

Abstract
Group A Streptococcus (GAS) is an historically important agent of puerperal infections and sepsis. The inception of hand-washing and improved hospital hygiene drastically reduced the incidence of puerperal sepsis, but recently the incidence and severity of postpartum GAS infections has been rising for uncertain reasons. Several epidemiological, host, and microbial factors contribute to the risk for GAS infection and mortality in postpartum women. These include the mode of delivery (vaginal vs. caesarean section), the location where labor and delivery occurred, exposure to GAS carriers, the altered immune status associated with pregnancy, the genetic background of the host, the virulence of the infecting GAS strain, and highly specialized immune responses associated with female reproductive tract tissues and organs. This review will discuss the complicated factors that contribute to the increased susceptibility to GAS after delivery and potential reasons for the recent increase observed in morbidity and mortality.
Keywords: Group A Streptococcus (GAS), postpartum sepsis, maternal immunology, female reproductive tract (FRT)
INTRODUCTION
Group A Streptococcus (GAS) is an historically important cause of puerperal infections and sepsis. Despite preventive measures, including antibiotic use and hospital sanitation efforts, GAS infections are re-emerging worldwide and remain the most common cause of severe puerperal infections [1-5]. The ability of GAS to establish infection in postpartum patients is influenced by numerous factors, including disrupted mucosal barriers, altered immune status of the mother, antibiotic administration during labor and delivery, delayed diagnosis, environmental exposures of the mother, and specific virulence factors utilized by GAS. The complex interactions of these potential risk determinants complicate our understanding of how and why postpartum GAS sepsis occurs. This review will discuss the complicated factors that contribute to the increased susceptibility to postpartum GAS and highlight topics in need of further study.
Methods
Manuscripts cited in this review were identified by searching the available English-language literature using PubMed (U.S. National Library of Medicine, National Institutes of Health, Bethesda MD) for all years available for the following terms or combination of terms: “Group A Streptococcus”, “GAS”, “Streptococcus”, “S. pyogenes”, “GAS virulence factors”, “STSS”, “Bacterial susceptibility”, “Maternal immunology”, “Maternal innate immunology”, “Vagina/Vaginal immunology”, “Uterine/Uterus immunology” “Female reproductive tract immunology”, “Pregnant/Pregnancy immunology”, “Prostaglandin E2”, “PGE2”, “Antimicrobial peptides”, “Neutrophils”, “Macrophages”, “Dendritic cells”, “Postpartum sepsis”, and “Puerperal sepsis”. Additional references were identified within bibliographies provided by PubMed-cited studies.The literature was reviewed through August 31, 2011.
Postpartum Sepsis
An Overview
Globally, puerperal infections cause morbidity in 5-10% of all pregnant women with over 75,000 deaths each year [6, 7]. Despite efforts to meet the United Nations Millennium Development Goal 5 (improve maternal health), the maternal mortality ratio has not improved and infections are an important reason [8]. Several bacterial pathogens can cause postpartum sepsis. While not the scope of this review, Group B Streptococcus is more prevalent than GAS, but typically causes less severe maternal disease [9]. Other causal organisms include staphylococci, Mycoplasma, Chlamydia, Clostridium sordellii, coliform bacteria, and bacteria associated with polymicrobial vaginosis [10]. However, GAS postpartum infections remain the most common cause of severe maternal postpartum infections and death worldwide [11, 12].
Following efforts by Semmelweis and others to popularize hand hygiene and raise the standards of hospital cleanliness, maternal postpartum infections decreased drastically (reviewed in [13]). Despite the dramatic and sustained decreases in postpartum GAS infections and sepsis experienced in the 20th century, the past two decades have witnessed an unexplained increase in severe postpartum GAS infections, resulting in greater numbers of maternal deaths worldwide [3, 8, 14]. This reemergence has placed a new urgency to better understand the host-microbial determinants of disease that might be targeted for improving preventive and therapeutic measures.
GAS is a ubiquitous human pathogen that causes a wide array of disease including cellulitis, pharyngitis, necrotizing soft tissue infections, scarlet fever and invasive puerperal infections. Puerperal infections present rapidly, within 2 to 48 hours postpartum and can be non-specific, delaying treatment. Primary symptoms include myalgias, fever, confusion, euphoria, dizziness, and abdominal pain [15]. Once GAS is diagnosed, the infection is often advanced. Notably, there does not appear to be an increase in GAS antibiotic resistance [16], so other factors must underlie the re-emergence of GAS postpartum infections.
Routes of maternal infection
GAS can be found in the normal biota of the female reproductive tract, but its colonization is considered to be relatively rare (0.03%) and its presence alone is not sufficient to cause disease [17]. However, GAS is asymptomatically carried on the skin or in the throat by 5-30% of the population and is easily spread by person-to-person contact or aerosolization [18]. The host and microbial factors that influence colonization progressing to infection remain unresolved, but it is apparent that postpartum and pregnant women are predisposed to bacterial infections in general (reviewed in [19]).
Women can be a source of contamination of their own reproductive tract. Some mothers with a recent history of sore throat succumb to GAS postpartum sepsis [9], suggesting that some women infect themselves after delivery, presumably through contamination of the perineum or through bacterial travel in the bloodstream from distal organ sites. Another frequent source of GAS exposure in the maternal environment is through interaction with children in the house or at work. In a recent report, all investigated patients who died from GAS postpartum sepsis had recent contact with children (frequent GAS carriers) in their home or work environment [9]. Lamagni et al. demonstrated that invasive GAS infections as a whole are on the rise in the general population [20], perhaps contributing to the increase in maternal exposure in the community.
Due to the presence of asymptomatic carriers, nosocomial infections are a significant potential route of maternal infection. The high incidence of healthcare-associated GAS infections in the time of Semmelweis was due to asymptomatic healthcare-worker carriers, resulting in sporadic postpartum GAS outbreaks in hospitals [6, 21]. Cesarean section has been called “the single most important risk factor” for postpartum maternal infection in a hospital and this may be due to several factors, but one obvious factor is the invasive nature of the surgery [21, 22]. Antibiotic administration during or after surgery significantly reduces the risk for postpartum infection [22] but is not 100% effective at preventing infections from progressing and rapidly causing maternal death [9]. It is easy to regard uncomplicated pregnancies with vaginal deliveries as low-risk for sepsis in a hospital setting, but there has been an increase in postpartum sepsis following these seemingly unremarkable deliveries [9]. The non-specific symptoms at the onset of GAS sepsis result in healthy women becoming critically ill and dying within a few hours or days [9, 23]. Regardless of delivery type, postpartum patients have a 20-fold increased incidence of GAS-induced disease compared to non-pregnant women [24]. Interestingly, this increased incidence is higher than that observed in adults over the age of 65 years, the typical age group associated with increased incidence of GAS infections [25]. The high incidence of asymptomatic carriage and multiple routes of inoculation (Figure 1) result in a significant risk for GAS postpartum infections.
Figure 1. Potential routes of GAS infection during pregnancy and postpartum.
