

Abstract
Background
All drugs of abuse, including nicotine, activate the mesocorticolimbic system that plays critical roles in nicotine reward and reinforcement development and triggers glutamatergic synaptic plasticity on the dopamine (DA) neurons in the ventral tegmental area (VTA). The addictive behavior and firing pattern of the VTA DA neurons are thought to be controlled by the glutamatergic synaptic input from prefrontal cortex (PFC). Interrupted functional input from PFC to VTA was shown to decrease the effects of the drug on the addiction process. Nicotine treatment could enhance the AMPA/NMDA ratio in VTA DA neurons, which is thought as a common addiction mechanism. In this study, we investigate whether or not the lack of glutamate transmission from PFC to VTA could make any change in the effects of nicotine.
Methods
We used the traditional AMPA/NMDA peak ratio, AMPA/NMDA area ratio, and KL (Kullback-Leibler) divergence analysis method for the present study.
Results
Our results using AMPA/NMDA peak ratio showed insignificant difference between PFC intact and transected and treated with saline. However, using AMPA/NMDA area ratio and KL divergence method, we observed a significant difference when PFC is interrupted with saline treatment. One possible reason for the significant effect that the PFC transection has on the synaptic responses (as indicated by the AMPA/NMDA area ratio and KL divergence) may be the loss of glutamatergic inputs. The glutamatergic input is one of the most important factors that contribute to the peak ratio level.
Conclusions
Our results suggested that even within one hour after a single nicotine injection, the peak ratio of AMPA/NMDA on VTA DA neurons could be enhanced.
Background
Nicotine is thought to be the biologically active substance that promotes tobacco use. Approximately a quarter of the global population uses tobacco products that cause health and economical problems. Unfortunately, nicotine dependence creates problems for smokers to quit. The dopamine (DA) neurons in the ventral tegmental area (VTA) and their projection areas, including prefrontal cortex (PFC), nucleus accumbens (NAc), and amygdala, are thought to be very important in the reward-driven behavior-induced process by the drugs of addiction [1-5].
Malenka et al established a model to assess the glutamate receptor (GluR) plasticity and altered synaptic function by examining in vitro VTA DA neurons from midbrain slice preparation following 24 hours of a single, systemic administration of several types of drugs of addiction [6,7]. Following administration, they found that the peak ratio of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor-mediated excitatory postsynaptic currents (EPSCs) to N-methyl-D-aspartate (NMDA) receptor-mediated EPSCs was enhanced, which reflects a glutamatergic synapse plastic alteration onto DA neurons in the VTA. This may underlie a common mechanism of neural adaptation to addictive drugs [7].
Previous reports have shown that DA is released to NAc and locomotion activity in vivo has peaked with nicotine injection within one hour [8]. Additionally, long term potentiation (LTP) was rapidly induced by afferent stimulation and lasted more than one hour in VTA slice [9]. Moreover, a positive correlation between glutamatergic synaptic enhancement and behavioral locomotion existed [10].
The firing activities of VTA DA neurons and addictive behavior of the animals are believed to be controlled, impartially, by the glutamatergic synaptic inputs from PFC [11-15]. Evidence has shown that, the functional input loss from PFC and/or NAc may reduce the effects of these drugs on the addiction process [14,16-18]. In VTA, the AMPA/NMDA receptors' ratio response of dopamine neurons was found to be enhanced only by the drugs of abuse, and the enhanced ratio was thought to be caused by the excitatory input increase, which mostly originate from PFC [6,7].
Previous studies showed that, the strengthening of input from PFC to VTA plays an important role in the development of behavioral sensitization, a well-known model for addiction [7,19-22]. We recently showed that in in vivo experiments, acute response of VTA to nicotine with PFC transection is significantly changed when compared to PFC intact subjects [23,24].
Thus far, it is still unknown how the AMPA/NMDA peak ratio changes without PFC projection. In our study, we investigate whether the synaptic strength would increase following only one hour after single nicotine administration by activating multiple molecular and cellular cascades.
