism of MHC class I chain related gene-A and type 1 diabetes mellitus in Sweden. Hum Immunol 64: 553–561 [DOI] [PubMed] [Google Scholar]
- Held W, Kunz B, Lowin-Kropf B, van de Wetering M, Clevers H 1999. Clonal acquisition of the Ly49A NK cell receptor is dependent on the trans-acting factor TCF-1. Immunity 11: 433–442 [DOI] [PubMed] [Google Scholar]
- Hinks A, Barton A, John S, Bruce I, Hawkins C, Griffiths CE, Donn R, Thomson W, Silman A, Worthington J 2005. Association between the PTPN22 gene and rheumatoid arthritis and juvenile idiopathic arthritis in a UK population: Further support that PTPN22 is an autoimmunity gene. Arthritis Rheum 52: 1694–1699 [DOI] [PubMed] [Google Scholar]
- Horn GT, Bugawan TL, Long CM, Erlich HA 1988. Allelic sequence variation of the HLA-DQ loci: Relationship to serology and to insulin-dependent diabetes susceptibility. Proc Natl Acad Sci 85: 6012–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howson JM, Walker NM, Clayton D, Todd JA 2009a. Confirmation of HLA class II independent type 1 diabetes associations in the major histocompatibility complex including HLA-B and HLA-A. Diabetes Obes Metab 11 (Suppl 1): 31–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howson JM, Walker NM, Smyth DJ, Todd JA 2009b. Analysis of 19 genes for association with type I diabetes in the Type I Diabetes Genetics Consortium families. Genes Immun 10 (Suppl 1): S74–S84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilonen J, Merivuori H, Reijonen H, Knip M, Akerblom HK, Pociot F, Nerup J 1992. Tumour necrosis factor-β gene RFLP alleles in Finnish IDDM haplotypes. The Childhood Diabetes in Finland (DiMe) Study Group. Scand J Immunol 36: 779–783 [DOI] [PubMed] [Google Scholar]
- Jenhani F, Bardi R, Gorgi Y, Ayed K, Jeddi M 1992. C4 polymorphism in multiplex families with insulin dependent diabetes in the Tunisian population: Standard C4 typing methods and RFLP analysis. J Autoimmun 5: 149–160 [DOI] [PubMed] [Google Scholar]
- Ladner MB, Bottini N, Valdes AM, Noble JA 2005. Association of the single nucleotide polymorphism C1858T of the PTPN22 gene with type 1 diabetes. Hum Immunol 66: 60–64 [DOI] [PubMed] [Google Scholar]
- Lechler R, Warrens A 1999. HLA in health and disease. Academic Press, London [Google Scholar]
- Lhotta K, Auinger M, Kronenberg F, Irsigler K, Konig P 1996. Polymorphism of complement C4 and susceptibility to IDDM and microvascular complications. Diabetes Care 19: 53–55 [DOI] [PubMed] [Google Scholar]
- Marcelli-Barge A, Poirier JC, Chantome R, Deschamps I, Hors J, Colombani J 1990. Marked shortage of C4B DNA polymorphism among insulin-dependent diabetic patients. Res Immunol 141: 117–128 [DOI] [PubMed] [Google Scholar]
- Meigs JB, Dupuis J, Herbert AG, Liu C, Wilson PW, Cupples LA 2005. The insulin gene variable number tandem repeat and risk of type 2 diabetes in a population-based sample of families and unrelated men and women. J Clin Endocrinol Metab 90: 1137–1143 [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Uno M, Uehira M, Kikutani H, Kishimoto T, Kimoto M, Nishimoto H, Miyazaki J, Yamamura K 1990. Direct evidence for the contribution of the unique I-ANOD to the development of insulitis in non-obese diabetic mice. Nature 345: 722–724 [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Kobayashi T, Murase T, Nakatsuji T, Inoko H, Tsuji K, Kosaka K 1993. Association of HLA-A24 with complete β-cell destruction in IDDM. Diabetes 42: 1086–1093 [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Kobayashi T, Inoko H, Tsuji K, Murase T, Kosaka K 1995. Residual β-cell function and HLA-A24 in IDDM. Markers of glycemic control and subsequent development of diabetic retinopathy. Diabetes 44: 1334–1339 [DOI] [PubMed] [Google Scholar]
- Nejentsev S, Reijonen H, Adojaan B, Kovalchuk L, Sochnevs A, Schwarts EI, Akerblom HK, Honen J 1997. The effect of HLA-B allele on the IDDM risk defined by DRB1 *04 subtypes and DQB1 *0302. Diabetes 46: 1888–1892 [DOI] [PubMed] [Google Scholar]
- Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, Reynolds P, Hardy M, King E, Masters J, et al. 2007. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 450: 887–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, Ryder LP, Nielsen LS, Thomsen M, Svejgaard A 1974. HL-A antigens and diabetes mellitus. Lancet 2: 864–866 [DOI] [PubMed] [Google Scholar]