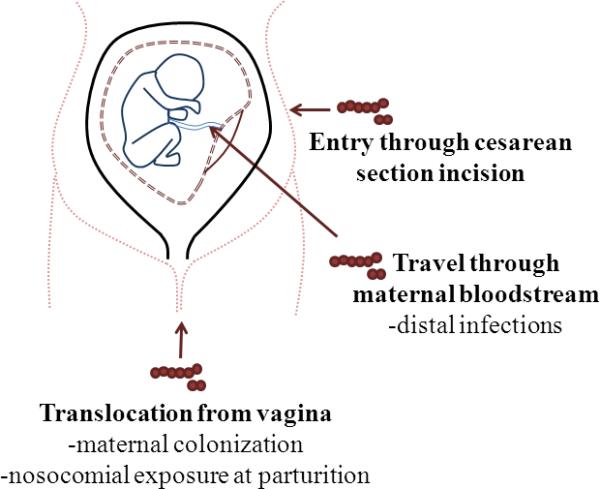
GAS can enter the incision made during cesarean section, leading to a quickly disseminating infection. GAS infections can also result from distal infections (i.e. pharyngitis) where the bacterium travels through the bloodstream, infecting the reproductive tract and developing fetus. The vagina is another source of GAS infections when maternal colonization is present or when the perineum is contaminated after environmental exposures to GAS.
Microbiology and immunology of GAS sepsis
GAS virulence factors and Streptococcal toxic shock syndrome (STSS)
GAS is a versatile human pathogen that utilizes numerous virulence factors to evade immune recognition or clearance. Several recent reviews describe in detail the microbial factors that contribute to GAS pathogenesis [26-30] and will not be discussed in detail. GAS virulence factors aid in evading phagocytosis and facilitate in adherence to host cells, leading to colonization and invasion of the host [26, 31-35]. In addition, GAS has a family of bacterial antigens that are associated with streptococcal toxic shock syndrome (STSS) [36, 37]. This family includes SpeA (Streptococcal pyogenic exotoxin A), SpeC, and others that bind to the MHC class II molecules and T cell receptors, resulting in an excessive release of immunomodulators that activate complement, coagulation, and fibrinolytic cascades, resulting in toxic shock and death. STSS has been reported with invasive GAS soft-tissue infections with a mortality rate of approximately 30% [3]. A recent study of 11 European countries showed a 13% incidence level for STSS from GAS infections with a mortality rate of up to 50% [38]. SpeA is the superantigen most commonly associated with GAS infections that result in STSS in the US [27, 39] and genome sequence comparisons of GAS patient isolates reveal new variants of speA, which may be contributing to the increased severity of these clinical strains in postpartum infections [40, 41].
Immune recognition of GAS
Despite diverse evasion strategies, GAS is recognized by the innate immune response. GAS is recognized by an unidentified MyD88-dependent receptor, which is independent of TLR2, TLR4 and TLR9 activation [42] and in vitro studies demonstrate GAS activation of p38 MAPK, NF-κB, TNFα, IL-6, and type 1 IFN production [42], indicating host immune activation. GAS was long considered an extracellular pathogen, but recent research has demonstrated GAS survival within multiple host cell types, including epithelial cells, neutrophils and macrophages [43-48]. Biopsies from patients with severe GAS tissue infections contained viable GAS within macrophages, confirming their intracellular survival ability [44]. GAS survival in epithelial cells may contribute to severe GAS postpartum infections by providing a location for systemic invasion or the initiation of STSS. The next few sections detail potential roles for the diverse cellular components of innate immunity in defense against reproductive tract GAS infection.
Epithelial cells and antimicrobial peptides
Epithelial cells (EC) play a pivotal role in maintaining maternal health by forming tight junctions that provide a physical barrier against potentially pathogenic microbes, through antimicrobial molecule release, and TLRs 1-9 expression [49, 50]. Studies of chemokine and cytokine production by EC during pregnancy indicate an overall immune hypo-responsiveness with reduced levels of IL-1β, IL-8 and IL-6 in cervical fluid [51]. The altered antimicrobial peptide production of epithelial cells in the FRT may play a role in the ability of clinical strains of GAS to cause more severe postpartum infections. Numerous endogenous antimicrobials actively protect the pregnant uterus including α- and β- defensins, found in healthy pregnant females (reviewed in [52, 53]). SLPI and elafin are two other antimicrobials present in the pregnant uterus [54] that have anti-protease and anti-inflammatory activities and are thought to regulate inflammation during pregnancy and labor [52]. However, certain pathogens, including GAS can degrade these antimicrobials [55, 56]. GAS might inhibit the innate immune response through molecules like SpeB that can cleave host molecules like LL-37, an antimicrobial peptide [20, 27]. LL-37 is found throughout the FRT and plays an important role in preventing infections, but LL-37 can be inhibited by PGE2, which is up-regulated at the end of pregnancy, which may contribute to susceptibility to GAS infections in the FRT [52, 57].
Macrophages
Macrophages are an important first line of defense against invading pathogens through phagocytosis, antigen presentation, and cytokine production [58-61]. Previous mouse studies demostrate that when macrophage populations are depleted during a sublethal systemic GAS infection, mice are significantly more susceptible [62]. Macrophages can also promote chemotaxis responses to GAS infections through the activation of transcription factors involved in cytokine signaling and chemokine expression [63, 64]. However, macrophages in the FRT have altered activity compared to macrophages found in other organ/tissue sites (reviewed in [65]).
Macrophages account for approximately 10% of the total leukocytes in the female reproductive tract [66] and display phenotypic changes and up-regulated intracellular reactive oxygen species during pregnancy [67-70]. Estrogen and progesterone levels alter the migration of macrophages in the FRT and there is cyclic variation of macrophage movement due to the hormonal regulation of cytokine and chemokine expression [71, 72]. The mechanism behind these cellular alterations remains unknown and controversial in the field and further work must be done to elucidate the role of hormone alterations, prostaglandins, stage of pregnancy, the indigenous microbiota of the reproductive tract, and other factors that may alter macrophage response to GAS infections in pregnant and postpartum women.
Dendritic cells
Dendritic cells (DC) are present throughout the FRT and within the epithelial layer [73, 74] and are the most potent antigen presenting cells [75]. DC play a vital role in maintaining Th1/Th2 balance [76-78] and secrete soluble immune modulators that alter DC cytokine production [79]. The ability of estradiol and progesterone to alter DC differentiation remains controversial, but GAS can inhibit DC maturation [80] and may be a potential mechanism of GAS colonization and disease in the FRT.
Neutrophils
Neutrophils are an essential part of the innate immune response to invading bacterial pathogens. Neutrophils efficiently phagocytose bacteria, activate the production of reactive oxygen species and neutrophil degranulation, and result in bacterial killing (reviewed in [81]). GAS utilizes several virulence factors to evade ingestion and cellular recruitment by neutrophils in soft tissue infections [26, 82, 83]. Upon neutrophil phagocytosis, GAS up-regulates genes involved in tempering oxidative stress, in cell envelope components and virulence factors [81, 84, 85], suggesting that GAS can effectively respond to different host environments to promote persistence. The rapid response to bacterial infections in soft tissue makes it likely that neutrophils will play a role in susceptibility to GAS postpartum sepsis.
