

Abstract
Background
Human serum transferrin (hTF) is a bilobal glycoprotein that reversibly binds Fe3+ and delivers it to cells by the process of receptor-mediated endocytosis. Despite decades of research, the precise events resulting in iron release from each lobe of hTF within the endosome have not been fully delineated.
Scope of Review
We provide an overview of the kinetics of iron release from hTF ± the transferrin receptor (TFR) at endosomal pH (5.6). A critical evaluation of the array of biophysical techniques used to determine accurate rate constants is provided.
General Significance
Delivery of Fe3+ by to actively dividing cells by hTF is essential; too much or too little Fe3+ directly impacts the well-being of an individual. Because the interaction of hTF with the TFR controls iron distribution in the body, an understanding of this process at the molecular level is essential.
Major Conclusions
Not only does TFR direct the delivery of iron to the cell through the binding of hTF, kinetic data demonstrate that it also modulates iron release from the N- and C-lobes of hTF. Specifically, the TFR balances the rate of iron release from each lobe, resulting in efficient Fe3+ release within a physiologically relevant time frame.
Keywords: transferrin, transferrin receptor, kinetics, fluorescence
1. Introduction
1.1 Overview of Iron and Human Serum Transferrin (hTF)
The one electron transfer between ferrous (Fe2+) and ferric (Fe3+) iron is accomplished with relative ease. Due to its inherent redox properties, iron is critical to a number of biological processes including oxygen and electron transport [1]. However, these same redox properties that provide versatility also make iron dangerous. In oxygen rich environments Fe3+ is extremely insoluble and Fe2+ can be toxic. Specifically, reduction of O2 by Fe2+ generates superoxide, which ultimately can lead to the formation of the hydroxyl radical, a powerful oxidant known to damage DNA, proteins and lipids [2]. To avoid Fenton chemistry, iron must be carefully chaperoned through the body by human serum transferrin (hTF) where it is solubilized and stabilized as Fe3+ or stored in ferritin.
An ~80 kDa bilobal protein, hTF is divided into four subdomains (N1, N2, C1 and C2) which form two lobes (termed N- and C-lobes). Each lobe of hTF binds one Fe3+ ion tightly (~1022 M−1), yet reversibly. In humans iron absorbed from the diet passes through duodenyl crypt cells into the serum where it is acquired by hTF. In both hTF lobes the Fe3+ is coordinated by identical ligands: two tyrosine residues, one aspartic acid and one histidine residue (Tyr95, Tyr188, Asp63 and His249 in the N-lobe; Tyr426, Tyr517, Asp392 and His585 in the C-lobe). The distorted octahedral coordination of the iron is completed by a synergistic anion, identified as carbonate, which is anchored in place by a conserved arginine residue (Arg124 in the N-lobe and Arg456 in the C-lobe).
Given the ability to bind iron in either or both lobes, hTF circulates in the blood as four different species differing only in iron content. The ~25-50 μM hTF in the serum is unevenly distributed between diferric (Fe2hTF, ~27%), monoferric N-lobe hTF (FeNhTF, ~23%), monoferric C-lobe hTF (FeChTF, ~11%) and iron-free hTF (apohTF, ~40%) [3]. At the pH of the serum (~7.4) iron-bearing hTF (either Fe2hTF, FeNhTF or FeChTF) binds with nM affinity to the specific transferrin receptor (TFR), located on the cell surface of all iron-requiring cells (Fig. 1). The hTF/TFR complex is endocytosed in a clathrin-dependent manner. Through the action of ATP-dependent H+ pumps, the pH within the endosome is lowered. The significantly lower pH (~5.6) in conjunction with salt and an unidentified chelator within the endosome initiate receptor stimulated iron release from hTF. Although the TFR influences the redox potential of Fe3+ bound to hTF [4], recent evidence suggests that the reduction of Fe3+ to Fe2+ may be accomplished by an endosomal ferrireductase (Steap3)[5], presumably following Fe3+ release from hTF. Critical to the hTF endocytic cycle, apohTF remains bound to the TFR with high affinity at endosomal pH, allowing it to return to the cell surface. ApohTF is released back into the serum, either through dissociation from the TFR or displacement by an iron-containing hTF [6], and free to bind more Fe3+. The hTF/TFR cycle has become the classic example of clathrin-dependent receptor mediated endocytosis and often serves as a positive control for other systems.
Figure 1.
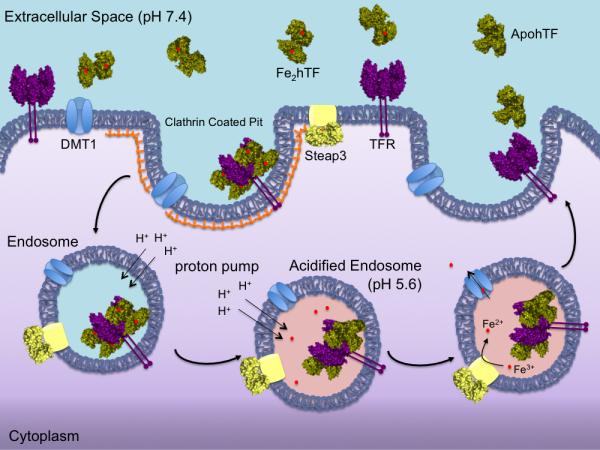
Endocytic hTF/TFR cycle. Iron bound hTF (green) in the blood binds to the specific TFR (purple) with nM affinity at the cell surface (pH 7.4). The hTF/TFR complex is endocytosed in a clathrin-coated pit. Within the endosome, the pH is lowered to ~5.6 causing iron to be released from hTF to an, as yet, unidentified chelator. Fe3+ is reduced to Fe2+ by the ferrireductase Steap3 (yellow) within the endosome. The Fe2+ can then be transported out of the endosome via the divalent metal transporter DMT1 (blue) for use throughout the cell. The apohTF remains tightly bound to the TFR at pH 5.6 and is recycled back to the cell surface. Upon exposure to the slightly basic pH (7.4), apohTF is released or displaced from the TFR and free to bind more Fe3+.
Although the iron binding ligands in each lobe of hTF are completely conserved, the mechanism for iron release from each lobe differs. This is largely due to differences in “second-shell” residues (residues that do not directly coordinate the Fe3+ but form an intricate hydrogen bonding network with the primary iron ligands). In part because of differences in the composition of the second shell residues, the two lobes differ in their response to pH [7-9], anions [9, 10], the TFR [11, 12], and the conformation of the other lobe.
The crystal structure of the N-lobe of hen ovotransferrin (a closely related family member of hTF) provided the first description of a potential iron release mechanism from the N-lobe of hTF. Attributed to an unusually low pKa value, two second shell lysine residues, Lys206 and Lys296, located in each subdomain on opposite sides of the cleft of the N-lobe are 3.0 Å apart, share a hydrogen bond and form what is referred to as the dilysine trigger [13]. When the pH is reduced, protonation of one of the lysine residues causes the positively charged lysines to repel each other (moving at least 9 Å apart in the apo N-lobe structure [14]) and literally triggers iron release. Studies have shown that mutation of either member of the dilysine trigger to a glutamate or alanine drastically slowed the rate of iron removal, validating the critical nature of the Lys206 and Lys296 interaction in the mechanism of iron release from the N-lobe of hTF [15].
