

Abstract
Background
Glutathione S-transferases (GSTs) control multidrug-resistance and are upregulated in many cancers including malignant gliomas. The diazeniumdiolate JS-K generates nitric oxide (NO) on enzymatic activation by glutathione and GST, showing promising NO-based anticancer efficacy.
Objective
To evaluate the role of NO-based antitumor therapy with JS-K in U87 gliomas in vitro and in vivo.
Methods
U87 glioma cells and primary glioblastoma cell lines were exposed to JS-K and a variety of inhibitors to study cell death by necrosis, apoptosis and other mechanisms. GST-expression was evaluated by immunocytochemistry, PCR and Western blot and NO release from JS-K using a NO assay. The growth-inhibitory effect of JS-K was studied in a U87 xenograft model in vivo.
Results
Dose-dependent inhibition of cell proliferation was observed in human U87 glioma cells and primary glioblastoma cells in vitro. Cell death was partially induced by caspase-dependent apoptosis which could be blocked by Z-VAD-FMK and Q-VD-OPH. GST-inhibition by sulfasalazine, cGMP inhibition by ODQ and MEK 1/2 inhibition by UO126 attenuated the antiproliferative effect of JS-K, suggesting the involvement of various intracellular death signalling pathways. Response to JS-K correlated with mRNA and protein expression of GST and the amount of NO released by the glioma cells. Growth of U87 xenografts was significantly reduced, with immunohistochemical evidence for increased necrosis, apoptosis and reduced proliferation.
Conclusion
Our data for the first time show the potent antiproliferative effect of JS-K in gliomas in vitro and in vivo. These findings warrant further investigation of this novel NO-releasing prodrug in gliomas.
Keywords: glioma, GST, JS-K, nitric oxide (NO), U87
BACKGROUND
Nitric oxide (NO) plays a key role in tumor biology, controlling tumor growth, migration, invasion, angiogenesis, tumor blood flow and vascular permeability1-5.
A strategy to modulate NO signalling might be a promising option in cancer treatment, especially in malignant gliomas, because they defy even multi-modal treatment due to insufficient delivery of therapeutic drugs across the blood-brain barrier and due to chemo- and radioresistance6. Some approaches aim at delivering NO to alter vascular permeability and blood flow to increase drug uptake by the tumor, others induce tumor cell killing or chemo- or radiosensitization using NO donors7-11.
NO donors such as O2-aryl diazeniumdiolates (NONOates), compounds of the structure R1R2NN(O)=NOAr3, allow controlled spontaneous NO generation in aqueous media at physiological pH with reproducible half-lives12. Antiproliferative and radiosensitizing effects on hypoxic tumor cells by the spontaneous NO donor diazeniumdiolates DEA/NO, SPER/NO and PAPA/NO have been described earlier13-15. Nitric oxide donors increase the efficiency of cytostatic therapy with cyclophosphamide, doxyrubicin, cisplatin and retard the development of drug resistance in intracerebral leukemia P388 transplantation and in B16 melanoma16. Our group could demonstrate a dose-dependent cytotoxic effect of DEA/NO and SPER/NO and chemosensitization to carboplatin in glioma cells in vitro17.
JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] generates NO upon reaction with glutathione (GSH), a reaction that is catalyzed by glutathione-S-transferase isoforms GST-α, GST -π and GST-μ18. It has a strong, concentration-dependent antiproliferative effect in HL-60 leukemia cells, prostate cancer (PPC-1), hepatoma (Hep 3B), multiple myeloma (MM) and lung cancer in vitro and in vivo18-24. Cell death is induced by various mechanisms including necrosis and apoptosis via activation of caspases -3, -8 and -9. Furthermore, JS-K induces mitochondrial cytochrome c release in leukemia cells24. The mechanism by which JS-K exerts its growth inhibitory effects includes induction of the mitogen-activated protein kinases (MAPK) ERK, JNK and p38 and arylation of GSH and other cellular nucleophiles22, 23. In addition to its intrinsic antiproliferative effect, JS-K increases cisplatin and arsenic cytolethality in hepatomas by increasing intracellular accumulation and activating MAPK pathways20.
Malignant gliomas might be suitable candidates for treatment with a GST-activated NO donor drug such as JS-K because they exhibit overexpression and genetic polymorphisms of the GST-gene which influence the malignancy of the tumor and its response to chemo- or radiotherapy25-29. While there is evidence that NO released by NO donors influences cell viability, apoptosis, response to chemotherapy and the permeability of the blood-tumor barrier in gliomas9, 30, 31, NO donor drugs have not been thoroughly investigated, and the effects of JS-K in malignant glioma cells have not been characterized to date.