GAS and host genetic susceptibility
Emerging data suggest that host genetics play a significant role in the outcome of GAS infections [86, 87]. Hypervirulent strains of GAS emerged globally in the 1980s and have persisted since, but despite the increase in virulence, there is a wide spectrum of clinical manifestations associated with these strains of GAS [88, 89], suggesting that host genetics are a factor [90-93]. The severity of the response to GAS varies by patient [93-95] and the level of host cytokine production is correlated with the severity of disease [93, 96-100]. Of patients with previous postpartum infections, there were significant changes in allele frequencies compared to control patients for TLR9, hsp70 and IL-1β, suggesting that innate immune response gene polymorphisms are associated with susceptibility to severe GAS puerperal sepsis [101]. These studies have helped to clarify some of the host immune factors that influence infection risk, but further research is needed to clarify genetic predispositions of pregnant and postpartum women to GAS infection (or its complications).
Postpartum physiology and immunology
The gravid female reproductive tract (FRT) environment is unique in its immunology (reviewed in [19]). The maternal immune system must be tolerant to the indigenous bacteria in the reproductive tract, to paternal antigens in sperm and to the immunologically-distinct fetus. Despite this immunological tolerance, the FRT must be able to detect and respond to potentially pathogenic organisms. Pregnancy takes place in a physiologically and immunologically distinct organ with its own mucosal barrier (uterus and decidua) and accommodates an allogeneic fetus [102]. In addition, hormonal products in the FRT alter the immune response, and the fetus progressively challenges the maternal immune system as its size and complexity increases. Prostaglandin (PG)E2, IL-4 and IL-10 are induced by pregnancy and suppress the maternal Th1 immune response (reviewed in [103]) and the systemic down-regulation of the Th1 response results in immune alterations that promote maternal susceptibility to infection [70]. Pregnancy has often been referred to as a Th2-type immune state, but pregnancy is a modulated immune state that is not simply anti-inflammatory, but is continually changing during fetal development [104-106]. Although much is known about immunomodulatory aspects of gestation, these findings have not been studied in the context of invasive GAS infections.
Prostaglandin E2
The lipid mediator PGE2 deserves special mention because it has emerged as an important modulator of host immunity, especially during pregnancy and the postpartum period [107-112]. PGE2 is an arachidonic acid-derived mediator that modulates cell behavior via the ligation of four distinct G protein coupled receptors called E prostanoid (EP) receptors, which are numbered EP1-4 (reviewed in [113]). Throughout gestation, PGE2 dampens maternal immune responses against fetal tissues [107-110, 114] and regulates cervical softening and uterine contractions during labor, where it is found at increased levels [111, 112, 115]. It is also a critical negative regulator of the host immune response, with the ability to down-regulate lymphocyte and neutrophil activity [116], to inhibit production of Th1 cytokines (IL-12 and IFNγ) and to enhance production of Th2 cytokines (IL-5 and IL-10) [117-119]. Elevation in PGE2 levels has previously been shown to play a role in host susceptibility to infections in many patient populations including pregnant women [114, 120-129].
The capacity for PGE2 to regulate host-microbial interactions is increasingly evident in the context of streptococcal infections [130-136]. In 1982, Short et al. demonstrated increased survival in animals when PGE2 synthesis was inhibited during Group B Streptococcus sepsis [137]. Prostaglandin endoperoxide synthase 2 (COX-2) is the enzyme that converts arachidonic acid into prostaglandin endoperoxide H2 (PGH2) before PGH2 is converted into other prostaglandins. In 2010 Goldman et al. demonstrated that COX-2 is up-regulated in human and mouse tissues infected by GAS [130, 138]. Using a mouse model of GAS bacteremia and in vitro studies of bone marrow-derived macrophages, they established that PGE2 signaling via EP2 receptors and cAMP elevation suppressed host defenses against GAS [130]. An unbiased systems genetics approach later identified two PGE2 synthase enzymes (mPGES-1 and -2) as key participants mediating susceptibility to GAS [86]. However, little is known about PGE2 and GAS in the FRT [139-141].
FRT mucus, pH, and the indigenous microbiota
Vaginal colonization by GAS appears to be an important preceding event in some cases of puerperal sepsis (following vaginal delivery), yet host-microbial interactions that determine the capacity for GAS to colonize and invade the mucosal surfaces of the FRT need further study. In addition, the innate immune mechanisms that prevent GAS from ascending through the cervical canal into the postpartum uterus remain incompletely understood. Mucus within the FRT protects epithelial cells from bacterial infections through several mechanisms. Mucus can physically trap potential pathogens and inhibit pathogen survival due to the low pH, immunoglobulins, and antimicrobial peptides [142, 143]. The changes in mucus in the postpartum FRT and its effect on GAS colonization and dissemination remain unknown.
The indigenous bacteria in the reproductive tract also provide pathogen resistance through several means, including competitive exclusion of pathogenic microbes and contributing to the acidic vaginal environment through lactic acid production. Lactobacillus spp. are the most common bacteria present across all ethnic groups and produce lactic acid in the FRT [142, 144]. Few studies have been done to investigate individual variation between women over time, but these preliminary studies suggest that the bacterial diversity is dynamic even amongst individuals and the vagina is implicated as a significant source of infectious organisms resulting in preterm labor [142, 145-149]. Membranes collected from healthy women following at term cesarean sections demonstrate bacterial DNA in up to 70% of samples indicating a dynamic host control of individual bacterial diversity in healthy pregnancies [150]. The role of the indigenous microbiota in the reproductive tract and its interactions with the host immune response during GAS postpartum infections remain unknown, and whether probiotic approaches would be successful at preventing puerperal GAS infections depends upon more research into how the microbiota creates a colonization and infection resistance against this pathogen.
Conclusions
Pregnancy is a highly immunomodulated state that permits implantation and development of the immunologically distinct fetus. This may result in an immunologically vulnerable FRT that is more easily infected after delivery. The immune changes that progress during pregnancy are complex and remain largely uncharacterized, but recent research suggests GAS infections are re-emerging and postpartum patients are particularly prone to severe GAS infections that result in death [1-4]. The mechanisms behind GAS bacterial virulence, postpartum susceptibility and the immune response to FRT infections remain poorly understood and future work must be done to address the increase in maternal mortality from postpartum GAS infections.
ACKNOWLEDGMENTS
This work was supported by a National Institutes of Health grant HD057176 (DMA) and a Burroughs Welcome Fund Investigators in the Pathogenesis in Infectious Diseases award (DMA).