The mechanism of iron release from the C-lobe differs from that of the N-lobe because the dilysine trigger is replaced by a triad of residues (Lys534, Arg632 and Asp634) [13]. Similar to the dilysine trigger, Lys534 and Arg632 in the C-lobe may share a hydrogen bond that is stabilized by Asp634, which upon protonation would trigger iron release from the lobe; however, in the structure of pig TF, the NZ and NE group of the homologous lysine (Lys543) and arginine (Arg641) are ~4.1 Å apart, too far to share a hydrogen bond [16]. Lacking a crystal structure of an iron-containing C-lobe of hTF, the precise mechanism by which the C-lobe triad triggers iron release remains unclear. However, it has been shown that mutation of Lys534 or Arg632 to an alanine severely retards iron release from that lobe, essentially locking iron in the C-lobe [17].
1.2 Anion Binding to hTF
It is well documented that synergistic anion binding is an absolute requirement for high affinity Fe3+ binding by hTF [18]. Although carbonate is the physiologically relevant synergistic anion [19], other molecules (oxalate, glycolate, malonate, etc.) can substitute for carbonate to promote high affinity Fe3+ binding to hTF [20]. Synergistic anions appear to follow an interlocking sites model [20] in which the anion contains a carboxylate group available to bind the anchoring arginine residue in each lobe of hTF, as well as a proximal electron donor group 1-2 carbon atoms away to complete the distorted octahedral coordination of the Fe3+[21].
Along with synergistic anion binding, non-synergistic anions also bind to hTF. By definition non-synergistic anions do not facilitate Fe3+ binding to hTF, but bind to other sites. As detailed by Folajtar and Chasteen [22] the binding of non-synergistic anions to hTF follows the lyotropic series (SCN− > ClO4− > PPi > ATP > Cl− >> BF4−) and is suggested to induce structural changes that perturb the Fe3+ binding center and thereby influence iron release from hTF. Moreover, Kretchmar and Raymond [23] clearly demonstrated that binding of non-synergistic anions to hTF is obligatory for iron release: iron release from hTF ceases as the ionic strength is extrapolated to zero at pH 7.4. To emphasize their critical allosteric effect on iron release from hTF, these non-synergistic anion binding sites were therefore named “kinetically significant anion binding” or KISAB sites by Egan et al. [24-27].
Numerous spectroscopic techniques, including EPR [22, 28-31], NMR [32] and UV-difference spectra [33-37], have been utilized to detect anion binding to hTF. Work by Moreton et al. [38] clearly demonstrated that salt retards the rate of iron release from the N-lobe while facilitating iron release from the C-lobe at pH 7.4. The Harris laboratory utilized UV-difference techniques to provide an estimate of the binding strength of various anions to apohTF [34, 37, 39]. The Sadler group has published a review with the equilibrium binding constants for thirteen non-synergistic anions to hTF [40].
It is important to note that many of these earlier studies monitoring anion binding to hTF were performed at neutral pH (~7.4), in the absence of the TFR. We propose that in order to be designated as kinetically significant, non-synergistic anion binding must correlate with low pH; i.e., anion binding to a KISAB site must exert maximal effect at pH ~5.6 which is not always the case (vide infra). Although it is well known that non-synergistic anions are required for iron release from hTF at pH 7.4, identification of the specific non-synergistic anion binding sites has remained elusive. Positively charged (lysine, arginine and histidine) residues are obvious choices for potential KISAB sites. For many years it was hypothesized that the most likely sites would be iron liganding residues, second-shell residues and residues in the hinging β-strands which facilitate opening and closing of each lobe of hTF [27, 29, 41]. It seems likely that anions bind to these residues (normally buried within the cleft of each lobe when Fe3+ is bound) only after iron is released and therefore do not qualify as legitimate KISAB sites, but may function to stabilize the apo conformation of each lobe.
1.3 The Transferrin Receptor (TFR)
Briefly, the TFR is a type II transmembrane homodimeric receptor comprised of a short cytoplasmic tail (residues 1-67) with an internalization motif, a membrane-spanning portion (residues 68-88), a stalk region (residues 89-120) and a large extracellular ectodomain (residues 121-760) [42]. The ectodomain is further subdivided into three domains: the protease-like domain (residues 121-188 and 384-606), the apical domain (residues 189-383) and the helical domain (residues (607-760) responsible for dimerization [43].
1.4 Techniques to Monitor Iron Release from hTF
A number of different methods have been utilized to monitor iron release from hTF. The interaction between the two tyrosinates and the coordinated Fe3+ in each lobe of hTF produces a ligand-to-metal charge transfer (LMCT) band centered at ~470 nm [44-46]. In the past, iron release from hTF was directly measured by monitoring the decrease in the visible absorbance maximum [47]. However, this technique is rather insensitive requiring substantial amounts of protein. Another technique to monitor iron release from hTF and hTF/TFR complexes was the polyethylene glycol (PEG) precipitation method developed by Aisen and coworkers [11, 48, 49]. These experiments required adding labeled 59Fe to apohTF. Because PEG allows precipitation of the 59Fe2hTF while the chelate-iron complex 59Fe-PPi remains in the soluble fraction, the 59Fe label released from hTF can be monitored during the iron removal process (at various pH values). More recently, intrinsic tryptophan fluorescence has become the method of choice to monitor iron release from hTF. Binding of Fe3+ to hTF results in disruption of the π-π* transition of the two tyrosine ligands, increasing the UV absorbance which overlaps with and strongly quenches the Trp fluorescence through radiationless transfer of excited-state energy (as first described by Lehrer) [44]. Therefore, a significant increase in the intrinsic tryptophan fluorescence of hTF is observed when iron is removed from hTF: iron removal from Fe2hTF, FeNhTF and FeChTF results in a 368%, 74% and 71% increase the fluorescence intensity, respectively [50]. Mutagenesis studies on both the N- and C-lobes of hTF have clearly determined which Trp residues (3 in the N-lobe and 5 in the C-lobe) contribute to the observed increase in fluorescence upon iron removal [51, 52]. Moreover, the utilization of Trp analogues established that the increase in the fluorescent signal that is observed when iron is removed from hTF/TFR complexes is probably attributable to the Trp residues in hTF and not the 22 Trp residues of the TFR homodimer (note that these experiments were carried out using the soluble portion of the TFR, designated sTFR) [53].
2. Kinetics of Iron Release from hTF
Although years of research effort have produced interesting findings on the kinetics of iron release from hTF at pH ~7.4 [11, 25, 39, 48, 54-59], with relevance to whole body chelator development and iron acquisition by bacterial siderophores during pathogenesis, this review will focus on the kinetics of iron release from hTF at endosomal pH ~5.6 with relevance to intracellular iron delivery.