OBJECTIVE
The objective of this study was to investigate the effect of JS-K on cell viability and apoptosis induction in human U87 glioma cells and primary glioblastoma cells in vitro and to verify these effects in a U87 xenograft model in vivo.
METHODS
Materials
Human U87 glioma cells and human fibroblasts were provided by American Tissue Type Collection (ATCC® HTB-14™, ATCC®-CRL-1634, Rockville, MD, USA). Standard cell line verification and testing for contamination were performed regularly. Primary glioblastoma cultures were generated from glioblastoma tissue obtained during brain tumor surgery after informed consent of the patients. The use of human glioblastoma tissue was approved by the Ethics Committee at the University Medical Center Freiburg, Germany, under protocol 281/04. The NO donor JS-K [O2-(2,4-dinitrophenyl)1 [(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] was synthesized as described earlier20, 32. Because of JS-K’s relative insolubility in aqueous media, a 5 mM stock solution was prepared in 100% DMSO which was stored at -20°C. JS-K stock solution was dissolved in culture media immediately prior to use. In the presence of 1 mM GSH alone, JS-K generates approximately 1.1 mol NO/mol JS-K within 43 min, reaching a plateau after 8 min at 37°C and pH 7.4. The catalysis of JS-K’s NO release is predominantly enhanced by interaction with the GST-α and GST-μ isoforms18. For the U87 flank tumor model JS-K was dissolved in 100% DMSO and then serially diluted with PBS (pH 6.5) to the final concentration and injection volume.
Nude rats (rnu/rnu) from Charles River, Germany, were used for the study at the age of 5-6 weeks. Institutional guidelines for animal welfare and experimental conduct were followed for all animal experiments, which were approved by the Institutional Animal Care and Use Committee and the Regional Administrative Authority under protocol G08/28. Food and water were provided ad libitum.
Methods
Cell Culture
Human U87 glioma cells were cultivated in Dulbecco’s modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum, penicillin and streptomycin at 37°C and 5% CO2. Human fibroblasts were cultured in DMEM supplemented with 10% FCS, penicillin, streptomycin and glutamine. Primary glioblastoma cell cultures were established from surgical specimens from 4 patients with glioblastoma multiforme (PJ, LT, PM, TG) after mechanical mincing and lysis in PDD dissociation medium (Papain 0.01%, Dispase 0.1%, DNAse 0.01%, MgSO4 12,4 M, HBSS). Primary cell lines were studied below passage 10.
U87 glioma cells and primary glioblastoma cells were exposed to increasing JS-K concentrations for 24 h after they reached 70-80% confluence. After 24 h, the compounds were removed from the culture and medium was changed in all groups. Cell viability was measured by MTT at 24 h (end of incubation), and 48 h and 72 h after the end of the exposure time to check the extent of cell death and potential recovery.
In order to elucidate the intracellular mechanisms involved in the growth-inhibitory of JS-K in glioma cells, cells were pretreated with the pancaspase-inhibitors Z-VAD-FMK (Z-Val-Ala-Asp(OMe)-fluoromethylketone, 50 μM18; Biomol, Hamburg, Germany) or Q-VD-OPH (Quinoline-Val-Asp-Difluorophenoxymethylketone, 20 μM; MP Biomedicals, Eschwege, Germany), the guanylyl cyclase inhibitor ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one, 100 μM33, 34; Sigma, München, Germany), the MEK 1+2 inhibitor U0126 (bis[amino[(2-aminophenyl)thio]methylene]butanedinitrile, 10 μM22, 35; Biomol, Hamburg, Germany) or the GST inhibitor sulfasalazine (5-[4-(2-pyridylsulfamoyl)phenylazo]salicylic acid, 50 μM19; Sigma, München, Germany) for 2 h prior to the 24-h incubation period.
Viability Assay
Cell viability was determined using the MTT Assay. Briefly, 20 μl MTT salt was added to each well of cells cultured in a 96-well plate and incubated for 2 h at 37°C. After removal of the supernatant, cells were lysed with 100 μl of a DMSO, acetic acid and SDS mixture to dissolve the formazan crystals. Plates were shaken on a plate shaker to allow equilibration of the colored solution throughout the well. Colorimetric assessment of cell viability was performed by ELISA at 570 nm with 630 nm as a reference wavelength. Cell viability was expressed in percent (%) of the cell viability in the control group set to 100%. The experiment was done in triplicates and is representative of a minimum of three independent studies.