REFERENCES
- 1.Maharaj D. Puerperal pyrexia: a review. Part I. Obstet Gynecol Surv. 2007;62(6):393–9. doi: 10.1097/01.ogx.0000265998.40912.5e. [DOI] [PubMed] [Google Scholar]
- 2.Castagnola DE, et al. Necrotizing cervical and uterine infection in the postpartum period caused by group A streptococcus. Obstet Gynecol. 2008;111(2 Pt 2):533–5. doi: 10.1097/01.AOG.0000284453.41550.66. [DOI] [PubMed] [Google Scholar]
- 3.Stevens DL, et al. Severe group A streptococcal infections associated with a toxic shock-like syndrome and scarlet fever toxin A. N Engl J Med. 1989;321(1):1–7. doi: 10.1056/NEJM198907063210101. [DOI] [PubMed] [Google Scholar]
- 4.van Dillen J, et al. Maternal sepsis: epidemiology, etiology and outcome. Curr Opin Infect Dis. 23(3):249–54. doi: 10.1097/QCO.0b013e328339257c. [DOI] [PubMed] [Google Scholar]
- 5.Cantwell R, et al. Saving Mothers’ Lives: Reviewing maternal deaths to make motherhood safer: 2006-2008. The Eighth Report of the Confidential Enquiries into Maternal Deaths in the United Kingdom. BJOG. 2011;118(Suppl 1):1–203. doi: 10.1111/j.1471-0528.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 6.WHO Maternal mortality in 2005: estimates developed by WHO, UNICEF, UNFPA, and the World Bank. 2007 ( http://www.who.int/whosis/mme_2005.pdf)
- 7.Seale AC, et al. Maternal and early onset neonatal bacterial sepsis: burden and strategies for prevention in sub-Saharan Africa. Lancet Infect Dis. 2009;9(7):428–38. doi: 10.1016/S1473-3099(09)70172-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.UNICEF The state of the world's children 2009: maternal and newborn health. 2008 ( http://www.unicef.org/publications/files/The_State_of_the_Worlds_Children_2009.pdf)
- 9.Harper A, editor. Chapter 7: Sepsis. Centre for Maternal and Child Enquiries (CMACE), BJOG. 2011 [Google Scholar]
- 10.Aronoff DM, Mulla ZD. Postpartum invasive group A streptococcal disease in the modern era. Infect Dis Obstet Gynecol. 2008;2008:796892. doi: 10.1155/2008/796892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saving Mothers’ Lives 2006-2008: Briefing on genital tract sepsis. Centre for Maternal and Child Enquiries 2010. Available from: ( http://www.cmace.org.uk/getdoc/c0942829-44bf-4de3-988c-6b7bda7be065/Microsoft-Word---GT_Sepsis_0508_final_v2-style.aspx)
- 12.Momoh MA, Ezugworie OJ, Ezeigwe HO. Causes and Management of Puerperal Sepsis: The Health Personnel View Point. Advances in Biological Research. 2010;4(3):154–158. [Google Scholar]
- 13.Nathan L, Leveno KJ. Group a streptococcal puerperal sepsis: historical review and 1990s resurgence. Infect Dis Obstet Gynecol. 1994;1(5):252–5. doi: 10.1155/S1064744994000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cone LA, et al. Clinical and bacteriologic observations of a toxic shock-like syndrome due to Streptococcus pyogenes. N Engl J Med. 1987;317(3):146–9. doi: 10.1056/NEJM198707163170305. [DOI] [PubMed] [Google Scholar]
- 15.Monif GRG, Baker DA, editors. Group A Beta-Hemolytic Streptococcus (Streptococcus Pyogenes). 6 ed. Informa Healthcare; London: 2008. Infectious Diseases in Obstetrics and Gynecology. [Google Scholar]
- 16.Moses AE, et al. Increased incidence and severity of Streptococcus pyogenes bacteremia in young children. Pediatr Infect Dis J. 1995;14(9):767–70. doi: 10.1097/00006454-199509000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Mead PB, Winn WC. Vaginal-rectal colonization with group A streptococci in late pregnancy. Infect Dis Obstet Gynecol. 2000;8(5-6):217–9. doi: 10.1155/S1064744900000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.HPA Interim UK guidelines for management of close community contacts of invasive group A streptococcal disease. Commun Dis Public Health. 2004;7:354–61. [PubMed] [Google Scholar]
- 19.Mor G, Cardenas I. The immune system in pregnancy: a unique complexity. Am J Reprod Immunol. 2010;63(6):425–33. doi: 10.1111/j.1600-0897.2010.00836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamagni TL, et al. Increase in invasive group A streptococcal infections in England, Wales and Northern Ireland, 2008-9. Euro Surveill. 2009;14(5) doi: 10.2807/ese.14.05.19110-en. [DOI] [PubMed] [Google Scholar]
- 21.Chuang I, et al. Population-based surveillance for postpartum invasive group a streptococcus infections, 1995-2000. Clin Infect Dis. 2002;35(6):665–70. doi: 10.1086/342062. [DOI] [PubMed] [Google Scholar]
- 22.Smaill FM, Gyte GM. Antibiotic prophylaxis versus no prophylaxis for preventing infection after cesarean section. Cochrane Database Syst Rev. 2010;(1):CD007482. doi: 10.1002/14651858.CD007482.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crum NF, et al. Group A streptococcal toxic shock syndrome developing in the third trimester of pregnancy. Infect Dis Obstet Gynecol. 2002;10(4):209–16. doi: 10.1155/S1064744902000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deutscher M, et al. Incidence and Severity of Invasive Streptococcus pneumoniae, Group A Streptococcus, and Group B Streptococcus Infections Among Pregnant and Postpartum Women. Clin Infect Dis. 2011;53(2):114–123. doi: 10.1093/cid/cir325. [DOI] [PubMed] [Google Scholar]
- 25.O'Loughlin RE, et al. The epidemiology of invasive group A streptococcal infection and potential vaccine implications: United States, 2000-2004. Clin Infect Dis. 2007;45(7):853–62. doi: 10.1086/521264. [DOI] [PubMed] [Google Scholar]
- 26.Bisno AL, Brito MO, Collins CM. Molecular basis of group A streptococcal virulence. Lancet Infect Dis. 2003;3(4):191–200. doi: 10.1016/s1473-3099(03)00576-0. [DOI] [PubMed] [Google Scholar]
- 27.Bohach GA, et al. Staphylococcal and streptococcal pyrogenic toxins involved in toxic shock syndrome and related illnesses. Crit Rev Microbiol. 1990;17(4):251–72. doi: 10.3109/10408419009105728. [DOI] [PubMed] [Google Scholar]
- 28.Lynskey NN, Lawrenson RA, Sriskandan S. New understandings in Streptococcus pyogenes. Curr Opin Infect Dis. 2011;24(3):196–202. doi: 10.1097/QCO.0b013e3283458f7e. [DOI] [PubMed] [Google Scholar]
- 29.Dmitriev AV, Chaussee MS. The Streptococcus pyogenes proteome: maps, virulence factors and vaccine candidates. Future Microbiol. 2010;5(10):1539–51. doi: 10.2217/fmb.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwinn LA, Nizet V. How group A Streptococcus circumvents host phagocyte defenses. Future Microbiol. 2007;2(1):75–84. doi: 10.2217/17460913.2.1.75. [DOI] [PubMed] [Google Scholar]
- 31.Fischetti VA. Streptococcal M protein. Sci Am. 1991;264(6):58–65. doi: 10.1038/scientificamerican0691-58. [DOI] [PubMed] [Google Scholar]