In addition to the AMPA/NMDA peak ratio measurement proposed by the other research groups, we performed analysis of the synaptic response by estimating the areas under the AMPA and NMDA EPSC waveforms [25,26]. This allows us to better understand the dynamics of the synaptic charge transfer. Moreover, we used the Kullback-Leibler (KL) divergence analysis method to quantitatively evaluate the difference between the shapes of the AMPA and NMDA signals [27].
Methods
Animals and treatment
We used Sprague Dawley rats (14-19 days old) for the experiments [9]. All experimental protocols and surgeries were approved by The Institutional Animal Care and Use Committee of Arizona State University. For PFC intact animals, the skin on the skull was cut to mock the surgery under anesthesia (isofluran USP) and was sutured after the manipulation. The subjects were given one hour to recover from the anesthesia effect before saline (volume matched to nicotine injection) or nicotine (0.5 mg/kg) was intraperitoneal (i.p.) injected.
The subjects in the PFC transected group were under anesthesia (isofluran USP) while bilateral transections were made immediate caudal to the PFC to disrupt the connection between PFC and VTA DA neuron with the skin on the head open. A slit was drilled in the skull around 1 mm anterior to bregma. A sharp blade was lowered to the base of skull, without damaging the main artery, to completely interrupt the connections between the PFC and the rest of the brain [28]. The post-surgical care and drug administration were identical to the PFC intact animals.
The disruption between PFC and VTA was observed at the time the brain was removed from the skull. Once the brain has been removed from the skull, the area immediate caudal to PFC has been observed to be cut. This indicates the PFC has completely lost its connection with the rest of the brain.
Electrophysiological recordings
One hour after single systemic injection of nicotine, animals were anesthetized by forane (isoflurane USP) and sacrificed. The remaining procedures were identical as previously described [6]. Briefly, horizontal midbrain slices (250 μm) were cut using a vibratome 1000 (Vibratom, St. Louis, MO). Slices were prepared in ice-cold artificial cerebrospinal fluid (ASCF) solution containing (in mM): 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2 , 18 NaHCO3 and 11 glucose. The slices were incubated for at least one hour in a holding chamber at room temperature (22-24°C) and continuously bubbled with 95% O2 and 5% CO2 carbogen in the same ACSF solution. Conventional whole-cell recordings were made using a patch clamp amplifier (Multiclamp 700B, Axon Instruments) under infrared-DIC microscopy (Axioskop2 FS Plus, Zeiss). Data acquisition and analysis were performed using a digitizer (DigiData 1440A, Axon Instruments) and the analysis software pClamp 10.2 (Axon Instruments). Signals were filtered at 2 kHz and sampled at 10 kHz. For presynaptic stimulation, a bipolar tungsten stimulation electrode (WPI, Sarasota, Florida) was placed 100-200 μm rostral to the recording electrode to stimulate excitatory afferents, stimulation pulse of 40 μs duration and 0.1 Hz frequency were applied. For measurements of the ratio of AMPA and NMDA receptor-mediated currents, the DA neuron was voltage-clamped at +40 mV. Picrotoxin (100 μM) was added to the bath solution to block GABAA-receptor-mediated inhibitory synaptic transmission. Initially, a stable baseline recording of total evoked EPSCs was obtained for 5 min. Then the NMDA receptor antagonist AP-V (50 μM) was applied to the bath for 10 min to obtain AMPA-receptor-mediated EPSCs. An average of 15 evoked EPSCs was collected for each type of EPSC. NMDA-receptor-EPSCs were obtained by digitally subtracting the AMPA-receptor-EPSCs from the total EPSCs from the same neuron. For the ratio experiments, the whole-cell recording pipette (3-6 MΩ) was filled with a solution containing (in mM): 117 cesium methansulfonic acid, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA-Cl, 2.5 MgATP and 0.25 GTP (pH 7.2-7.4 with CsOH). Series resistance was monitored throughout the whole-cell recording. Only two slices were obtained from each animal and a single cell was examined from each slice. All values are expressed as mean ± SEM. Statistical significance was assessed using two-tailed Student's t- tests.
All recordings were performed at 31 ± 1°C [6,7]. The DA neuron was identified by large hyperpolarization-activated current (Ih) as shown in Figure 1[29,30]. All drugs were obtained from Sigma, unless otherwise specified.