5’-Bromodeoxyuridine Incorporation Assay
Cell proliferation was monitored by 5’-bromodeoxyuridine (BrdU) incorporation assay (Roche Diagnostics, Mannheim, Germany). 5×103 U87 glioma cells and primary glioblastoma cells (PJ, LT, PM, TG) were plated on glass cover-slips (Ø 12 mm) and cultured for three days. Cells were treated with increasing JS-K concentrations (1 - 10 μM) for 24 h. Treatment was terminated by exchanging the medium. 48 h after treatment, BrdU was added to the cell cultures for 1 h at a final concentration of 10 μM. Cells were fixed with 70% ethanol (in 50 mM glycine buffer, pH2) for 30 min. Detection of DNA-incorporated BrdU was performed according to manufacturer’s instructions. Nuclei were counterstained with 4’-,6’-diamidino-2-phenylindole dihydrochloride (DAPI 10 μg/ml, Sigma-Aldrich, München, Germany). The percentage of proliferating cells out of 100% vital cells was determined for each treatment group with a fluorescence microscope (Axio Observer microscope, Zeiss, Jena, Germany). Experiments were performed in triplicate.
Apoptosis Assay
Apoptosis induction was studied with a caspase-3 assay kit assaying DEVD-dependent caspase activity using a fluorescence microtiter plate reader (Promega, Heidelberg, Germany). 3000 cells were plated in 96 wells and exposed to JS-K and/or the inhibitors for 24 h. Quantification of caspase-3 activity was performed at the end of the incubation period, after 24, 48 and 72 h. Cells were exposed to lysis buffer on ice for 10 min, and then exposed to DTT reaction buffer and the DEVD-AFC substrate (50 μM) for 2 h at 37°C. Samples were read with a 400-nm excitation and a 505-nm emission filter, and the fold-increase in caspase-3 activity was determined compared to untreated controls.
Guanosine 3’,5’cyclic Monophosphate (cGMP) Analysis
An enzyme-linked immunosorbent assay (ELISA) was used to determine the accumulation of intracellular cGMP. 2×106 U87 cells were grown overnight. The next day, cells were treated with JS-K (5, 15 μM) for 2 h. Cells were then lysed in 0.1 M trichloroacetic acid for 20 min at room temperature and scraped off the cell culture flask. Cell culture extracts were homogenized thoroughly and centrifuged at 1,000 ×g for 10 min. Total protein concentration of the supernatants was determined according to Bradford to assure comparability of the samples. Probes (2.5 mg total protein/ml) were assayed for cGMP by a cGMP competitive enzyme immunoassay (cGMP-EIA Kit, Cayman Chemical Company, Ann Arbor, MI, USA). ELISA and statistical analyses were performed according to the manufacturer’s instruction. Spectrophotometric readings (λ=410 nm) were performed using the Tecan i-Control infinite 200 photometer and software (Tecan, Männedorf, Switzerland).
Immunocytochemistry
Expression of GST-α (Calbiochem, Darmstadt, Germany) and GST-π (MBL, MA, USA) was assessed by immunocytochemistry. U87 cells, primary glioblastoma cell lines (LT, PJ, PM, TG), fibroblasts and astrocytes were cultured on glass cover-slips (Ø 12 mm). After removal of the medium, cells were fixed with 4% PFA (in PBS) for 30 min on ice. Cells were washed three times with PBS and subsequently permeabilized with acetone for 10 min at -20°C. Accessible epitopes were blocked with 10% normal goat serum (in PBS) for 1 h at room temperature. Binding of primary antibodies (GST-α, 1:100 and GST- π 1:500 in PBS containing 0.05% Tween 20) was performed overnight at 4°C. Afterwards, cells were washed in PBS and incubated in presence of secondary antibodies (1:400 in PBS, 0.05% Tween 20, donkey anti-rabbit IgG-Alexa568, Invitrogen, Darmstadt, Germany) for 1 h at room temperature. Cell nuclei were counterstained with DAPI (Sigma, München, Germany). Cells were then repeatedly washed in PBS and mounted in Fluorescence Mounting Medium (Dako, Glostrup, Denmark). Immunofluorescence was documented using AxioVision software (Zeiss, Jena, Germany).