- 32.Wessels MR, et al. Hyaluronic acid capsule is a virulence factor for mucoid group A streptococci. Proc Natl Acad Sci U S A. 1991;88(19):8317–21. doi: 10.1073/pnas.88.19.8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanski E, Caparon M. Protein F, a fibronectin-binding protein, is an adhesin of the group A streptococcus Streptococcus pyogenes. Proc Natl Acad Sci U S A. 1992;89(13):6172–6. doi: 10.1073/pnas.89.13.6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanski E, Horwitz PA, Caparon MG. Expression of protein F, the fibronectin-binding protein of Streptococcus pyogenes JRS4, in heterologous streptococcal and enterococcal strains promotes their adherence to respiratory epithelial cells. Infect Immun. 1992;60(12):5119–25. doi: 10.1128/iai.60.12.5119-5125.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ofek I, et al. Cell membrane-binding properties of group A streptococcal lipoteichoic acid. J Exp Med. 1975;141(5):990–1003. doi: 10.1084/jem.141.5.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tilanus AM, et al. Severe group A streptococcal toxic shock syndrome presenting as primary peritonitis: a case report and brief review of the literature. Int J Infect Dis. 2010;14(Suppl 3):e208–12. doi: 10.1016/j.ijid.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 37.Lappin E, Ferguson AJ. Gram-positive toxic shock syndromes. Lancet Infect Dis. 2009;9(5):281–90. doi: 10.1016/S1473-3099(09)70066-0. [DOI] [PubMed] [Google Scholar]
- 38.Lamagni TL, et al. Epidemiology of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol. 2008;46(7):2359–67. doi: 10.1128/JCM.00422-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hauser AR, et al. Molecular analysis of pyrogenic exotoxins from Streptococcus pyogenes isolates associated with toxic shock-like syndrome. J Clin Microbiol. 1991;29(8):1562–7. doi: 10.1128/jcm.29.8.1562-1567.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musser JM, et al. Geographic and temporal distribution and molecular characterization of two highly pathogenic clones of Streptococcus pyogenes expressing allelic variants of pyrogenic exotoxin A (Scarlet fever toxin). J Infect Dis. 1993;167(2):337–46. doi: 10.1093/infdis/167.2.337. [DOI] [PubMed] [Google Scholar]
- 41.Nelson K, et al. Characterization and clonal distribution of four alleles of the speA gene encoding pyrogenic exotoxin A (scarlet fever toxin) in Streptococcus pyogenes. J Exp Med. 1991;174(5):1271–4. doi: 10.1084/jem.174.5.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gratz N, et al. Group A streptococcus activates type I interferon production and MyD88-dependent signaling without involvement of TLR2, TLR4, and TLR9. J Biol Chem. 2008;283(29):19879–87. doi: 10.1074/jbc.M802848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Staali L, et al. Streptococcus pyogenes expressing M and M-like surface proteins are phagocytosed but survive inside human neutrophils. Cell Microbiol. 2003;5(4):253–65. doi: 10.1046/j.1462-5822.2003.00272.x. [DOI] [PubMed] [Google Scholar]
- 44.Thulin P, et al. Viable group A streptococci in macrophages during acute soft tissue infection. PLoS Med. 2006;3(3):e53. doi: 10.1371/journal.pmed.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306(5698):1037–40. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 46.Osterlund A, Engstrand L. Intracellular penetration and survival of Streptococcus pyogenes in respiratory epithelial cells in vitro. Acta Otolaryngol. 1995;115(5):685–8. doi: 10.3109/00016489509139387. [DOI] [PubMed] [Google Scholar]
- 47.LaPenta D, et al. Group A streptococci efficiently invade human respiratory epithelial cells. Proc Natl Acad Sci U S A. 1994;91(25):12115–9. doi: 10.1073/pnas.91.25.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greco R, et al. Invasion of cultured human cells by Streptococcus pyogenes. Res Microbiol. 1995;146(7):551–60. doi: 10.1016/0923-2508(96)80561-4. [DOI] [PubMed] [Google Scholar]
- 49.Schaefer TM, et al. Innate immunity in the human female reproductive tract: antiviral response of uterine epithelial cells to the TLR3 agonist poly(I:C). J Immunol. 2005;174(2):992–1002. doi: 10.4049/jimmunol.174.2.992. [DOI] [PubMed] [Google Scholar]
- 50.Schaefer TM, et al. Toll-like receptor (TLR) expression and TLR-mediated cytokine/chemokine production by human uterine epithelial cells. Immunology. 2004;112(3):428–36. doi: 10.1111/j.1365-2567.2004.01898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simhan HN, et al. Decreased cervical proinflammatory cytokines permit subsequent upper genital tract infection during pregnancy. Am J Obstet Gynecol. 2003;189(2):560–7. doi: 10.1067/s0002-9378(03)00518-0. [DOI] [PubMed] [Google Scholar]
- 52.Wira CR, et al. Innate immunity in the human female reproductive tract: endocrine regulation of endogenous antimicrobial protection against HIV and other sexually transmitted infections. Am J Reprod Immunol. 2011;65(3):196–211. doi: 10.1111/j.1600-0897.2011.00970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King AE, et al. Expression of natural antimicrobials by human placenta and fetal membranes. Placenta. 2007;28(2-3):161–9. doi: 10.1016/j.placenta.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 54.King AE, et al. Innate immune defences in the human uterus during pregnancy. Placenta. 2007;28(11-12):1099–106. doi: 10.1016/j.placenta.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 55.Moreau T, et al. Multifaceted roles of human elafin and secretory leukocyte proteinase inhibitor (SLPI), two serine protease inhibitors of the chelonianin family. Biochimie. 2008;90(2):284–95. doi: 10.1016/j.biochi.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Johansson L, et al. Cathelicidin LL-37 in severe Streptococcus pyogenes soft tissue infections in humans. Infect Immun. 2008;76(8):3399–404. doi: 10.1128/IAI.01392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chamorro CI, et al. The human antimicrobial peptide LL-37 suppresses apoptosis in keratinocytes. J Invest Dermatol. 2009;129(4):937–44. doi: 10.1038/jid.2008.321. [DOI] [PubMed] [Google Scholar]
- 58.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 59.Keshav S, Chung LP, Gordon S. Macrophage products in inflammation. Diagn Microbiol Infect Dis. 1990;13(5):439–47. doi: 10.1016/0732-8893(90)90016-o. [DOI] [PubMed] [Google Scholar]
- 60.Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79(2):319–26. doi: 10.1172/JCI112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sansonetti P. Phagocytosis of bacterial pathogens: implications in the host response. Semin Immunol. 2001;13(6):381–90. doi: 10.1006/smim.2001.0335. [DOI] [PubMed] [Google Scholar]