Figure 1.

Example of Ih currents were observed for midbrain DA neurons under voltage clamp. The holding potential was -70 mV and was given -10 mV step size to reach -150 mV (calibration bars: 20 pA/50 ms).
AMPA/NMDA area ratio
The area under AMPA and NMDA EPSC curves was estimated. This area represents the synaptic charge transfer [25,26]. For each pair AMPA/NMDA of each VTA DA neuron the area ratios were computed for 50 ms length segments along the signal. This approach allows us to estimate the way charge transfer dynamically evolves along the synaptic response. At the same time it enables us to compare the changes brought to the synaptic charge transfer by different experimental conditions (PFC intact vs transected; saline treated vs nicotine treated).
Kullback-Leibler divergence
The Kullback-Leibler (KL) divergence method originates from information theory and is a quantitative measure of the difference between two probability distributions [27]. The KL divergence of distribution q(x) from distribution p(x), KL(p||q), is mathematically defined as:
| (1) |
From its properties, the KL divergence satisfies KL(p||q) ≥ 0 with equality if and only if p(x) = q(x), and is asymmetric quantity, i.e. KL(p||q) ≠ KL(q||p).
In this analysis, we used the following measure,KL(p, q), as a KL divergence to treat it as symmetric quantity [31].
| (2) |
In information theory, p(x) and q(x) are assumed as probability distributions. In the present study, however, we assumed that p(x) and q(x) correspond to AMPA and NMDA receptor-mediated EPSCs, respectively. Under this assumption, we can quantitatively evaluate the difference between the shapes of the AMPA and NMDA signals using the KL divergence. This measure provides information on the whole area of synaptic response and not just the maximum response value, as a measure based on the peak ratio would.
Since all the recording data of AMPA and NMDA signals is sampled and has discrete values, we need to transform the KL divergence in Eq.(2) to a discrete format as below:
| (3) |
where ximeans an i -th discrete signal and N is the number of recording data. Before calculating the KL divergence using Eq.(3), we need to preprocess the signals to take positive values and their sums equal to one, i.e. because of an original restriction on the probability distribution in information theory.
We calculated the KL divergence for each pair of AMPA and NMDA signals, under different experimental conditions (nicotine and saline and also with PFC intact and transected rats). Subsequently, the statistical significance of the difference between AMPA and NMDA signals under the different conditions was assessed using two-tailed Student's t-tests.
Results
AMPA/NMDA peak ratio
The measurement of glutamatergic synaptic strength was applied exactly as previously described [6,7], in which, the AMPA receptor-mediated EPSCs was normalized to NMDA receptor-mediated EPSCs to obtain the peak ratio of AMPA/NMDA as seen in Figure 2. In the nicotine treated group, one hour after single injection of nicotine with PFC intact, the AMPA/NMDA peak ratio was 0.68 ± 0.04 (n = 6), while in saline group, that was 0.46 ± 0.035 (n = 7) as seen in Figure 3A. This significant enhancement induced by nicotine treatment (p < 0.01) is consistent with another previous report that 24 hours after a single, systemic administration of nicotine enhances the excitatory synapse strength on VTA DA neurons by enhancement of postsynaptic AMPA receptors [7].
Figure 2.

Example recordings of evoked NMDA and AMPA EPSCs from midbrain VTA DA neurons of rats one hour after treatment of saline and nicotine with PFC intact and PFC transection (calibration bars: 20 pA/15 ms).
Figure 3.

Summary of AMPA/NMDA peak ratios obtained from rats with different treatments. (A) Summary of AMPA/NMDA peak ratio obtained from rats treated with saline and nicotine with PFC intact (** indicates p < 0.01). (B) Summary of AMPA/NMDA peak ratio obtained from rats treated with saline and nicotine with PFC transected (** indicates p < 0.01). (C) Summary of AMPA/NMDA peak ratio obtained from rats treated with saline with PFC intact and PFC transected. (D) Summary of AMPA/NMDA peak ratio obtained from rats treated with nicotine with PFC intact and PFC transacted.