2.1 Experimental Challenges
A great deal of effort has been devoted to understanding the mechanism and kinetics of iron removal from hTF. However, numerous experimental challenges exist due to the complexity of the hTF system. A particular challenge has been the isolation and assignment of the individual rate constants for each kinetic event (iron release and associated conformational changes). Previous studies utilized hTF constructs in which iron had been selectively removed from one iron-binding site [9, 60], or site-specifically loaded with kinetically inert cobalt allowing isolation of some of these microscopic rate constants. However, uncertainty with regard to the homogeneity of the samples has been a concern.
More recently, the use of recombinant technology has provided the means to produce hTFs that are either incapable of binding (mutation of the two liganding Tyr residues to Phe precludes iron binding in one lobe to create authentic FeNhTF or FeChTF constructs) [61] or releasing iron from one of the two lobes (mutation of residues in the dilysine trigger or C-lobe triad prevent iron removal to create LockNhTF or LockChTF constructs) [17, 50, 62].
The initial studies measuring iron release from hTF in the presence of the receptor utilized full-length TFR isolated from placenta. Solubilization of the membrane bound TFR required harsh purification techniques and the presence of detergent micelles, resulting in low yields of TFR [63, 64]. Again, the homogeneity of such TFR preparations has been a serious concern. Nevertheless, a number of significant findings emerged from the early studies with placental-derived TFR (described below) that served as the groundwork for subsequent work.
The recombinant production of the soluble portion of the TFR (sTFR, residues 121-706) [43, 65, 66] has overcome most of the challenges of working with the full-length placental-derived TFR. The use of the sTFR eliminates the need for detergent, and although two disulfide bonds in the stalk region (Cys89 and Cys98) are formed as a result of TFR dimerization, the homodimer is maintained even in their absence (as in the sTFR) due to the strong interaction between the helical domains of each monomer.
A daunting experimental challenge has been to capture all kinetic events, including conformational changes during the process of iron removal from hTF. Although the kinetics of iron release from hTF at pH 5.6 are relatively slow in comparison to most enzymatic processes, many techniques monitoring iron release rates (PEG precipitation, absorbance studies and steady-state Trp fluorescence) were unable to measure early kinetic events in the process. This challenge has recently been overcome by the use of stopped-flow absorbance and fluorescence [51, 52, 62]. The use of the stopped-flow fluorescence method to measure the kinetics of iron release not only captures rapid kinetic events, but is also very sensitive (requiring only nM concentrations of protein) and is reproducible [62].
2.2 Early Studies on the Kinetics of Iron Release from hTF ± TFR at pH 5.6
Some of the first mechanistic insights into the role of the TFR on the kinetics of iron release from hTF at pH 5.6 were gained from the works of the Aisen laboratory[11, 49]. Specifically, PEG precipitation studies with the TFR provided the initial suggestion that the TFR hinders iron release from hTF at pH 7.4 while facilitating iron release from hTF at pH 5.6 [64]. Additionally, Bali and Aisen [11, 49], determined that in the absence of the TFR, iron is released first from the N-lobe followed by the C-lobe; importantly they also suggested that the presence of the TFR reversed this order. Further studies utilizing a time-based steady-state tryptophan fluorescence technique clearly demonstrated that the TFR enhanced the rate of iron release from the C-lobe of hTF at pH 5.6 [12].
Studies from the laboratory of el Hage Chahine used stopped-flow absorbance and pH-jump chemical relaxation studies to monitor iron release from Fe2hTF and FeChTF in the presence of detergent-solubilized placental-derived TFR [67, 68]. The Aisen laboratory previously noted the absolute requirement of an iron chelator, even at low pH, to achieve reasonable rates for iron removal from hTF, i.e., to remove 50% of iron from an hTF/TFR complex at pH 5.6 in the absence of an iron chelator required hours [64]. Contrary to our findings (discussed below) and the findings of the Aisen laboratory, the el Hage Chahine laboratory concluded that iron is preferentially removed from the N-lobe of hTF first, both in the absence and presence of the detergent solubilized TFR. However, in these studies acetate was used as the competing ligand for the iron. Recently, we have shown that at pH 5.6, acetate alone is ineffective in removing iron from hTF in the presence or absence of the sTFR [62]. However, when the same acetate buffer was supplemented with a chelator (EDTA), complete iron release was achieved [62]. Given the absence of a strong chelator in the kinetic studies from the el Hage Chahine laboratory, it is unclear what was actually being measured and the validity of their findings is unclear.
2.3 A Cautionary Tale
Our laboratory has recently published the most comprehensive kinetic analysis of iron release from hTF in the absence and presence of the sTFR using an optimized stopped-flow iron removal assay (see below) [62]. Briefly, to perform these experiments, hTF, alone or in complex with the sTFR, is rapidly mixed with our standard iron removal buffer (100 mM MES, pH 5.6 containing 300 mM KCl and 4 mM EDTA). Use of the stopped-flow spectrofluorometer allows capture of all events leading to iron release. In combination with the ability to produce recombinant hTF with specific properties of iron binding and/or release, a comprehensive determination of all kinetic steps is now possible. Crucially, such a scheme (Fig. 2A) must also include assessment of the release properties of hTF constructs bound to the TFR (Fig. 2B). It is important to re-emphasize that in our previous work and in that of others using a steady-state format, crucial kinetic events were missed. Examples include the updated and far more detailed analysis of the kinetics of iron release from a series of six mutants in which the histidine at position 349 in the C-lobe was changed to another amino acid [69]. We suggest that in this case although the conclusions of the earlier work were correct [70], the stopped-flow format has allowed capture of additional events providing a better understanding of the role of His349. Additionally, the sensitivity of the spectrofluorometer requires lower concentrations of sample, facilitating multiple determinations and an excellent signal to noise ratio.
Figure 2.
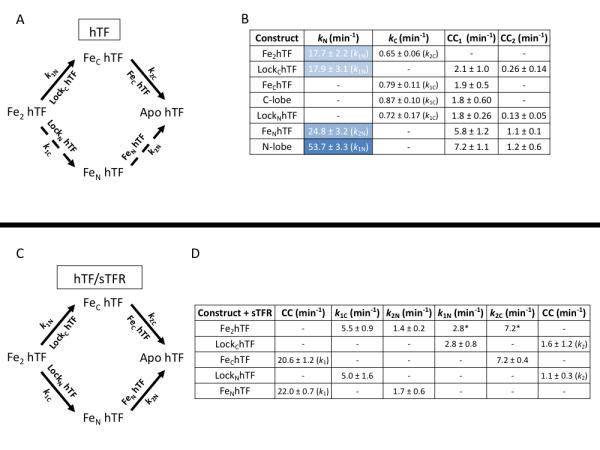
Kinetics of iron release at pH 5.6 from hTF ± sTFR. (A) Iron release in the absence of the sTFR. The primary pathway of iron release is indicated as solid arrows while the alternative pathway is indicated as broken arrows. Specific constructs used to isolate the rates are indicated by the arrows. (B) Rates of iron release from various hTF constructs. Rates of iron release from the N-lobe are highlighted in blue (differing shades are utilized to highlight the cooperativity of the N-lobe). Conformational changes (CC1 and CC2) that occur following iron release are also reported. (C) Iron release in the presence of the sTFR. (D) Rates of iron release from hTF/sTFR complexes. As previously reported [62], all values are rate constants ± errors at the 95% confidence interval and are averages of multiple kinetic runs. *k1N and k2C are held fixed at the values for LockChTF and FeChTF, respectively, during fitting of Fe2hTF/sTFR data.