Reverse Transcription with Polymerase Chain Reaction
Total RNA from U-87 cells, human primary glioblastoma cell lines (PM, PJ, LT, TG) and human fibroblasts was isolated by using the Qiagen Rneasy kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Reverse transcription (RT) was carried out on 0.8 μg RNA in 30 μl reactions containing 1 μl oligo(dT)primers (0.1 nM, Invitrogen, Karlsruhe, Germany) and 2 μl 10 mM dNTP mix (Fermentas, St. Leon-Roth, Germany). After denaturation for 10 min at 70°C and annealing on ice, 6×RT buffer, 14 U M-MLV reverse transcriptase, and 10 U RNAse inhibitor (Fermentas, St. Leon-Roth, Germany) were added. The reaction was performed for 60 min at 42°C, followed by a terminal denaturation for 10 min at 95°C. Parallel reactions without M-MLV reverse transcriptase were used as negative controls. For polymerase chain reaction (PCR), the following primers were used: GST-π (NM000852.3) forward 5’-aaagcctcctgcctatacgg-3’, GST-π reverse 5’-tcagcgaaggagatctggtct-3’, β-actin (NM001101.3) forward 5’-caccacaccttctacaatgag-3’ and β-actin reverse 5’-ctccttaatgtcacgcacga-3’, generating amplification products of 226 bp and 383 bp, respectively. The PCR was performed in 30 μl reactions containing 0.1 μM specific primers (Invitrogen, Karlsruhe, Germany), 200 μM of each deoxynucleoside-triphosphate, 10x reaction buffer, 2 mM MgCl2, 1.5 U Taq polymerase (Fermentas, St. Leon-Roth, Germany), and 2 μl of the RT reaction as template. Amplification was carried out under the following conditions: initial denaturation for 10 min at 95°C, denaturation for 1 min at 95°C, primer annealing for 30 s at 53°C, and elongation for 1 min at 72°C. After 35 cycles, the amplification was terminated with a final elongation for 10 min at 72°C, followed by rapid cooling to 8°C. Amplification products were separated by agarose gel electrophoresis and visualized by GelRed™ staining (Invitrogen, Karlsruhe, Germany).
Western Blot
U-87 cells treated with JS-K (15 μM) were cultured for 24 h. They were lysed in lysis buffer (50 mM Tris-HCl pH 8.8, 150 mM NaCl, 1% Triton-X with protease inhibitor cocktail PI) (Roche, Mannheim, Germany) and centrifuged at 15,000 rpm for 15 min at 4°C. Protein lysates (10-50 μg) were loaded on a 10 % polyacrylamide gel to determine activity and expression of GST-π. The protein was transferred to a PVDF membrane using an electroblotting apparatus (BioRad, Munich, Germany). The membrane was blocked with 5% dry milk in TBST (20 mM Tris-HCl, 500 mM NaCl, 0.05% Tween 20, pH 7.5) for 1 h at room temperature and incubated with antibodies against GST-π (1:2000, Biozol, Eching, Germany) overnight at 4°C. The membrane was washed three times for 10 min each time with TBST and incubated with horseradish peroxidase (HRP)-conjugate goat anti-rabbit IgG (1:3000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at room temperature. The membrane was again washed three times with TBST for 10 min. Protein bands were visualized by enhanced chemiluminescence using the Immun-Star Western C Chemiluminescent Kit (BioRad, Munich, Germany), analyzed by the Bio-1D application program (Peqlab, Erlangen, Germany) and compared by t-test.
Nitric Oxide Assay
Nitric oxide release induced by JS-K was determined using a fluorescence-based assay with the nitric oxide-sensitive fluorophore 4-amino-5-methylamino-2’,7’-difluorescein diacetate (DAF-FM, Invitrogen, Carlsbad, USA)36. 3000 U87 cells were plated in 96 wells in triplicates as previously described. They were exposed to a 10 μM solution of DAF-FM in warm HBSS at 37°C for 30 min in the dark. After removal of the DAF-FM solution, HBSS was added and the cells were treated with JS-K (5-30 μM) with sulfasalazine (50 μM) or with 1% DMSO in HBSS (controls). Fluorescence measurements for the estimated levels of intracellular NO were performed after 1, 2, 4, 6, and 8 h with an excitation wave length of 495 nm and an emission wave length of 525 nm. NO release measurements were given as relative fluorescence in % with reference to the controls which were run in parallel in the absence of the corresponding drug. Standard errors (SEM) were derived from fluorescence measurements in triplicates from 3 independent experiments.
In vivo Xenograft Experiments
The rats (n = 14) were injected with 106 U87 cells in 100 μl PBS with 30% Matrigel into both flanks under isoflurane anesthesia. Tumor size and body weight were measured three times a week by a researcher blinded to the treatment group. The tumor volume was calculated according to the formula Length x Width2/2. Treatment was initiated when the flank tumors reached a volume of 8000-9000 mm3. Rats were evenly distributed between the treatment groups with reference to tumor volume at the beginning of the treatment. Group A (n=6) was untreated controls and group B (n=8) received JS-K at a dose of 6 mg/kg/d subcutaneously (s.c.) for 7 consecutive days. Subcutaneous injections were given distant from the flank tumors into the neck. Two animals per group underwent MRI scanning on day 29 (T2 weighted imaging, Bruker BioSpec High Field MRI). Tumor growth was expressed as the difference in tumor volume in mm3 compared to the tumor volume at day 0 set to 0. The animals were sacrificed according to established criteria (tumor diameter > 5 cm, tumor volume > 50,000 mm3, ulceration of tumors, body weight loss >20%) or when they reached the study end point at day 43 after treatment.