- Nishimura M, Obayashi H, Mizuta I, Hara H, Adachi T, Ohta M, Tegoshi H, Fukui M, Hasegawa G, Shigeta H, et al. 2003. TNF, TNF receptor type 1, and allograft inflammatory factor-1 gene polymorphisms in Japanese patients with type 1 diabetes. Hum Immunol 64: 302–309 [DOI] [PubMed] [Google Scholar]
- Noble JA, Valdes AM, Cook M, Klitz W, Thomson G, Erlich HA 1996. The role of HLA class II genes in insulin-dependent diabetes mellitus: Molecular analysis of 180 Caucasian, multiplex families. Am J Hum Genet 59: 1134–1148 [PMC free article] [PubMed] [Google Scholar]
- Noble JA, Valdes AM, Thomson G, Erlich HA 2000. The HLA class II locus DPB1 can influence susceptibility to type 1 diabetes. Diabetes 49: 121–125 [DOI] [PubMed] [Google Scholar]
- Noble JA, Valdes AM, Bugawan TL, Apple RJ, Thomson G, Erlich HA 2002. The HLA class I A locus affects susceptibility to type 1 diabetes. Hum Immunol 63: 657–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble JA, Valdes AM, Lane JA, Green AE, Erlich HA 2006. Linkage disequilibrium with predisposing DR3 haplotypes accounts for apparent effects of tumor necrosis factor and lymphotoxin-α polymorphisms on type 1 diabetes susceptibility. Hum Immunol 67: 999–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble JA, Martin A, Valdes AM, Lane JA, Galgani A, Petrone A, Lorini R, Pozzilli P, Buzzetti R, Erlich HA 2008. Type 1 diabetes risk for HLA-DR3 haplotypes depends on genotypic context: Association of DPB1 and HLA class I loci among DR3 and DR4 matched Italian patients and controls. Hum Immunol 69: 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble JA, Valdes AM, Varney MD, Carlson JA, Moonsamy P, Fear AL, Lane JA, Lavant E, Rappner R, Louey A, et al. 2010. HLA class I and genetic susceptibility to type 1 diabetes: Results from the Type 1 Diabetes Genetics Consortium. Diabetes 59: 2972–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JH, MacLean LD 1975. Diseases associated with specific HL-A antigens. Can Med Assoc J 112: 1315–1318 [PMC free article] [PubMed] [Google Scholar]
- Onengut-Gumuscu S, Ewens KG, Spielman RS, Concannon P 2004. A functional polymorphism (1858C/T) in the PTPN22 gene is linked and associated with type I diabetes in multiplex families. Genes Immun 5: 678–680 [DOI] [PubMed] [Google Scholar]
- Perez C, Gonzalez FE, Pavez V, Araya AV, Aguirre A, Cruzat A, Contreras-Levicoy J, Dotte A, Aravena O, Salazar L, et al. 2004. The -308 polymorphism in the promoter region of the tumor necrosis factor-α (TNF-α) gene and ex vivo lipopolysaccharide-induced TNF-α expression in patients with aggressive periodontitis and/or type 1 diabetes mellitus. Eur Cytokine Netw 15: 364–370 [PubMed] [Google Scholar]
- Permutt MA, Chirgwin J, Rotwein P, Giddings S 1984. Insulin gene structure and function: A review of studies using recombinant DNA methodology. Diabetes Care 7: 386–394 [DOI] [PubMed] [Google Scholar]
- Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, Nierras CR, Todd JA, Rich SS, Nerup J 2010. Genetics of type 1 diabetes: What's next? Diabetes 59: 1561–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese A 2005. The insulin gene in type 1 diabetes. IUBMB Life 57: 463–468 [DOI] [PubMed] [Google Scholar]
- Qu H, Tessier MC, Hudson TJ, Polychronakos C 2005. Confirmation of the association of the R620W polymorphism in the protein tyrosine phosphatase PTPN22 with type 1 diabetes in a family based study. J Med Genet 42: 266–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu HQ, Bradfield JP, Li Q, Kim C, Frackelton E, Grant SF, Hakonarson H, Polychronakos C 2010. In silico replication of the genome-wide association results of the Type 1 Diabetes Genetics Consortium. Hum Mol Genet 19: 2534–2538 [DOI] [PubMed] [Google Scholar]
- Reijonen H, Nejentsev S, Tuokko J, Koskinen S, Tuomilehto-Wolf E, Akerblom HK, Ilonen J 1997. HLA-DR4 subtype and -B alleles in DQB1*0302-positive haplotypes associated with IDDM. The Childhood Diabetes in Finland Study Group. Eur J Immunogenet 24: 357–363 [DOI] [PubMed] [Google Scholar]
- Rich S, O’Neill G, Dalmasso AP, Nerl C, Barbosa J 1985. Complement and HLA. Further definition of high-risk haplotypes in insulin-dependent diabetes. Diabetes 34: 504–509 [DOI] [PubMed] [Google Scholar]
- Rich SS, Concannon P, Erlich H, Julier C, Morahan G, Nerup J, Pociot F, Todd JA 2006. The Type 1 Diabetes Genetics Consortium. Ann NY Acad Sci 1079: 1–8 [DOI] [PubMed] [Google Scholar]