In contrast, we incorrectly suggested that when Asp634, a member of the C-lobe triad (Lys534, Arg632 and Asp634), was mutated to alanine the rate of iron release was extremely slow [17]. Using the decrease in absorbance in the absence of the sTFR and the steady-state tryptophan fluorescence format in the presence of the sTFR, we reported that the D634A mutant had the slowest rate of iron release compared to the other mutants K534A and R632A. More recently, kinetic analysis with the stopped-flow spectrofluorometer reveals that iron release from the D634A FeChTF construct (± sTFR) is so fast that we missed it completely in the earlier study (Fig. 3B). Notably, in the absence of the sTFR, the D634A FeChTF mutant releases iron very rapidly (Fig. 3A, ~83-fold faster than the FeChTF control). Of interest, the rate of the conformational change normally following iron release in the FeChTF construct is unaffected in the D634A FeChTF mutant (Fig. 3B). In the presence of the sTFR, both the rate for the conformational change and iron release are affected. The rate of iron release is increased only 2-fold by the mutation (as opposed to ~83-fold in the absence of the sTFR) and the preceding conformational change is increased ~5-fold. It is now clear from the stopped-flow data that mutation of Asp634 does not hinder, but rather accelerates iron release from the C-lobe. The rapid release of iron from the D634A mutant is consistent with the suggestion that Asp634 stabilizes the interaction between Lys534 and Arg632.
Figure 3.
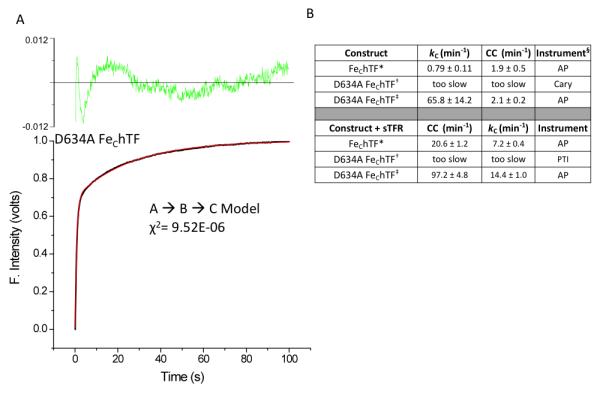
Effects on iron release when Asp634 is mutated to alanine. (A) Normalized iron release curve from the D634A FeChTF mutant (black line) and fit (red line). The residuals are indicated (green). (B) Values for iron release and conformational changes are reported as averages ± 95% confidence intervals. * From [62]. † From [17]. ‡ Previously unpublished results. § Measurements were carried out using a Varian Cary 100 dual beam spectrophotometer (Cary), a Photon Technology International QuantaMaster (PTI) spectrofluorometer, or an Applied Photophysics (AP) SX.18MV stopped-flow spectrofluorometer as indicated.
Using this new stopped-flow iron release assay, kinetic rate laws have been derived for iron release from all control constructs (± the sTFR) under our standard conditions [62]. With all of the proper controls and equations in place we are confident in our ability to extract accurate kinetic constants that provide important information with regard to the detailed mechanism of iron release. We are currently in the process of revisiting available mutants that were evaluated using the steady-state format. Understanding this complex system, especially with regard to the role of the TFR, is crucial to ultimately discovering how to manipulate it.
2.4 Iron Release from hTF at Endosomal pH in the Absence of the TFR
As indicated above, it is clear from decades of research that iron release from hTF is complex and involves a number of factors including pH, anion binding, chelator, lobe-lobe cooperativity and binding to the TFR. Under pseudo-first order conditions, iron release from Fe2hTF can occur via two pathways (Fig. 2A) giving rise to four microscopic rate constants, (k1C, k2N, k1N, and k2C) [49, 71]. The use of diferric locked constructs (LockNhTF and LockChTF) allows the specific assignment of k1C and k1N, while the use of authentic monoferric constructs (FeNhTF and FeChTF) allows the specific assignment of k2N and k2C, respectively. Curve fitting of Fe2hTF shows that in the absence of the sTFR iron release from the N-lobe is very rapid (17.7 ± 2.2 min−1) and relatively slow from the C-lobe (0.65 ± 0.06 min−1) (Fig. 2B). These data indicate that iron is released from Fe2hTF almost exclusively (~96% of the time) via the upper pathway (Fig. 2A, k1N→ k2C, solid arrows), precluding the need to consider the negligible lower pathway (Fig. 2A, k1C→ k2N, broken arrows) in the analysis of Fe2hTF data. The validity of these values is further supported by the independent measures of k1N and k2C through analysis of the LockChTF and FeChTF constructs.
It has long been suggested that cooperativity exists between the two iron binding sites of hTF. Kinetic analysis of FeNhTF and the isolated N-lobe, along with k1N values obtained from the Fe2hTF and LockChTF constructs, indicates that the rate of iron release from the N-lobe (kN) is impacted not only by the presence, but also the iron occupancy of the C-lobe (Fig 2B). However, in the absence of the TFR the rate of iron release from the C-lobe of hTF (kC) is obviously unaffected by the presence or iron status of the N-lobe (Fig 2B). Kinetic cooperativity factors have often been used as a measure of the cooperative effect of iron release in one lobe on the other lobe [49, 62]. Given as a ratio of k2/k1 of a lobe, a number greater than one indicates positive cooperativity between the two lobes, whereas a number equal to or less than one indicates a lack of cooperativity. The kinetic cooperativity figures based on these determined kinetic values (k2N/k1N = 24.8/17.9 = 1.4 and k2C/k1C = 0.79/072 = 1.1) indicate that iron release from the N-lobe is accelerated by the absence of iron in the C-lobe as well as by the complete absence of the C-lobe, whereas iron occupancy of the N-lobe has little affect on the kinetics of iron release from the C-lobe.
Intrinsic tryptophan fluorescence allows monitoring of not only iron release rates from hTF, but conformational changes as well. Two conformational changes (CC1 and CC2) are observed following iron release from the two locked constructs (LockNhTF and LockChTF), the N-lobe and FeNhTF, whereas only one conformational change is observed following iron release from the C-lobe and FeChTF constructs. These conformational changes have been ascribed to structural changes that occur in hTF to adopt the final apo conformation following iron release [62]. Interestingly, inclusion of the rates for the conformational changes observed in the monoferric constructs does not significantly affect the rate obtained during the analysis of Fe2hTF data and therefore are excluded as detailed in the Supplementary Data of reference [62].