Histology/Immunohistochemistry
After sacrifice of the animals tumor implants were excised, frozen in liquid nitrogen, sliced into 5 μm sections and analyzed histologically for tumor necrosis using standard H&E staining. Immunofluorescence staining was performed for markers of cell proliferation (Ki-67, Dako, Glostrup, Denmark) and angiogenesis (CD31, BD Pharmingen, Heidelberg, Germany). Cryosections (10 μm) were fixed with acetone (-20°C) and rehydrated in PBS. Epitopes were blocked with 2% FCS in PBS (Ki-67) or 10% BSA in PBS (CD-31) for 1 h at room temperature. Subsequently, sections were incubated with primary antibody Ki-67 (1:150) or CD31 (1:100) in blocking solution overnight (4°C). Unbound antibodies were removed by multiple washing steps in PBST (PBS; 0.1% Triton X-100). Goat-anti-mouse IgG-Alexa 488 (Invitrogen, Darmstadt, Germany) was used as secondary antibody (1:400 in PBS, 1 h at room temperature). Slices were washed in PBST and cell nuclei were stained with DAPI (Sigma-Aldrich, Hamburg, Germany). After repeated washing steps, slices were mounted in Fluorescence Mounting Medium (Dako, Glostrup, Denmark). Immunofluorescence was viewed with a fluorescence microscope (Zeiss, Jena, Germany) and representative photomicrographs were taken at 20× magnification.
Statistical Analysis
Results were expressed as means of multiple experiments with SEM. Multiple comparisons of cell viability and apoptosis data were performed by ANOVA and Student Newman-Keul’s test. The results of the NO assay were analyzed by repeated measures ANOVA. Protein bands in the Western blot and differences in growth rate of s.c. tumors between the treatment groups were compared by Student’s t-test. Probability (p) values of < .05 were considered significant.
RESULTS
Antiproliferative and proapoptotic activity of JS-K in glioma cells
JS-K showed a dose-dependent cytotoxic effect in U87 and primary glioblastoma cells as assessed by MTT assay (Fig. 1A+B). The greatest growth inhibitory effect in U87 cells was observed 48 h after the end of the JS-K exposure. The IC50 for JS-K was 15 μM or less in U87 and primary glioblastoma cells (p=.011). At later time points, surviving cells started to regrow. Low JS-K concentrations (≤5 μM) showed no growth inhibition or even a tendency for enhanced tumor growth. Significant inhibition of glioma cell proliferation could be demonstrated in a dose-dependent fashion by BrdU incorporation assay in all glioma cell lines in vitro (Fig. 2A). This reduction in BrdU incorporation can be demonstrated in representative immunofluorescence images of U87 glioma cells 48 h after treatment with increasing concentrations of JS-K (Fig. 2B). JS-K (15 μM) had a proapoptotic effect in U87 glioma cells, inducing strong caspase-3 activation (Fig. 3B). The pan-caspase inhibitor Z-VAD-FMK (50 μM) partially blocked cell death by JS-K (p= .03). This was achieved by significant inhibition of caspase-3 activation when JS-K-treated cells were preincubated with Z-VAD-FMK (from 227±26% to 35±1% after 24 h, p< .0001). Q-VD-OPH (20 μM) significantly blocked caspase-3 activation in response to JS-K and thus reduced the cytotoxic effect of JS-K assessed by MTT (p< .0001 each).
Figure 1.