- 62.Goldmann O, et al. Role of macrophages in host resistance to group A streptococci. Infect Immun. 2004;72(5):2956–63. doi: 10.1128/IAI.72.5.2956-2963.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miettinen M, et al. Lactobacilli and Streptococci activate NF-kappa B and STAT signaling pathways in human macrophages. J Immunol. 2000;164(7):3733–40. doi: 10.4049/jimmunol.164.7.3733. [DOI] [PubMed] [Google Scholar]
- 64.Veckman V, et al. Lactobacilli and streptococci induce inflammatory chemokine production in human macrophages that stimulates Th1 cell chemotaxis. J Leukoc Biol. 2003;74(3):395–402. doi: 10.1189/jlb.0402212. [DOI] [PubMed] [Google Scholar]
- 65.Wira CR, et al. Innate and adaptive immunity in female genital tract: cellular responses and interactions. Immunol Rev. 2005;206:306–35. doi: 10.1111/j.0105-2896.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- 66.Givan AL, et al. Flow cytometric analysis of leukocytes in the human female reproductive tract: comparison of fallopian tube, uterus, cervix, and vagina. Am J Reprod Immunol. 1997;38(5):350–9. doi: 10.1111/j.1600-0897.1997.tb00311.x. [DOI] [PubMed] [Google Scholar]
- 67.Sacks GP, et al. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol. 1998;179(1):80–6. doi: 10.1016/s0002-9378(98)70254-6. [DOI] [PubMed] [Google Scholar]
- 68.Koumandakis E, et al. Enhanced phagocytosis of mononuclear phagocytes in pregnancy. Br J Obstet Gynaecol. 1986;93(11):1150–4. doi: 10.1111/j.1471-0528.1986.tb08636.x. [DOI] [PubMed] [Google Scholar]
- 69.Shibuya T, et al. Study on nonspecific immunity in pregnant women: increased chemiluminescence response of peripheral blood phagocytes. Am J Reprod Immunol Microbiol. 1987;15(1):19–23. doi: 10.1111/j.1600-0897.1987.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 70.Naccasha N, et al. Phenotypic and metabolic characteristics of monocytes and granulocytes in normal pregnancy and maternal infection. Am J Obstet Gynecol. 2001;185(5):1118–23. doi: 10.1067/mob.2001.117682. [DOI] [PubMed] [Google Scholar]
- 71.DeLoia JA, et al. Effects of exogenous estrogen on uterine leukocyte recruitment. Fertil Steril. 2002;77(3):548–54. doi: 10.1016/s0015-0282(01)03062-x. [DOI] [PubMed] [Google Scholar]
- 72.Kamat BR, Isaacson PG. The immunocytochemical distribution of leukocytic subpopulations in human endometrium. Am J Pathol. 1987;127(1):66–73. [PMC free article] [PubMed] [Google Scholar]
- 73.Iijima N, Thompson JM, Iwasaki A. Dendritic cells and macrophages in the genitourinary tract. Mucosal Immunol. 2008;1(6):451–9. doi: 10.1038/mi.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iijima N, et al. Dendritic cells and B cells maximize mucosal Th1 memory response to herpes simplex virus. J Exp Med. 2008;205(13):3041–52. doi: 10.1084/jem.20082039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–96. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 76.Nishioka Y, et al. Human monocyte-derived and CD83(+) blood dendritic cells enhance NK cell-mediated cytotoxicity. Eur J Immunol. 2001;31(9):2633–41. doi: 10.1002/1521-4141(200109)31:9<2633::aid-immu2633>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 77.Osada T, et al. Peripheral blood dendritic cells, but not monocyte-derived dendritic cells, can augment human NK cell function. Cell Immunol. 2001;213(1):14–23. doi: 10.1006/cimm.2001.1858. [DOI] [PubMed] [Google Scholar]
- 78.Bradley LM, et al. Availability of antigen-presenting cells can determine the extent of CD4 effector expansion and priming for secretion of Th2 cytokines in vivo. Eur J Immunol. 2002;32(8):2338–46. doi: 10.1002/1521-4141(200208)32:8<2338::AID-IMMU2338>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 79.Ochiel DO, et al. Human uterine epithelial cell secretions regulate dendritic cell differentiation and responses to TLR ligands. J Leukoc Biol. 2010;88(3):435–44. doi: 10.1189/jlb.1009700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cortes G, Wessels MR. Inhibition of dendritic cell maturation by group A Streptococcus. J Infect Dis. 2009;200(7):1152–61. doi: 10.1086/605696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Voyich JM, Musser JM, DeLeo FR. Streptococcus pyogenes and human neutrophils: a paradigm for evasion of innate host defense by bacterial pathogens. Microbes Infect. 2004;6(12):1117–23. doi: 10.1016/j.micinf.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 82.Ji Y, et al. C5a peptidase alters clearance and trafficking of group A streptococci by infected mice. Infect Immun. 1996;64(2):503–10. doi: 10.1128/iai.64.2.503-510.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cleary PP, et al. Streptococcal C5a peptidase is a highly specific endopeptidase. Infect Immun. 1992;60(12):5219–23. doi: 10.1128/iai.60.12.5219-5223.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Voyich JM, et al. Genome-wide protective response used by group A Streptococcus to evade destruction by human polymorphonuclear leukocytes. Proc Natl Acad Sci U S A. 2003;100(4):1996–2001. doi: 10.1073/pnas.0337370100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Medina E, Rohde M, Chhatwal GS. Intracellular survival of Streptococcus pyogenes in polymorphonuclear cells results in increased bacterial virulence. Infect Immun. 2003;71(9):5376–80. doi: 10.1128/IAI.71.9.5376-5380.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abdeltawab NF, et al. An unbiased systems genetics approach to mapping genetic loci modulating susceptibility to severe streptococcal sepsis. PLoS Pathog. 2008;4(4):e1000042. doi: 10.1371/journal.ppat.1000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nooh MM, et al. Individual genetic variations directly effect polarization of cytokine responses to superantigens associated with streptococcal sepsis: implications for customized patient care. J Immunol. 2011;186(5):3156–63. doi: 10.4049/jimmunol.1002057. [DOI] [PubMed] [Google Scholar]
- 88.Chatellier S, et al. Genetic relatedness and superantigen expression in group A streptococcus serotype M1 isolates from patients with severe and nonsevere invasive diseases. Infect Immun. 2000;68(6):3523–34. doi: 10.1128/iai.68.6.3523-3534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson DR, et al. A comparison of group A streptococci from invasive and uncomplicated infections: are virulent clones responsible for serious streptococcal infections? J Infect Dis. 2002;185(11):1586–95. doi: 10.1086/340576. [DOI] [PubMed] [Google Scholar]