Clearly, a complex relationship exists between anion binding to KISAB sites and iron release from hTF. At pH 5.6, anion concentration most dramatically affects the rate of iron release from the N-lobe (increasing it ~4-fold from 50-600 mM [Cl−]) in the absence of the TFR [62, 72], while the rate of iron release from the C-lobe is relatively unaffected [62]. Recently, the first authentic KISAB site in the N-lobe, residue Arg143, was definitively identified [72]. Mutation of Arg143 to alanine completely eliminates iron release from the N-lobe of hTF at all tested [Cl−] (50-600 mM) in the absence of the TFR and may (at least in part) account for the salt sensitivity of iron release from the N-lobe of hTF. Additionally, mutation of residue Lys569 in the Fe2hTF background was previously found to slow iron release by 15-20 fold and inhibit chloride enhancement of iron release from the C-lobe [73]. Therefore, Lys569 was deemed essential for iron release from the C-lobe of hTF and putatively identified as a KISAB site. However, using our standard conditions, our kinetic data shows that mutation of Lys569 to alanine (K569A) has little effect on the rate of iron release from Fe2hTF in the absence (Fe2hTF k2C = 0.65 ± 0.06 min−1 versus K569A Fe2hTF k2C = 0.56 ± 0.06 min−1) or presence of the sTFR (Fe2hTF/sTFR k1C = 5.5 ± 0.9 min−1 versus K569A Fe2hTF/sTFR k1C = 6.6 ± 0.8 min−1, previously unpublished data). These data coincide with the finding that the rate of iron release from the C-lobe is relatively unaffected by anion concentration. The discrepancy between more recent and previously published data again highlights the limitations of the steady-state tryptophan fluorescence technique of earlier work.
2.5 Iron Release from hTF/sTFR Complex at Endosomal pH
The TFR plays an integral role in physiologic iron release from hTF. Therefore, although more complex, it is critical that iron release from hTF also be monitored in the presence of the TFR. Corroborating previous findings [11, 49], under our standard conditions, the presence of the sTFR induces a switch in the order of iron release, such that iron is preferentially removed from the C-lobe first followed by the N-lobe (Fig. 2C, k1C → k2N) [62]. However, the fits indicate that this is not the case 100% of the time. The upper pathway (k1N → k2C) is reduced from 96% in the absence of the sTFR to 35% in the presence of the sTFR. Thus, it is clear that both pathways are physiologically relevant and must be included in the fits of Fe2hTF/sTFR complex (Fig. 2C). In order to limit the number of variables while analyzing Fe2hTF/sTFR complex kinetic data, the values for the upper pathway (k1N and k2C) are held constant based on kinetic values obtained from the LockChTF/sTFR and FeChTF/sTFR complexes (Fig. 2D). Again, the values obtained for the lower pathway from the LockNhTF/sTFR and FeNhTF/sTFR complexes (k1C = 5.0 ± 1.6 and k2N = 1.7 ± 0.6) corroborate the validity of the Fe2hTF/sTFR complex analysis (Fig. 2D).
The TFR has previously been shown to enhance the rate of iron release from the C-lobe of hTF [11]. Under our standard conditions, the sTFR enhances iron release from the C-lobe by ~7-11 fold (k1C, complex/ k1C, alone and k2C, complex/ k2C, alone) and retards iron release from the N-lobe by 6-15 fold (k1N, complex/ k1N, alone and k2N, complex/ k2N, alone) (Fig. 2B and 2D) [62]. Thus, not only does binding to the sTFR switch the order of iron release from hTF, but also makes the rates of iron release from the two lobes more equivalent than in the absence of the sTFR. Because the TFR serves to balance the rates of iron release from the lobes of hTF the calculated cooperativity factors of each lobe are much different in the presence of the sTFR (N-lobealone = 1.4 versus N-lobecomplex = 0.61 and C-lobealone = 1.1 versus C-lobecomplex =1.5) [62]. Therefore, in the presence of the sTFR, the rate of iron release from the C-lobe is slowed by the presence of iron in the N-lobe, while for the N-lobe the reverse is true where iron occupancy in the C-lobe accelerates iron release from the N-lobe.
As stated above, intrinsic tryptophan fluorescence allows not only iron release rates from hTF/sTFR complexes to be monitored, but conformational changes as well. Interestingly, a rapid conformational change with k = 22.0 min−1 or 20.6 min−1 is observed before iron release in both monoferric complexes FeNhTF/sTFR and FeChTF/sTFR, respectively (Fig. 2D). Conversely, a slow conformational change (k = 1.1 min−1 or 1.6 min−1) is observed after iron release in both locked complexes (LockNhTF/sTFR and LockChTF/sTFR) (Fig. 2D). The similar values of these conformational changes in the monoferric and locked complexes, respectively, suggest that these may represent the same conformational change in the hTF molecule or a similar conformational change in each lobe of hTF. Obviously further study is required to establish the exact nature and origin of these conformational changes.
A critical component to the endocytic cycle and iron delivery by hTF is endosomal pH (~5.6). Based on the inflection point of a pH titration, iron release from the FeChTF/TFR complex titrates with an apparent pKa of ~5.8-5.9 [69]. This value is strikingly similar to the pKa of histidine residues. As previously mentioned, residue His349 in the C-lobe of hTF has been a residue of particular interest for some time [70]. Interestingly, mutation of His349 to alanine or lysine completely abolishes the pH dependence of iron release from the FeChTF/sTFR complex [69]. This led to the proposal that His349 must act as a pH-inducible switch that accelerates iron release from the C-lobe of hTF. More recently, the effect of the H349A mutation in the Fe2hTF background has been investigated. Curiously, H349A Fe2hTF/sTFR complex displays three kinetic rates. The first rate (k1 = 23.7 ± 4.6 min−1) has been ascribed to a rapid conformational change that is again very similar to the conformational change observed in both monoferric complexes (Fig. 2B). Iron release from the H349A Fe2hTF/sTFR complex follows only the upper pathway (k1N → k2C) and the rate of iron release from the N-lobe is about doubled (k1N = 6.7 ± 0.3 min−1) compared to the Fe2hTF/sTFR complex (k1N = 2.8 ± 0.8 min−1). Interestingly, iron release from the C-lobe of the H349A Fe2hTF/sTFR complex is greatly retarded (k2C = 0.61 ± 0.02 min−1) such that the effect of receptor stimulation on iron release from the C-lobe is completely abrogated in the presence of the H349A mutation (unpublished results).
In contrast to the findings of Egan et al. [27], at pH 5.6 the effect of anion concentration on iron release from hTF in the presence of the sTFR is significantly muted [62]. Therefore, although salt slightly accelerates iron removal from the hTF/TFR complex at pH 5.6, the overriding kinetic effect of the TFR on iron release appears to outweigh any kinetic anion effects. In accordance with these findings [62], the effect of the Arg143 mutation (described above) is largely abolished in the presence of the TFR [72].
3. Conclusions
Our use of recombinant constructs has, for the first time, provided access to discrete steps in the pathway of iron removal from Fe2hTF to apohTF. Crucially, measurements in the absence and presence of the recombinant sTFR under identical experimental conditions highlight the definitive role of the TFR in the process of iron release from hTF. Given this comprehensive kinetic scheme, we are now well positioned to evaluate the contribution of individual residues in both hTF and the sTFR to the mechanism. Future studies must address in a detailed manner how the Fe3+ released from hTF within the endosome is distributed within the cell.
Highlights.