- Robinson WP, Barbosa J, Rich SS, Thomson G 1993. Homozygous parent affected sib pair method for detecting disease predisposing variants: Application to insulin dependent diabetes mellitus. Genet Epidemiol 10: 273–288 [DOI] [PubMed] [Google Scholar]
- Rodey GE, White N, Frazer TE, Duquesnoy RJ, Santiago JV 1979. HLA-DR specificities among black Americans with juvenile-onset diabetes. New Engl J Med 301: 810–812 [DOI] [PubMed] [Google Scholar]
- Rotwein P, Yokoyama S, Didier DK, Chirgwin JM 1986. Genetic analysis of the hypervariable region flanking the human insulin gene. Am J Hum Genet 39: 291–299 [PMC free article] [PubMed] [Google Scholar]
- Singal DP, Blajchman MA 1973. Histocompatibility (HL-A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes 22: 429–432 [DOI] [PubMed] [Google Scholar]
- Singer SM, Tisch R, Yang XD, Sytwu HK, Liblau R, McDevitt HO 1998. Prevention of diabetes in NOD mice by a mutated I-Ab transgene. Diabetes 47: 1570–1577 [DOI] [PubMed] [Google Scholar]
- Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, Vella A, Nutland S, Rance HE, Maier L, et al. 2004. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 53: 3020–3023 [DOI] [PubMed] [Google Scholar]
- Solow H, Hidalgo R, Singal DP 1979. Juvenile-onset diabetes HLA-A, -B, -C, and -DR alloantigens. Diabetes 28: 1–4 [PubMed] [Google Scholar]
- Steck AK, Rewers MJ 2011. Genetics of type 1 diabetes. Clin Chem 57: 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuchlikova M, Kantarova D, Michalkova D, Barak L, Buc M 2006. Association of HLA-DPB1 alleles with type I diabetes mellitus in Slovak population. Bratisl Lek Listy 107: 73–75 [PubMed] [Google Scholar]
- Tait BD, Boyle AJ 1986. DR4 and susceptibility to type I diabetes mellitus: Discrimination of high risk and low risk DR4 haplotypes on the basis of TA10 typing. Tissue Antigens 28: 65–71 [DOI] [PubMed] [Google Scholar]
- Tait BD, Colman PG, Morahan G, Marchinovska L, Dore E, Gellert S, Honeyman MC, Stephen K, Loth A 2003. HLA genes associated with autoimmunity and progression to disease in type 1 diabetes. Tissue Antigens 61: 146–153 [DOI] [PubMed] [Google Scholar]
- Tewhey R, Bansal V, Torkamani A, Topol EJ, Schork NJ 2011. The importance of phase information for human genomics. Nat Rev 12: 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsby E 1974. The human major histocompatibility system. Transplant Rev 18: 51–129 [PubMed] [Google Scholar]
- Todd JA, Bell JI, McDevitt HO 1987. HLA-DQ β gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature 329: 599–604 [DOI] [PubMed] [Google Scholar]
- Valdes AM, Noble JA, Genin E, Clerget-Darpoux F, Erlich HA, Thomson G 2001. Modeling of HLA class II susceptibility to type I diabetes reveals an effect associated with DPB1. Genet Epidemiol 21: 212–223 [DOI] [PubMed] [Google Scholar]
- Valdes AM, Erlich HA, Noble JA 2005. Human leukocyte antigen class I B and C loci contribute to type 1 diabetes (T1D) susceptibility and age at T1D onset. Hum Immunol 66: 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes AM, Thomson G, Barcellos LF 2010. Genetic variation within the HLA class III influences T1D susceptibility conferred by high-risk HLA haplotypes. Genes Immun 11: 209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varney MD, Valdes AM, Carlson JA, Noble JA, Tait BD, Bonella P, Lavant E, Fear AL, Louey A, Moonsamy P, et al. 2010. HLA DPA1, DPB1 alleles and haplotypes contribute to the risk associated with type 1 diabetes: Analysis of the type 1 diabetes genetics consortium families. Diabetes 59: 2055–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velaga MR, Wilson V, Jennings CE, Owen CJ, Herington S, Donaldson PT, Ball SG, James RA, Quinton R, Perros P, et al. 2004. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves’ disease. J Clin Endocrinol Metab 89: 5862–5865 [DOI] [PubMed] [Google Scholar]
- Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, Hebert M, Jones KN, Shu Y, Kitzmiller K, et al. 2007. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): Low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet 80: 1037–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, She JX 2005. Genetic association between a lymphoid tyrosine phosphatase (PTPN22) and type 1 diabetes. Diabetes 54: 906–908 [DOI] [PubMed] [Google Scholar]