To investigate whether PFC transection would cause the AMPA/NMDA peak ratio to be different, we repeated the same experiments in PFC transection rats. In response to the PFC transection, the saline group has peak ratio of 0.48 ± 0.035 (n = 7), while the nicotine group exhibited 0.74 ± 0.035 (n = 7). The results show that nicotine treatment still could increase the AMPA/NMDA peak ratio significantly (p < 0.01), even without intact inputs from PFC as seen in Figure 3B.
After confirming the nicotine's enhancing effects in both PFC intact and PFC transection rats, we investigated whether there is any difference in the effects of nicotine between these two groups. As showed in Figure 3D, the peak ratio for PFC intact with nicotine is 0.68 ± 0.04 (n = 6), while PFC transection with nicotine is increased to 0.74 ± 0.035 (n = 7). However, these changes are not significant.
AMPA/NMDA area ratio
We also performed analysis of the synaptic response by estimating the areas under the AMPA and NMDA EPSC curves. This allows us to better understand the dynamics of the synaptic charge transfer and provides more information than traditional measures based on only peak ratios. Specifically, as mentioned in the method section, we estimated areas under AMPA and NMDA curves on consecutive 50 ms segments. For each segment the AMPA/NMDA area ratio was computed. From Figure 4A, we observe that nicotine treatment induced a significant difference (p < 0.01) on the synaptic charge and hence on the AMPA/NMDA area ratio, when compared to saline on the first 50 ms of the synaptic response. The difference continues to be significant (p < 0.05) up to 100 ms, while subsequently, the synaptic charge transfer seems to be unaffected by nicotine.
Figure 4.
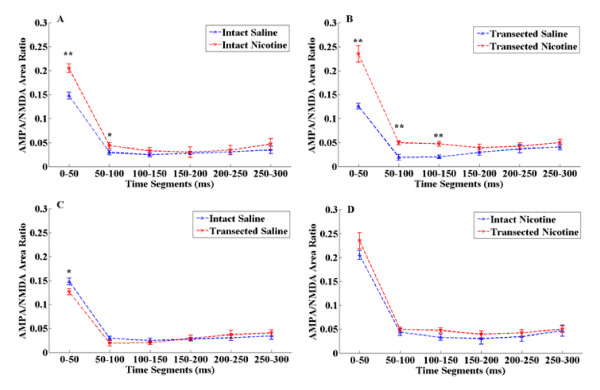
Summary of AMPA/NMDA area ratio obtained from rats with different treatments. (A) Summary of AMPA/NMDA area ratio obtained from rats treated with saline and nicotine with PFC intact (** indicates p < 0.01, * indicates p < 0.05). (B) Summary of AMPA/NMDA area ratio obtained from rats treated with saline and nicotine with PFC transected (** indicates p < 0.01). (C) Summary of AMPA/NMDA area ratio obtained from rats treated with saline with PFC intact and PFC transected (* indicates p < 0.05). (D) Summary of AMPA/NMDA area ratio obtained from rats treated with nicotine with PFC intact and PFC transacted.
A similar trend is also apparent when comparing the effect of nicotine treatment after PFC transection occurred. However, in this case the transection seems to prolong the differences in the synaptic charge induced by nicotine, when compared to saline. As observed in Figure 4B, the area ratios are significantly different up to 150 ms (p < 0.01).
The area ratio measure offered a substantial different view than the peak ratio when comparing the saline treated PFC intact and PFC transected responses. When using the peak ratio method, we could not identify any difference in the synaptic responses. However, the area ratio shows us there is a significant effect induced by PFC transection on the first 50 ms (p < 0.05) of saline treated group as seen in Figure 4C.
KL Divergence
Additionally, we estimated the KL divergence for signals treated with nicotine and saline and also with PFC intact and transected rats throughout the whole current response. The EPSCs are assumed as probability distribution. The advantages of the analysis based on KL divergence arise from the higher level information provided, since it allows us to estimate differences in the shapes of the AMPA and NMDA currents. From Figure 5A, one hour after single injection of nicotine for PFC intact rats, the KL divergence was 0.54 ± 0.036, while in saline group, that was 0.48 ± 0.024. We observed there is significant difference (p < 0.05) between nicotine treated and saline treated PFC intact group.
Figure 5.