Because the interaction of transferrin with the receptor controls iron distribution, an understanding of this process essential.
The transferrin receptor modulates iron release from the N- and C-lobes of human serum transferrin.
The transferrin receptor balances the rate of iron release from each lobe, resulting in efficient Fe3+ release from transferrin.
Acknowledgments
We are very grateful for the invitation to write this review. We thank our many wonderful colleagues who have made contributions to this work. Robert C. Woodworth who got us hooked on transferrin, previous graduate students, Peter J. Halbrooks and Nicholas G. James, as well as the laboratories of Ross T.A. MacGillivray and coworker Valerie C. Smith, Igor A. Kaltashov and coworker Cedric E. Bobst and Pamela Björkman and her former graduate student Anthony Giannetti. This work was supported by USPHS Grant R01 DK 21739 (ABM) and R01 GM-20194 (NDC). ANS is currently funded by an AHA Predoctoral Fellowship (10PRE4200010).
1 Abbreviations
- hTF
human serum transferrin
- TFR
transferrin receptor
- Fe2hTF
diferric hTF
- FeNhTF
hTF with iron bound only in the N-lobe
- FeChTF
hTF with iron bound only in the C-lobe
- LockNhTF
Fe2hTF that cannot release iron from the N-lobe
- LockChTF
Fe2hTF that cannot release iron from the C-lobe
- sTFR
soluble portion of the TFR
- KISAB
kinetically significant anion binding
- LMCT
ligand to metal charge transfer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Inter. J. of Biochem. Cell. Biol. 2001;33:940–959. doi: 10.1016/s1357-2725(01)00063-2. [DOI] [PubMed] [Google Scholar]
- [2].Park CH, Bacon BR, Brittenham GM, Tavill AS. Pathology of dietary carbonyl iron overload in rats. Lab. Invest. 1987;57:555–563. [PubMed] [Google Scholar]
- [3].Williams J, Moreton K. The distribution of iron between the metal-binding sites of transferrin human serum. Biochem. J. 1980;185:483–488. doi: 10.1042/bj1850483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dhungana S, Taboy CH, Zak O, Larvie M, Crumbliss AL, Aisen P. Redox properties of human transferrin bound to its receptor. Biochemistry. 2004;43:205–209. doi: 10.1021/bi0353631. [DOI] [PubMed] [Google Scholar]
- [5].Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005;37:1264–1269. doi: 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leverence R, Mason AB, Kaltashov IA. Noncanonical interactions between serum transferrin and transferrin receptor evaluated with electrospray ionization mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2010;107:8123–8128. doi: 10.1073/pnas.0914898107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Princiotto JV, Zapolski EJ. Difference between the two iron-binding sites of transferrin. Nature. 1975;255:87–88. doi: 10.1038/255087a0. [DOI] [PubMed] [Google Scholar]
- [8].Lestas AN. The effect of pH upon human transferrin: selective labelling of the two iron-binding sites. Br. J. Haematol. 1976;32:341–350. doi: 10.1111/j.1365-2141.1976.tb00937.x. [DOI] [PubMed] [Google Scholar]
- [9].Baldwin DA, de Sousa DM. The effect of salts on the kinetics of iron release from N-terminal and C terminal monoferric transferrins. Biochem. Biophys. Res. Commun. 1981;99:1101–1107. doi: 10.1016/0006-291x(81)90732-4. [DOI] [PubMed] [Google Scholar]
- [10].Baldwin DA. The kinetics of iron release from human transferrin by EDTA. Effect of salts and detergents. Biochim. Biophys. Acta. 1980;623:183–198. doi: 10.1016/0005-2795(80)90020-3. [DOI] [PubMed] [Google Scholar]
- [11].Bali PK, Aisen P. Receptor modulated iron release from transferrin: Differential effects on N- and C-terminal sites. Biochemistry. 1991;30:9947–9952. doi: 10.1021/bi00105a019. [DOI] [PubMed] [Google Scholar]
- [12].Zak O, Aisen P. Iron release from transferrin, its C-lobe, and their complexes with transferrin receptor: presence of N-lobe accelerates release from C-lobe at endosomal pH. Biochemistry. 2003;42:12330–12334. doi: 10.1021/bi034991f. [DOI] [PubMed] [Google Scholar]
- [13].Dewan JC, Mikami B, Hirose M, Sacchettini JC. Structural evidence for a pH-sensitive dilysine trigger in the hen ovotransferrin N-lobe: implications for transferrin iron release. Biochemistry. 1993;32:11963–11968. doi: 10.1021/bi00096a004. [DOI] [PubMed] [Google Scholar]
- [14].Jeffrey PD, Bewley MC, MacGillivray RT, Mason AB, Woodworth RC, Baker EN. Ligand-induced conformational change in transferrins: crystal structure of the open form of the N-terminal half-molecule of human transferrin. Biochemistry. 1998;37:13978–13986. doi: 10.1021/bi9812064. [DOI] [PubMed] [Google Scholar]
- [15].He Q-Y, Mason AB. Molecular aspects of release of iron from transferrin. D.M. Templeton; New York: 2002. [Google Scholar]
- [16].Hall DR, Hadden JM, Leonard GA, Bailey S, Neu M, Winn M, Lindley PF. The crystal and molecular structures of diferric porcine and rabbit serum transferrins at resolutions of 2.15 and 2.60 Å, respectively. Acta. Crystallogr. D Biol. Crystallogr. 2002;58:70–80. doi: 10.1107/s0907444901017309. [DOI] [PubMed] [Google Scholar]
- [17].Halbrooks PJ, He QY, Briggs SK, Everse SJ, Smith VC, MacGillivray RT, Mason AB. Investigation of the mechanism of iron release from the C-lobe of human serum transferrin: mutational analysis of the role of a pH sensitive triad. Biochemistry. 2003;42:3701–3707. doi: 10.1021/bi027071q. [DOI] [PubMed] [Google Scholar]
- [18].Price EM, Gibson JF. A re-interpretation of bicarbonate-free ferric transferrin E.P.R. spectra. Biochem. Biophys. Res. Commun. 1972;46:646–651. doi: 10.1016/s0006-291x(72)80189-x. [DOI] [PubMed] [Google Scholar]
- [19].Schade AL, Reinhart RW, Levy H. Carbon dioxide and oxygen in complex formation with iron and siderophilin, the iron-binding component of human plasma. Arch. Biochem. 1949;20:170–172. [PubMed] [Google Scholar]
- [20].Schlabach MR, Bates GW. The synergistic binding of anions and Fe3+ by transferrin. Implications for the interlocking sites hypothesis. J. Biol. Chem. 1975;250:2182–2188. [PubMed] [Google Scholar]
- [21].Baker EN. Structure and reactivity of transferrins. Adv. Inorg. Chem. 1994;41:389–463. [Google Scholar]