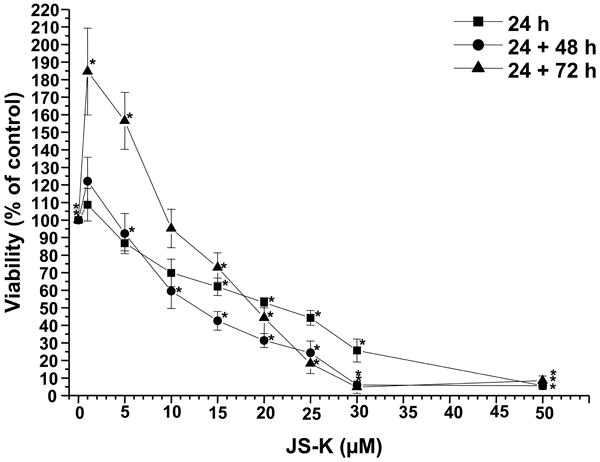
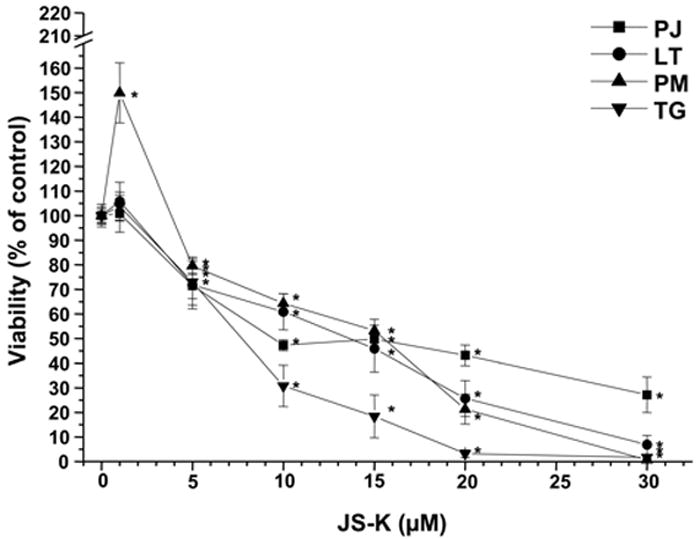
Cytotoxic effect of JS-K on U87 cells (Fig. 1A) and primary glioblastoma cells (PJ, LT, PM, TG, Fig. 1B) in vitro. Cells were cultured with JS-K for 24 hours at the indicated concentrations. Cell viability was determined using MTT assay at the end of the incubation, 48 and 72 hours after the end of the JS-K exposure (means ± SEM of three separate triplicate experiments). Induction of cell death by JS-K was plotted relative to the viability of untreated controls set to 100%. Viability data for JS-K in U87 gliomas is presented for all three time points (Fig. 1A), viability data for glioblastoma cells was assessed 48 hours after the end of exposure (Fig. 1B). Asterisks (*, p< .05) indicate a significant difference between JS-K and the controls.
Figure 2.
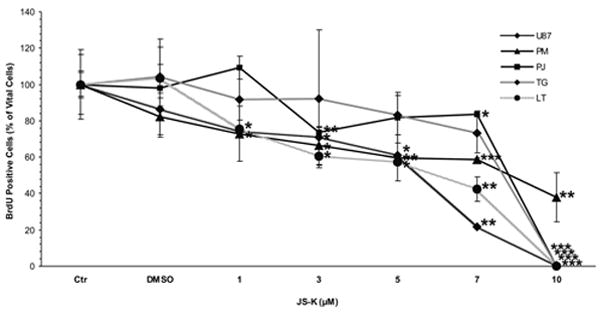
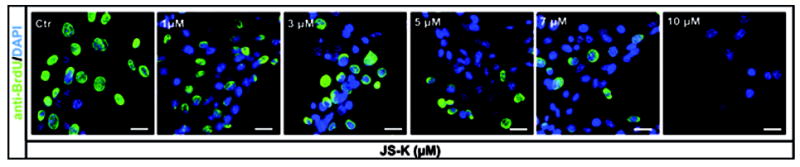
JS-K reduces proliferation of U87 and primary glioblastoma cells in a dose-dependent fashion in vitro. A. Cell proliferation was assessed by 5’-bromodeoxyuridine incorporation 48 h after treatment of glioma cells with JS-K (1-10 μM) for 24 h. Percentage of proliferating cells out of 100% vital cells was assessed by fluorescence microscopy. Data are expressed as means ± SEM from three independent experiments and analyzed by t-test. Asterisks (*, p< .05; **, p< .01; ***, p< .001) indicate a significant difference between JS-K and the controls. B. Representative images of U87 cells treated with increasing concentrations of JS-K (1-10 μM) for 24 h. Photomicrographs taken on a Zeiss Axio Observer Microscope, scale bar 25 μm.
Figure 3.