Summary of KL divergence obtained from rats with different treatments. (A) Summary of KL divergence obtained from rats treated with saline and nicotine with PFC intact (* indicates p < 0.05). (B) Summary of KL divergence obtained from rats treated with saline and nicotine with PFC transected (** indicates p < 0.01). (C) Summary of KL divergence obtained from rats treated with saline with PFC intact and PFC transected (* indicates p < 0.05). (D)Summary of KL divergence obtained from rats treated with nicotine with PFC intact and PFC transacted.
In Figure 5B, the saline group has KL divergence of 0.38 ± 0.030, while the nicotine group exhibited 0.52 ± 0.041. The results show that nicotine treatment still could increase the KL divergence significantly (p < 0.01), even without intact inputs from PFC.
However, the KL divergence analysis measure also offered a significant different view than the peak ratio when the saline treated PFC intact and PFC transected responses was compared. As seen in Figure 5C, results using KL divergence analysis method showed there is a significant difference (p < 0.05) with PFC transection that is not observed using the peak ratio method.
It is worth noting that, with PFC intact and PFC transected saline treatment, there is significant difference in the responses as seen in Figures 4C and 5C. Whereas PFC intact and PFC transected nicotine treatment has no significant difference. This result led us to believe that with PFC transection, the VTA DA neurons are more sensitive to nicotine exposure.
Discussion
The VTA in horizontal midbrain slices is identified and recognized as the area medial to the substantia nigra compacta and medial to terminal nucleus of the accessory optic tract. Additionally, a clear hyperpolarization-activated cation current (Ih) emerges after hyperpolarizing the VTA DA neuron from -70 to -150 mv, in 10 mv step size, immediately after break-in was observed in each recorded neuron. Ih was shown to be a reliable marker for VTA DA neurons [32-34]. A recent report has questioned the identification of VTA DA neurons using Ih [35]. However, in previous studies [6,7,34,36] and in the present study, this criterion was sufficient to provide necessary identification.
The disruption between PFC and VTA was observed at the time the brain was removed from the skull. Once the brain has been removed from the skull, the area immediately caudal to PFC has been observed to be cut. This indicates the PFC has completely lost its connection with the rest of the brain.
Previous studies had reported that single nicotine injection could enhance the peak ratio of VTA DA neurons AMPA/NMDA receptors responses within 24 hours, even after nicotine metabolized [7]. Moreover, Mansvelder and McGehee reported brief nicotine application rapidly induced LTP and maintained for more than one hour in VTA slice [9]. Additionally, the positive correlation between glutamatergic synaptic enhancement and behavioral locomotion were well described [10]. In vivo experiments also showed that following nicotine application, dopamine release to NAc and the locomotion activity of rats both would peak within one hour [8]. These evidences led us to predict that, within only one hour after single nicotine injection, the postsynaptic response will be changed. Our hypotheses are supported by the results, since we found that even within one hour after a single nicotine injection, the peak ratio of AMPA/NMDA on VTA DA neurons could be enhanced. However, the peak ratio increase observed within one hour was lower in comparison to 24 hours after a single nicotine injection [7]. This suggests that, after one hour of a single systemic nicotine injection, not all subset of synapses were potentiated.
This finding was also supported by previous reports that the AMPA/NMDA ratio enhancement was observed two hours after a single cocaine injection [37] and observed in another study two hours after a single amphetamine injection [36].
The PFC is a key structure for executive functions of the brain [38,39] and has been shown to regulate the firing pattern of dopamine (DA) neurons in the VTA. Gao et al [28] stated that there is an indirect coupling between PFC and VTA. Thus, PFC stimulation increases burst firing in DA neurons, whereas PFC inactivation produces the opposite effect [40-44].
Glutamate transmission from PFC to VTA is important in controlling VTA DA neurons firing activities and animal behavior [11-15]. Treatments of most drugs of abuse has been reported to increase excitatory inputs to the midbrain, which is thought to contribute, impartially, to the enhancement of VTA DA neurons AMPA/NMDA ratio. All the evidences suggested it is a common mechanism of addiction through neural adaptation [6,7].