- [22].Folajtar DA, Chasteen ND. Measurment of nonsynergistic anion binding to transferrin by EPR difference spectroscopy. J. Am. Chem. Soc. 1982;104:5775–5780. [Google Scholar]
- [23].Kretchmar SA, Raymond KN. Effects of ionic strength on iron removal from the monoferric transferrins. Inorg. Chem. 1988;27:1436–1441. [Google Scholar]
- [24].Marques HM, Egan TJ, Pattrick G. The non-reductive removal of iron from human serum N-terminal monoferric transferrin by pyrophosphate. S. Afr. J. Sci. 1990;86:21–24. [Google Scholar]
- [25].Marques HM, Watson DL, Egan TJ. Kinetics of iron removal from human serum monoferric transferrins by citrate. Inorg. Chem. 1991;30:3758–3762. [Google Scholar]
- [26].Egan TJ, Ross DC, Purves LR, Adams PA. Mechanism of iron release from human serum C-terminal monoferric transferrin to pyrophosphate: kinetic discrimination between alternative mechanisms. Inorg. Chem. 1992;31:1994–1998. [Google Scholar]
- [27].Egan TJ, Zak O, Aisen P. The anion requirement for iron release from transferrin is preserved in the receptor-transferrin complex. Biochemistry. 1993;32:8162–8167. doi: 10.1021/bi00083a016. [DOI] [PubMed] [Google Scholar]
- [28].Chasteen ND. Transferrin: a perspective. Adv. Inorg. Biochem. 1983;5:201–233. [PubMed] [Google Scholar]
- [29].Grady JK, Mason AB, Woodworth RC, Chasteen ND. The effect of salt and site-directed mutations on the iron(III)-binding site of human serum transferrin as probed by EPR spectroscopy. Biochem. J. 1995;309(Pt 2):403–410. doi: 10.1042/bj3090403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Thompson CP, McCarty BM, Chasteen ND. The effects of salts and amino group modification on the iron binding domains of transferrin. Biochim. Biophys. Acta. 1986;870:530–537. doi: 10.1016/0167-4838(86)90262-1. [DOI] [PubMed] [Google Scholar]
- [31].Price EM, Gibson JF. Electron paramagnetic resonance evidence for a distinction between the two iron-binding sites in transferrin and in conalbumin. J. Biol. Chem. 1972;247:8031–8035. [PubMed] [Google Scholar]
- [32].Kubal G, Mason AB, Patel SU, Sadler PJ, Woodworth RC. Oxalate- and Ga(3+)-induced structural changes in human serum transferrin and its recombinant N-lobe. 1H NMR detection of preferential C-lobe Ga3+ binding. Biochemistry. 1993;32:3387–3395. doi: 10.1021/bi00064a024. [DOI] [PubMed] [Google Scholar]
- [33].Pecoraro VL, Harris WR, Carrano CJ, Raymond KN. Siderophilin metal coordination. Difference ultraviolet spectroscopy of di-, tri-, and tetravalent metal ions with ethylenebis[(o-hydroxyphenyl)glycine] Biochemistry. 1981;20:7033–7039. doi: 10.1021/bi00527a040. [DOI] [PubMed] [Google Scholar]
- [34].Harris WR. Thermodynamics of anion binding to human serum transferrin. Biochemistry. 1985;24:7412–7418. doi: 10.1021/bi00346a057. [DOI] [PubMed] [Google Scholar]
- [35].Harris WR, Bali PK. Effects of anions on the removal of iron from transferrin by phosphonic acids and pyrophosphate. Inorg. Chem. 1988;27:2687–2691. [Google Scholar]
- [36].Harris WR, Cafferty AM, Abdollahi S, Trankler K. Binding of monovalent anions to human serum transferrin. Biochim. Biophys. Acta. 1998;1383:197–210. doi: 10.1016/s0167-4838(97)00152-0. [DOI] [PubMed] [Google Scholar]
- [37].Harris WR, Nesset-Tollefson D, Stenback JZ, Mohamed-Hani N. Site selectivity in the binding of inorganic anions to serum transferrin. J. Inorg. Biochem. 1990;38:175–183. doi: 10.1016/0162-0134(90)84011-d. [DOI] [PubMed] [Google Scholar]
- [38].Williams J, Chasteen ND, Moreton K. The effect of salt concentration on the iron-binding properties of human transferrin. Biochem. J. 1982;201:527–532. doi: 10.1042/bj2010527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bali PK, Harris WR. Site-specific rate constants for iron removal from diferric transferrin by nitrilotris(methylenephosphonic acid) and pyrophosphate. Arch. Biochem. Biophys. 1990;281:251–256. doi: 10.1016/0003-9861(90)90440-a. [DOI] [PubMed] [Google Scholar]
- [40].Sun H, Li H, Sadler PJ. Transferrin as a metal ion mediator. Chem. Rev. 1999;99:2817–2842. doi: 10.1021/cr980430w. [DOI] [PubMed] [Google Scholar]
- [41].Harris WR, Nesset-Tollefson D. Binding of phosphonate chelating agents and pyrophosphate to apotransferrin. Biochemistry. 1991;30:6930–6936. doi: 10.1021/bi00242a017. [DOI] [PubMed] [Google Scholar]
- [42].McClelland A, Kuhn LC, Ruddle FH. The human transferrin receptor gene: genomic organization, and the complete primary structure of the receptor deduced from a cDNA sequence. Cell. 1984;39:267–274. doi: 10.1016/0092-8674(84)90004-7. [DOI] [PubMed] [Google Scholar]
- [43].Lawrence CM, Ray S, Babyonyshev M, Galluser R, Borhani DW, Harrison SC. Crystal structure of the ectodomain of human transferrin receptor. Science. 1999;286:779–782. doi: 10.1126/science.286.5440.779. [DOI] [PubMed] [Google Scholar]
- [44].Lehrer SS. Fluorescence and absorption studies of the binding of copper and iron to transferrin. J. Biol. Chem. 1969;244:3613–3617. [PubMed] [Google Scholar]
- [45].Gaber BP, Miskowski V, Spiro TG. Resonance Raman scattering from iron(III)- and copper(II)-transferrin and an iron(III) model compound. A spectroscopic interpretation of the transferrin binding site. J. Am. Chem. Soc. 1974;96:6868–6873. doi: 10.1021/ja00829a010. [DOI] [PubMed] [Google Scholar]
- [46].Patch MG, Carrano CJ. The Origin of the Visible Absorption in Metal Transferrins. Inorganica Chimica Acta. 1981;56:L71–L73. [Google Scholar]
- [47].Harris WR, Rezvani AB, Bali PK. Removal of iron from transferrin by pyrophosphate and tripodal phosphonate ligands. Inorg. Chem. 1986;26:2711–2716. [Google Scholar]
- [48].Bali PK, Harris WR. Cooperativity and heterogeneity between the two binding sites of diferric transferrin during iron removal by pyrophosphate. J. Am. Chem. Soc. 1989;111:4457–4461. [Google Scholar]
- [49].Bali PK, Aisen P. Receptor-induced switch in site-site cooperativity during iron release by transferrin. Biochemistry. 1992;31:3963–3967. doi: 10.1021/bi00131a011. [DOI] [PubMed] [Google Scholar]