- 90.Aziz RK, Kotb M. Rise and persistence of global M1T1 clone of Streptococcus pyogenes. Emerg Infect Dis. 2008;14(10):1511–7. doi: 10.3201/eid1410.071660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kansal RG, et al. Dissection of the molecular basis for hypervirulence of an in vivo-selected phenotype of the widely disseminated M1T1 strain of group A Streptococcus bacteria. J Infect Dis. 2010;201(6):855–65. doi: 10.1086/651019. [DOI] [PubMed] [Google Scholar]
- 92.Basma H, et al. Risk factors in the pathogenesis of invasive group A streptococcal infections: role of protective humoral immunity. Infect Immun. 1999;67(4):1871–7. doi: 10.1128/iai.67.4.1871-1877.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kotb M, et al. An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat Med. 2002;8(12):1398–404. doi: 10.1038/nm1202-800. [DOI] [PubMed] [Google Scholar]
- 94.Kotb M. Bacterial pyrogenic exotoxins as superantigens. Clin Microbiol Rev. 1995;8(3):411–26. doi: 10.1128/cmr.8.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Norrby-Teglund A, Lustig R, Kotb M. Differential induction of Th1 versus Th2 cytokines by group A streptococcal toxic shock syndrome isolates. Infect Immun. 1997;65(12):5209–15. doi: 10.1128/iai.65.12.5209-5215.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Norrby-Teglund A, et al. Host variation in cytokine responses to superantigens determine the severity of invasive group A streptococcal infection. Eur J Immunol. 2000;30(11):3247–55. doi: 10.1002/1521-4141(200011)30:11<3247::AID-IMMU3247>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 97.Norrby-Teglund A, et al. Evidence for superantigen involvement in severe group a streptococcal tissue infections. J Infect Dis. 2001;184(7):853–60. doi: 10.1086/323443. [DOI] [PubMed] [Google Scholar]
- 98.Norrby SR, Norrby-Teglund A. Infections due to group A streptococcus: new concepts and potential treatment strategies. Ann Acad Med Singapore. 1997;26(5):691–3. [PubMed] [Google Scholar]
- 99.Norrby-Teglund A, Ihendyane N, Darenberg J. Intravenous immunoglobulin adjunctive therapy in sepsis, with special emphasis on severe invasive group A streptococcal infections. Scand J Infect Dis. 2003;35(9):683–9. doi: 10.1080/00365540310015944. [DOI] [PubMed] [Google Scholar]
- 100.Medina E, Lengeling A. Genetic regulation of host responses to group A streptococcus in mice. Brief Funct Genomic Proteomic. 2005;4(3):248–57. doi: 10.1093/bfgp/4.3.248. [DOI] [PubMed] [Google Scholar]
- 101.Davis SM, et al. The association of innate immune response gene polymorphisms and puerperal group A streptococcal sepsis. Am J Obstet Gynecol. 2010;202(3):308, e1–8. doi: 10.1016/j.ajog.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 102.Gaunt G, Ramin K. Immunological tolerance of the human fetus. Am J Perinatol. 2001;18(6):299–312. doi: 10.1055/s-2001-17861. [DOI] [PubMed] [Google Scholar]
- 103.Sacks G, Sargent I, Redman C. An innate view of human pregnancy. Immunol Today. 1999;20(3):114–8. doi: 10.1016/s0167-5699(98)01393-0. [DOI] [PubMed] [Google Scholar]
- 104.Jin LP, Fan DX, Li DJ. Regulation of costimulatory signal in maternal-fetal immune tolerance. Am J Reprod Immunol. 2011;66(2):76–83. doi: 10.1111/j.1600-0897.2010.00982.x. [DOI] [PubMed] [Google Scholar]
- 105.Veenstra van Nieuwenhoven AL, Heineman MJ, Faas MM. The immunology of successful pregnancy. Hum Reprod Update. 2003;9(4):347–57. doi: 10.1093/humupd/dmg026. [DOI] [PubMed] [Google Scholar]
- 106.Mor G, et al. Inflammation and pregnancy: the role of the immune system at the implantation site. Ann N Y Acad Sci. 2011;1221:80–7. doi: 10.1111/j.1749-6632.2010.05938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kvirkvelia N, et al. Placentally derived prostaglandin E2 acts via the EP4 receptor to inhibit IL-2-dependent proliferation of CTLL-2 T cells. Clin Exp Immunol. 2002;127(2):263–9. doi: 10.1046/j.1365-2249.2002.01718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Parhar RS, Lala PK. Prostaglandin E2-mediated inactivation of various killer lineage cells by tumor-bearing host macrophages. J Leukoc Biol. 1988;44(6):474–84. doi: 10.1002/jlb.44.6.474. [DOI] [PubMed] [Google Scholar]
- 109.Scodras JM, et al. Prostaglandin-mediated inactivation of natural killer cells in the murine decidua. Cell Immunol. 1990;127(2):352–67. doi: 10.1016/0008-8749(90)90138-h. [DOI] [PubMed] [Google Scholar]
- 110.Tawfik OW, Hunt JS, Wood GW. Implication of prostaglandin E2 in soluble factor-mediated immune suppression by murine decidual cells. Am J Reprod Immunol Microbiol. 1986;12(4):111–7. doi: 10.1111/j.1600-0897.1986.tb00075.x. [DOI] [PubMed] [Google Scholar]
- 111.Hertelendy F, Woods R, Jaffe BM. Prostaglandin E levels in peripheral blood during labor. Prostaglandins. 1973;3(2):223–7. doi: 10.1016/0090-6980(73)90090-7. [DOI] [PubMed] [Google Scholar]
- 112.Lee SE, et al. Amniotic fluid prostaglandin concentrations increase before the onset of spontaneous labor at term. J Matern Fetal Neonatal Med. 2008;21(2):89–94. doi: 10.1080/14767050701830514. [DOI] [PubMed] [Google Scholar]
- 113.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu W, et al. COX-2 inhibition affects growth rate of Chlamydia muridarum within epithelial cells. Microbes Infect. 2006;8(2):478–86. doi: 10.1016/j.micinf.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 115.Olson DM, Ammann C. Role of the prostaglandins in labour and prostaglandin receptor inhibitors in the prevention of preterm labour. Front Biosci. 2007;12:1329–43. doi: 10.2741/2151. [DOI] [PubMed] [Google Scholar]
- 116.Hoedemaker M, Lund LA, Wagner WC. Influence of arachidonic acid metabolites and steroids on function of bovine polymorphonuclear neutrophils. Am J Vet Res. 1992;53(9):1534–9. [PubMed] [Google Scholar]
- 117.Fabricius D, et al. Prostaglandin E2 inhibits IFN-alpha secretion and Th1 costimulation by human plasmacytoid dendritic cells via E-prostanoid 2 and E-prostanoid 4 receptor engagement. J Immunol. 2010;184(2):677–84. doi: 10.4049/jimmunol.0902028. [DOI] [PubMed] [Google Scholar]
- 118.Kalinski P, et al. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol. 1997;159(1):28–35. [PubMed] [Google Scholar]
- 119.Harizi H, et al. Cyclooxygenase-2-issued prostaglandin e(2) enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J Immunol. 2002;168(5):2255–63. doi: 10.4049/jimmunol.168.5.2255. [DOI] [PubMed] [Google Scholar]
- 120.Ballinger MN, et al. Critical role of prostaglandin E2 overproduction in impaired pulmonary host response following bone marrow transplantation. J Immunol. 2006;177(8):5499–508. doi: 10.4049/jimmunol.177.8.5499. [DOI] [PubMed] [Google Scholar]