The disconnection of the functional pathway between PFC and VTA could significantly reduce the effects of drugs of addiction, including nicotine [9,28,45]. We recently have demonstrated in in vivo experiments that the responses of main VTA DA neurons to acute nicotine injection are greatly changed after the PFC transaction [23,24]. Based on these, in this study, we transected the PFC and examined the changes of AMPA/NMDA peak ratio of VTA DA neurons. Interestingly, without the intact input from PFC, the AMPA/NMDA ratio was still enhanced by nicotine injection.
Like LTP, AMPA/NMDA ratio alteration reflects the plasticity change in synapse. Normally these changes are caused by either increase in excitatory input or decrease in inhibitory input. In VTA, DA neurons receive excitatory inputs from PFC and the inhibitory inputs from GABAergic interneuron in VTA and/or NAc, which also should have functional coupling with PFC. The openings of GluR were changed after nicotine treatments, via the regulation from PFC to VTA DA neurons, that induced EPSCs. After PFC transection, the signals induced by nicotine could not be transferred from PFC to VTA adequately, it may be the result of the different alteration to GluR on VTA DA neurons. Measurement of AMPA/NMDA peak ratio only takes into account the maximum GluR response. However, the AMPA and NMDA curves represent the whole GluR response with respect to time. Therefore, we estimated the AMPA/NMDA area ratio and KL divergence to better understand the dynamics of AMPA and NMDA signals since they took into consideration the whole current response rather than just the peak response. With these two methods, we found that there is statistical significance between PFC intact and PFC transected rats with saline treatments as seen in Figures 4C and 5C. This is not observed when measuring the AMPA/NMDA peak ratio. One possible reason for the observed differences may be due to the loss of glutamatergic inputs from PFC, which is one of the most important factors that contribute to the ratio level [46].
The use of traditional analysis method of AMPA/NMDA peak ratio suggests the PFC is not a "must" area and the ratio enhancement could occur locally in VTA. Previous studies showed that in vitro exposure of VTA slices to amphetamine did not enhance AMPA/NMDA ratio [36]. However, local injection of amphetamine to the VTA in vivo triggered sensitization [47,48]. This suggests that the enhancement effects should be triggered in VTA and need other areas to provide functional feedback, increase the excitatory input and/or reduce the inhibitory input.
However, the use of different analysis methods based on AMPA/NMDA area ratio and KL divergence, show that PFC is suggested to play an important role in affecting the VTA DA neurons. These two methods took into account the dynamics of AMPA and NMDA signals of the complete response and not just the maximum, making them more suitable in understanding the effects caused by nicotine and PFC transection.
Conclusions
We demonstrated that the ratio of AMPA/NMDA responses of VTA DA neuron could be enhanced by single nicotine injection even within one hour, and AMPA/NMDA area ratio and the KL divergence analysis method are able to provide a more complete understanding of AMPA and NMDA responses, and may be better fit for the analysis of other neurological signals.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TC performed experiments, the data analysis, and helped to write the manuscript, DZ helped to write the manuscript, AD contributed to the data analysis and helped to write the manuscript, KK helped with the data analysis, YMA helped with the experiments, helped to interpret the data and write the paper. MA oversaw the data collection, the data analysis, helped to interpret the results and write the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ting Chen, Email: ting.y.chen@gmail.com.
Die Zhang, Email: gnahzeid@gmail.com.
Andrei Dragomir, Email: andrei.drag@gmail.com.
Kunikazu Kobayashi, Email: koba@yamaguchi-u.ac.jp.
Yasemin Akay, Email: ymakay@uh.edu.
Metin Akay, Email: makay@uh.edu.
Acknowledgements
We would like to thank Ms. Jessica Diefenderfer for her editing the manuscript.