- [50].Byrne SL, Mason AB. Human serum transferrin: a tale of two lobes. Urea gel and steady state fluorescence analysis of recombinant transferrins as a function of pH, time, and the soluble portion of the transferrin receptor. J. Biol. Inorg. Chem. 2009;14:771–781. doi: 10.1007/s00775-009-0491-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].James NG, Berger CL, Byrne SL, Smith VC, MacGillivray RT, Mason AB. Intrinsic fluorescence reports a global conformational change in the N-lobe of human serum transferrin following iron release. Biochemistry. 2007;46:10603–10611. doi: 10.1021/bi602425c. [DOI] [PubMed] [Google Scholar]
- [52].James NG, Byrne SL, Steere AN, Smith VC, MacGillivray RT, Mason AB. Inequivalent contribution of the five tryptophan residues in the C-lobe of human serum transferrin to the fluorescence increase when iron is released. Biochemistry. 2009;48:2858–2867. doi: 10.1021/bi8022834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].James NG, Byrne SL, Mason AB. Incorporation of 5-hydroxytryptophan into transferrin and its receptor allows assignment of the pH induced changes in intrinsic fluorescence when iron is released. Biochim. Biophys. Acta. 2008;1794:532–540. doi: 10.1016/j.bbapap.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bali PK, Harris WR, Nesset-Tollefson D. Kinetics of iron removal from monoferric and cobalt-labeled monoferric human serum transferrin by nitilotris(methylenephosphonic acid) and nitrilotriacetic acid. Inorg. Chem. 1991;30:502–508. [Google Scholar]
- [55].Turcot I, Stintzi A, Xu J, Raymond KN. Fast biological iron chelators: kinetics of iron removal from human diferric transferrin by multidentate hydroxypyridonates. J. Biol. Inorg. Chem. 2000;5:634–641. doi: 10.1007/s007750000149. [DOI] [PubMed] [Google Scholar]
- [56].Harris WR, Wang Z, Brook C, Yang B, Islam A. Kinetics of metal ion exchange between citric acid and serum transferrin. Inorg. Chem. 2003;42:5880–5889. doi: 10.1021/ic034009o. [DOI] [PubMed] [Google Scholar]
- [57].Hemadi M, Miquel G, Kahn PH, El Hage Chahine JM. Aluminum exchange between citrate and human serum transferrin and interaction with transferrin receptor 1. Biochemistry. 2003;42:3120–3130. doi: 10.1021/bi020627p. [DOI] [PubMed] [Google Scholar]
- [58].Brook C, Harris WR, Spilling CD, Peng W, Harburn JJ, Srisung S. Effect of ligand structure on pathways for iron release from human serum transferrin. Inorg. Chem. 2005;44:5183–5191. doi: 10.1021/ic050411m. [DOI] [PubMed] [Google Scholar]
- [59].Kumar R, Mauk AG. Atypical effects of salts on the stability and iron release kinetics of human transferrin. J. Phys. Chem. B. 2009;113:12400–12409. doi: 10.1021/jp903257c. [DOI] [PubMed] [Google Scholar]
- [60].Harris DC. Functional equivalence of iron bound to human transferrin at low pH or high pH. Biochim. Biophys. Acta. 1977;496:563–565. doi: 10.1016/0304-4165(77)90338-5. [DOI] [PubMed] [Google Scholar]
- [61].Mason AB, Halbrooks PJ, Larouche JR, Briggs SK, Moffett ML, Ramsey JE, Connolly SA, Smith VC, MacGillivray RT. Expression, purification, and characterization of authentic monoferric and apo-human serum transferrins. Protein. Expr. Purif. 2004;36:318–326. doi: 10.1016/j.pep.2004.04.013. [DOI] [PubMed] [Google Scholar]
- [62].Byrne SL, Chasteen ND, Steere AN, Mason AB. The unique kinetics of iron-release from transferrin: The role of receptor, lobe-lobe interactions and salt at endosomal pH. J. Mol. Biol. 2010;396:130–140. doi: 10.1016/j.jmb.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Turkewitz AP, Amatruda JF, Borhani D, Harrison SC, Schwartz AL. A high yield purification of the human transferrin receptor and properties of its major extracellular fragment. J. Biol. Chem. 1988;263:8318–8325. [PubMed] [Google Scholar]
- [64].Bali PK, Zak O, Aisen P. A new role for the transferrin receptor in the release of iron from transferrin. Biochemistry. 1991;30:324–328. doi: 10.1021/bi00216a003. [DOI] [PubMed] [Google Scholar]
- [65].Lebron JA, Bjorkman PJ. The transferrin receptor binding site on HFE, the class I MHC-related protein mutated in hereditary hemochromatosis. J. Mol. Biol. 1999;289:1109–1118. doi: 10.1006/jmbi.1999.2842. [DOI] [PubMed] [Google Scholar]
- [66].Byrne SL, Leverence R, Klein JS, Giannetti AM, Smith VC, MacGillivray RT, Kaltashov IA, Mason AB. Effect of glycosylation on the function of a soluble, recombinant form of the transferrin receptor. Biochemistry. 2006;45:6663–6673. doi: 10.1021/bi0600695. [DOI] [PubMed] [Google Scholar]
- [67].Hemadi M, Ha-Duong NT, el Hage Chahine JM. The mechanism of iron release from the transferrin-receptor 1 adduct. J. Mol. Biol. 2006;358:1125–1136. doi: 10.1016/j.jmb.2006.02.055. [DOI] [PubMed] [Google Scholar]
- [68].el Hage Chahine JM, Pakdaman R. Transferrin, a mechanism for iron release. Eur. J. Biochem. 1995;230:1102–1110. doi: 10.1111/j.1432-1033.1995.tb20661.x. [DOI] [PubMed] [Google Scholar]
- [69].Steere AN, Byrne SL, Chasteen ND, Smith VC, MacGillivray RTA, Mason AB. Evidence that His349 acts as a pH-inducible switch to accelerate receptor-mediated iron release from the C-lobe of human transferrin. J. Biol. Inorg. Chem. 2010;15:1341–1352. doi: 10.1007/s00775-010-0694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Giannetti AM, Halbrooks PJ, Mason AB, Vogt TM, Enns CA, Bjorkman PJ. The molecular mechanism for receptor-stimulated iron release from the plasma iron transport protein transferrin. Structure. 2005;13:1613–1623. doi: 10.1016/j.str.2005.07.016. [DOI] [PubMed] [Google Scholar]
- [71].Harris WR, Bali PK, Crowley MM. Kinetics of iron removal from monoferric and cobalt-labeled monoferric transferrins by diethylene-triaminepenta (methylenephosphonic acid) and diethylenetriaminepentaacetic acid. Inorg. Chem. 1992;31:2700–2705. [Google Scholar]
- [72].Byrne SL, Steere AN, Chasteen ND, Mason AB. Identification of a kinetically significant anion binding (KISAB) site in the N-lobe of human serum transferrin. Biochemistry. 2010;49:4200–4207. doi: 10.1021/bi1003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zak O, Tam B, MacGillivray RT, Aisen P. A kinetically active site in the C-lobe of human transferrin. Biochemistry. 1997;36:11036–11043. doi: 10.1021/bi970628v. [DOI] [PubMed] [Google Scholar]