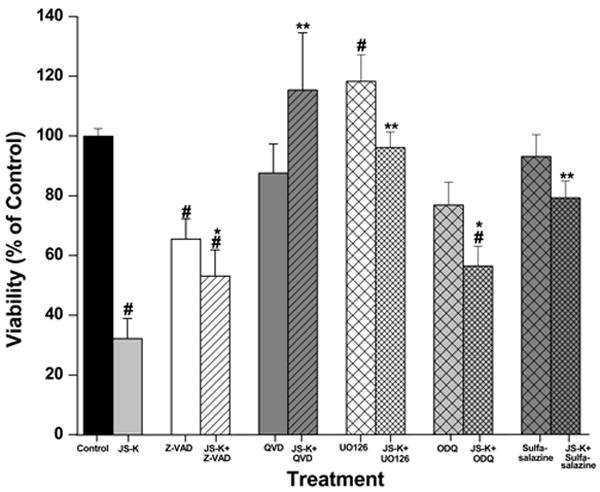
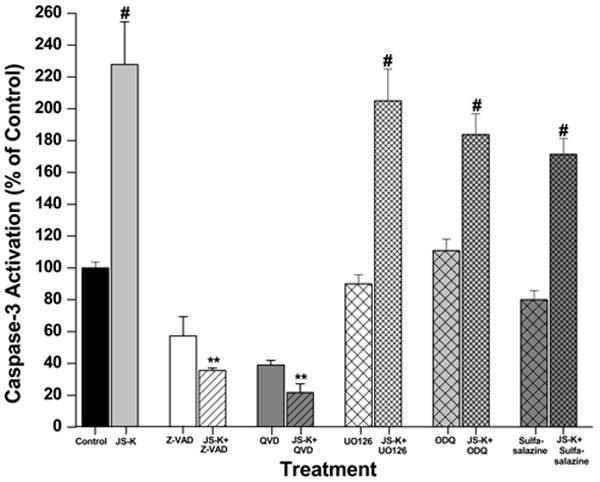
Induction of cell death by JS-K involves regulation by several signalling pathways leading to apoptosis. A. The cytotoxic effect of JS-K in U87 cells can be modified by pre-treatment with Z-VAD-FMK (50 μM), QVD-OPH (20μM), U0126 (10 μM), ODQ (100 μM) or sulfasalazine (50 μM) for 2 hours prior to the 24 h incubation period with JS-K (15μM). Cell viability was assessed by MTT test. B. JS-K induces apoptosis in U87 cells associated with caspase-3 activation. Cells were treated with JS-K (15 μM) for 24 hours, and caspase-3 activity was assessed at the end of the incubation. Data from MTT and caspase-3 assays are expressed as means ± SEM from three independent experiments. # indicates a significant difference compared to the control (p< .05), * and ** refer to statistical testing against JS-K monotherapy with p-values of p< .05 and p< .0001, respectively.
Reduction in cell viability in response to JS-K could be completely blocked by pre-treatment with the MEK1/2 inhibitor U0126 (10 μM, p< .0001; Fig. 3A). However, JS-K-induced apoptotic cell death was not affected by MEK inhibition (p= .27). Inhibition of the cGMP pathway by pre-treatment with ODQ (100 μM) partially reduced the cytotoxic effect of JS-K in U87 cells, increasing cell viability from 32±7% to 56±6% (p= .01). This protective effect was not mainly induced by apoptosis inhibition, as ODQ only moderately affected caspase-3 activation induced by JS-K (p= .03). JS-K predominantly induces cell death in U87 cells by mechanisms other than activation of the cGMP pathway, as treatment with JS-K (5, 15 μM) only showed a trend towards increased intracellular cGMP levels, which was not statistically significant (data not shown). Pre-treatment with sulfasalazine to inhibit GST blocked JS-K induced cell death (p< .0001). Sulfasalazine alone did not reduce cell viability and did not affect caspase-3 activation (Fig. 3A+B).
Glioma Cells Express GST
Higher expression of GST-α and GST-π was confirmed by immunocytochemistry in U87 cells, and four primary GBM cell lines (LT, TG, PJ, PM) compared to astrocytes and fibroblasts as non-neoplastic controls (Fig. 4). There is a certain variability in the expression of the GST-α and GST-π isoforms among the glioma cell lines. U87 cells showed strong GST-π protein expression (Fig. 5A). Treatment with JS-K (15 μM) did not alter the level of protein expression of GST-π compared to untreated cells. All glioma cell lines expressed high levels of GST-π mRNA compared to fibroblasts, which were tested as controls (Fig. 5B). This might account for the selectivity and the degree of the antiproliferative response to JS-K treatment, which requires reaction of the compound with GSH for intracellular NO release in tumor cells.
Figure 4.