References
- Kalivas PW. Interactions between dopamine and excitatory amino acids in behavioral sensitization to psychostimulants. Drug Alcohol Depend. 1995;37:95–100. doi: 10.1016/0376-8716(94)01063-Q. [DOI] [PubMed] [Google Scholar]
- Tong ZY, Overton PG, Martinez-Cue C, Clark D. Do non-dopaminergic neurons in the ventral tegmental area play a role in the responses elicited in A10 dopaminergic neurons by electrical stimulation of the prefrontal cortex? Exp. Brain Res. 1998;118:466–476. doi: 10.1007/s002210050303. [DOI] [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/S0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/S0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Bjorklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202. doi: 10.1016/j.tins.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/S0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ. The effects of acute and repeated nicotine treatment on nucleus accumbens dopamine and locomotor activity. Br J Pharmacol. 1992;105:849–856. doi: 10.1111/j.1476-5381.1992.tb09067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-Term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/S0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. Neurotransmitter regulation of dopamine neurons in the ventral tegmental area. Brain Res Rev. 1993;18:75–113. doi: 10.1016/0165-0173(93)90008-N. [DOI] [PubMed] [Google Scholar]
- White FJ. Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci. 1996;19:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res Rev. 1997;25:312–334. doi: 10.1016/S0165-0173(97)00039-8. [DOI] [PubMed] [Google Scholar]
- Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, White FJ. Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci. 2005;25:936–940. doi: 10.1523/JNEUROSCI.4715-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Wolf M. Dopamine alters AMPA receptor synaptic expression and subunit composition in dopamine neurons of the ventral tegmental area cultured with prefrontal cortex neurons. J Neurosci. 2007;27:14275–14285. doi: 10.1523/JNEUROSCI.2925-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Vartanian AJ, White FJ, Xue XJ, Wolf ME. Effects of the AMPA receptor antagonist NBQX on the development and expression of behavioral sensitization to cocaine and amphetamine. Psychopharmacology (Berl) 1997;134:266–276. doi: 10.1007/s002130050449. [DOI] [PubMed] [Google Scholar]
- Li Y, Hu XT, Berney TG, Vartanian AJ, Stine CD, Wolf ME, White FJ. Both glutamate receptor antagonists and prefrontal cortex lesions prevent induction of cocaine sensitization and associated neuroadaptations. Synapse. 1999;34:169–180. doi: 10.1002/(SICI)1098-2396(19991201)34:3<169::AID-SYN1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol. 2001;63:241–320. doi: 10.1016/S0301-0082(00)00033-2. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction. 2000;95(Suppl 2):S91–117. doi: 10.1080/09652140050111681. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Wolf ME. Psychomotor stimulant addiction: a neural systems perspective. J Neurosci. 2002;22:3312–3320. doi: 10.1523/JNEUROSCI.22-09-03312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Grace AA. Cortico-Basal ganglia reward network: Microcircuitry. Neuropsychopharmacology (Reviews) 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology (Reviews) 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TY, Zhang D, Dragomir A, Akay YM, Akay M. Complexity of VTA DA neural activities in response to PFC transection in nicotine treated rats. J Neuroengineering and Rehabilitation. 2011;8:13. doi: 10.1186/1743-0003-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TY, Zhang D, Dragomir A, Akay YM, Akay M. The effects of nicotine exposure and PFC transaction on the time-frequency distribution of VTA DA neurons' firing activities. Med Biol Eng Comput. 2011;49(5):605–612. doi: 10.1007/s11517-011-0759-6. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Lisman JE. Pathway-specific properties of AMPA and NMDA-mediated transmission in CA1 hippocampal pyramidal cells. J Neurosci. 2002;22:1199–1207. doi: 10.1523/JNEUROSCI.22-04-01199.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Kroener S, Zaitsev AV, Povysheva NV, Krimer LS, Barrionuevo G, Lewis D. Functional maturation of excitatory synapses in layer 3 pyramidal neurons during postnatal development of the primate prefrontal cortex. Cerebral Cortex. 2008;18:626–637. doi: 10.1093/cercor/bhm095. [DOI] [PubMed] [Google Scholar]