- 121.Chaimoff C, Malachi T, Halbrecht I. Prostaglandin E2 and cyclic nucleotides in plasma and urine of colonic cancer patients. J Cancer Res Clin Oncol. 1985;110(2):153–6. doi: 10.1007/BF00402730. [DOI] [PubMed] [Google Scholar]
- 122.Fraifeld V, et al. Increased prostaglandin E2 production by concanavalin A-stimulated splenocytes of old mice. Gerontology. 1995;41(3):129–33. doi: 10.1159/000213673. [DOI] [PubMed] [Google Scholar]
- 123.Ramis I, et al. Cyclooxygenase and lipoxygenase arachidonic acid metabolism by monocytes from human immune deficiency virus-infected drug users. J Chromatogr. 1991;557(1-2):507–13. doi: 10.1016/s0021-9673(01)87159-4. [DOI] [PubMed] [Google Scholar]
- 124.Anstead GM, Zhang Q, Melby PC. Malnutrition promotes prostaglandin over leukotriene production and dysregulates eicosanoid-cytokine crosstalk in activated resident macrophages. Prostaglandins Leukot Essent Fatty Acids. 2009;81(1):41–51. doi: 10.1016/j.plefa.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 125.el-Sharabasy MM, el-Naggar MM. Prostaglandin E2 in renal transplant recipients. Int Urol Nephrol. 1992;24(4):447–51. doi: 10.1007/BF02550640. [DOI] [PubMed] [Google Scholar]
- 126.Cayeux SJ, et al. Elevated plasma prostaglandin E2 levels found in 14 patients undergoing autologous bone marrow or stem cell transplantation. Bone Marrow Transplant. 1993;12(6):603–8. [PubMed] [Google Scholar]
- 127.Bernheim HA. Is prostaglandin E2 involved in the pathogenesis of fever? Effects of interleukin-1 on the release of prostaglandins. Yale J Biol Med. 1986;59(2):151–8. [PMC free article] [PubMed] [Google Scholar]
- 128.Bernheim J, et al. Renal prostaglandins E2 and F2 alpha throughout normal human pregnancy. Eur J Clin Invest. 1986;16(2):113–6. doi: 10.1111/j.1365-2362.1986.tb01317.x. [DOI] [PubMed] [Google Scholar]
- 129.Aronoff DM, et al. Misoprostol impairs female reproductive tract innate immunity against Clostridium sordellii. J Immunol. 2008;180(12):8222–30. doi: 10.4049/jimmunol.180.12.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Goldmann O, et al. Inducible cyclooxygenase released prostaglandin E2 modulates the severity of infection caused by Streptococcus pyogenes. J Immunol. 2010;185(4):2372–81. doi: 10.4049/jimmunol.1000838. [DOI] [PubMed] [Google Scholar]
- 131.Maloney CG, et al. Induction of cyclooxygenase-2 by human monocytes exposed to group B streptococci. J Leukoc Biol. 2000;67(5):615–21. doi: 10.1002/jlb.67.5.615. [DOI] [PubMed] [Google Scholar]
- 132.Medeiros AI, et al. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med. 2009;206(1):61–8. doi: 10.1084/jem.20082058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.N'Guessan PD, et al. Streptococcus pneumoniae induced p38 MAPK- and NF-kappaB-dependent COX-2 expression in human lung epithelium. Am J Physiol Lung Cell Mol Physiol. 2006;290(6):L1131–8. doi: 10.1152/ajplung.00383.2005. [DOI] [PubMed] [Google Scholar]
- 134.Stables MJ, et al. Priming innate immune responses to infection by cyclooxygenase inhibition kills antibiotic-susceptible and -resistant bacteria. Blood. 2010;116(16):2950–9. doi: 10.1182/blood-2010-05-284844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bennett PR, et al. Preterm labor: stimulation of arachidonic acid metabolism in human amnion cells by bacterial products. Am J Obstet Gynecol. 1987;156(3):649–55. doi: 10.1016/0002-9378(87)90070-6. [DOI] [PubMed] [Google Scholar]
- 136.Rayon JI, et al. The fatty acid composition of maternal diet affects lung prostaglandin E2 levels and survival from group B streptococcal sepsis in neonatal rat pups. J Nutr. 1997;127(10):1989–92. doi: 10.1093/jn/127.10.1889. [DOI] [PubMed] [Google Scholar]
- 137.Short BL, Miller MK, Fletcher JR. Improved survival in the suckling rat model of group B streptococcal sepsis after treatment with nonsteroidal anti-inflammatory drugs. Pediatrics. 1982;70(3):343–7. [PubMed] [Google Scholar]
- 138.Goldmann O, et al. Transcriptome analysis of murine macrophages in response to infection with Streptococcus pyogenes reveals an unusual activation program. Infect Immun. 2007;75(8):4148–57. doi: 10.1128/IAI.00181-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Aronoff DM, Bloch KC. Assessing the relationship between the use of nonsteroidal antiinflammatory drugs and necrotizing fasciitis caused by group A streptococcus. Medicine (Baltimore) 2003;82(4):225–35. doi: 10.1097/01.md.0000085060.63483.bb. [DOI] [PubMed] [Google Scholar]
- 140.Barnham M, Anderson AW. Non-steroidal anti-inflammatory drugs (NSAIDs). A predisposing factor for streptococcal bacteraemia? Adv Exp Med Biol. 1997;418:145–7. doi: 10.1007/978-1-4899-1825-3_35. [DOI] [PubMed] [Google Scholar]
- 141.Hamilton SM, et al. Muscle injury, vimentin expression, and nonsteroidal anti-inflammatory drugs predispose to cryptic group A streptococcal necrotizing infection. J Infect Dis. 2008;198(11):1692–8. doi: 10.1086/593016. [DOI] [PubMed] [Google Scholar]
- 142.Ravel J, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4680–7. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ming L, et al. Purification of antimicrobial factors from human cervical mucus. Hum Reprod. 2007;22(7):1810–5. doi: 10.1093/humrep/dem128. [DOI] [PubMed] [Google Scholar]
- 144.Witkin SS, Linhares IM, Giraldo P. Bacterial flora of the female genital tract: function and immune regulation. Best Pract Res Clin Obstet Gynaecol. 2007;21(3):347–54. doi: 10.1016/j.bpobgyn.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 145.Zhou X, et al. Recent advances in understanding the microbiology of the female reproductive tract and the causes of premature birth. Infect Dis Obstet Gynecol. 2010;2010:737425. doi: 10.1155/2010/737425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brotman RM, et al. The effect of vaginal douching cessation on bacterial vaginosis: a pilot study. Am J Obstet Gynecol. 2008;198(6):628, e1–7. doi: 10.1016/j.ajog.2007.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.DiGiulio DB, et al. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PLoS One. 2008;3(8):e3056. doi: 10.1371/journal.pone.0003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N Engl J Med. 2000;342(20):1500–7. doi: 10.1056/NEJM200005183422007. [DOI] [PubMed] [Google Scholar]
- 149.Han YW, et al. Uncultivated bacteria as etiologic agents of intra-amniotic inflammation leading to preterm birth. J Clin Microbiol. 2009;47(1):38–47. doi: 10.1128/JCM.01206-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Steel JH, et al. Bacteria and inflammatory cells in fetal membranes do not always cause preterm labor. Pediatr Res. 2005;57(3):404–11. doi: 10.1203/01.PDR.0000153869.96337.90. [DOI] [PubMed] [Google Scholar]