- Kullback S, Leibler RA. On information and sufficiency. Annals of Mathematical Statistics. 1951;22:79–86. doi: 10.1214/aoms/1177729694. [DOI] [Google Scholar]
- Gao M, Liu CL, Yang S, Jin GZ, Bunney BS, Shi WX. Functional coupling between the prefrontal cortex and dopamine neurons in the ventral tegmental area. J Neurosci. 2007;27:5414–5421. doi: 10.1523/JNEUROSCI.5347-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci. 2003;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayyad GM, Samiuddin M, Al-Harbey AA. On parametric density estimation. Biometrika. 1989;76:343–348. doi: 10.1093/biomet/76.2.343. [DOI] [Google Scholar]
- Cameron DL, Wessendorf MW, Williams JT. A subset of ventral tegmental area neurons is inhibited by dopamine, 5-hydroxytryptamine and opioids. Neuroscience. 1997;77:155–166. doi: 10.1016/S0306-4522(96)00444-7. [DOI] [PubMed] [Google Scholar]
- Neuhoff H, Neu A, Liss B, Roeper J. I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J Neurosci. 2002;22:1290–1302. doi: 10.1523/JNEUROSCI.22-04-01290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586:2157–2170. doi: 10.1113/jphysiol.2007.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faleiro LJ, Jones S, Kauer JA. Rapid synaptic plasticity of glutamatergic synapses on dopamine neurons in the ventral tegmental area in response to acute amphetamine injection. Neuropsychopharmacology. 2004;29:2115–2125. doi: 10.1038/sj.npp.1300495. [DOI] [PubMed] [Google Scholar]
- Argilli E, Sibley DR, Malenka RC, England PM, Bonci A. Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci. 2008;28:9092–9100. doi: 10.1523/JNEUROSCI.1001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- Miller EK, Freedman DJ, Wallis JD. The prefrontal cortex: categories, concepts and cognition. Philos Trans R Soc Lond B Biol Sci. 2002;357:1123–1136. doi: 10.1098/rstb.2002.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariano RF, Groves PM. Burst firing induced in midbrain dopamine neurons by stimulation of the medial prefrontal and anterior cingulate cortices. Brain Res. 1988;462:194–198. doi: 10.1016/0006-8993(88)90606-3. [DOI] [PubMed] [Google Scholar]
- Svensson TH, Tung CS. Local cooling of pre-frontal cortex induces pacemaker-like firing of dopamine neurons in rat ventral tegmental area in vivo. Acta Physiol Scand. 1989;136:135–136. doi: 10.1111/j.1748-1716.1989.tb08640.x. [DOI] [PubMed] [Google Scholar]
- Murase S, Grenhoff J, Chouvet G, Gonon FG, Svensson TH. Prefrontal cortex regulates burst firing and transmitter release in rat mesolimbic dopamine neurons studied in vivo. Neurosci Lett. 1993;157:53–56. doi: 10.1016/0304-3940(93)90641-W. [DOI] [PubMed] [Google Scholar]
- Overton PG, Tong ZY, Clark D. A pharmacological analysis of the burst events induced in midbrain dopaminergic neurons by electrical stimulation of the prefrontal cortex in the rat. J Neural Transm. 1996;103:523–540. doi: 10.1007/BF01273151. [DOI] [PubMed] [Google Scholar]
- Tong ZY, Overton PG, Clark D. Stimulation of the prefrontal cortex in the rat induces patterns of activity in midbrain dopaminergic neurons which resemble natural burst events. Synapse. 1996;22:195–208. doi: 10.1002/(SICI)1098-2396(199603)22:3<195::AID-SYN1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Schilstrom B, Nomikos GG, Nisell M, Hertel P, Svensson TH. N-methyl-D-aspartate receptor antagonism in the ventral tegmental area diminishes the systemic nicotine-induced dopamine release in the nucleus accumbens. Neuroscience. 1998;82:781–789. doi: 10.1016/s0306-4522(97)00243-1. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nature Reviews Neuroscience. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Vezina P. Amphetamine injected into the ventral tegmental area sensitizes the nucleus accumbens dopaminergic response to systemic amphetamine: an in vivo microdialysis study in the rat. Brain Res. 1993;605:332–337. doi: 10.1016/0006-8993(93)91761-G. [DOI] [PubMed] [Google Scholar]
- Cador M, Bjijou Y, Cailhol S, Stinus L. D-amphetamine-induced behavioral sensitization: implication of a glutamatergic medial prefrontal cortex-ventral tegmental area innervation. Neuroscience. 1999;94:705–721. doi: 10.1016/S0306-4522(99)00361-9. [DOI] [PubMed] [Google Scholar]