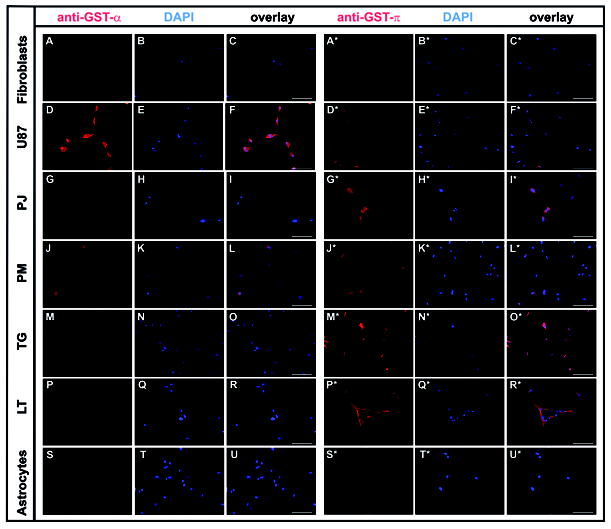
Expression of GST-α and -π in U87 glioma cells, four selected primary glioblastoma cell lines (PJ, PM, TG, LT), and in fibroblasts and astrocytes was assessed by immunocytochemistry. Cells were fixed, permeabilized, and incubated with specific primary antibodies and fluorescence labelled secondary antibodies for imaging as detailed in Materials and Methods. Photomicrographs taken on a Zeiss Axio Observer Microscope, 20X magnification, scale bar represents 100 μm. Photomicrographs in columns 1 and 4 show immunostaining for GST- α and -π, respectively, columns 2 and 5 show nuclear staining with DAPI, columns 3 and 6 composites with overlay of both images.
Figure 5.
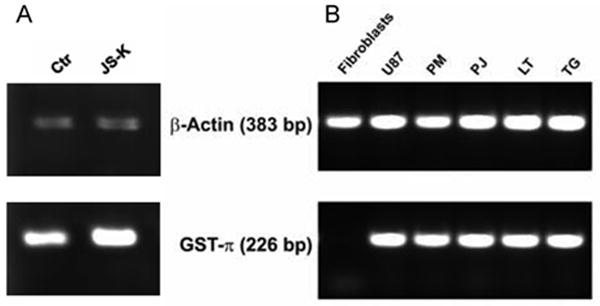
Gliomas express GST–π. A. GST-π protein expression is strong in U87. Treatment with 15 μM JS-K for 24 h did not significantly modify protein expression as analyzed by Western blot. B. All glioma cell lines exhibit strong GST-π mRNA expression compared to fibroblasts which were used as normal controls.
JS-K Induces NO Release from U87 Cells
Exposure of U87 cells to JS-K (5-30 μM) resulted in a dose-dependent rapid intracellular generation of NO after addition of JS-K compared to untreated controls (p< .0001 for all concentrations) (Fig. 6). As these experiments were performed in HBSS instead of media, NO measurements were only done in the first 8 h as viability of the cells was significantly reduced after 24 h due to the absence of culture media. Very little NO release could be detected after pre-treatment with sulfasalazine (50 μM), possibly as a result of GST inhibition (p< .0001 for all concentrations).
Figure 6.
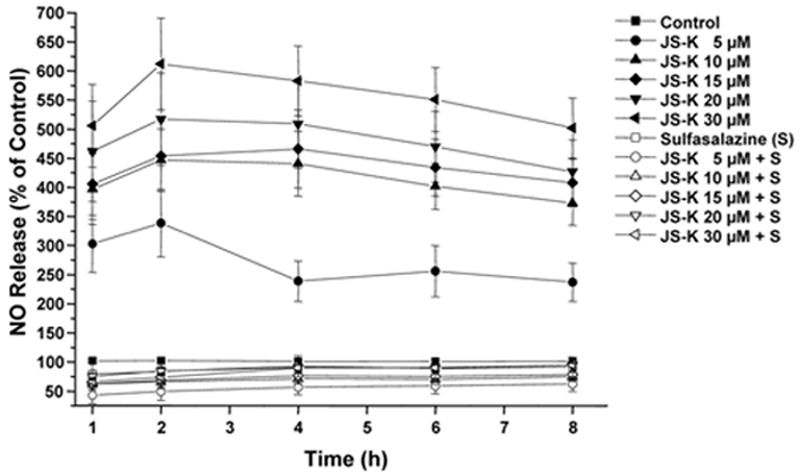
NO release from JS-K in glioma cells can be blocked by sulfasalazine.
Nitric oxide release measured using NO-sensitive DAF-FM diacetate in U87 cells over 8 h. Incubation with JS-K (5-30 μM) results in rapid nitric oxide production in U87 cells compared to untreated controls. A significant reduction of NO release was observed after pre-treatment with sulfasalazine (50 μM) for 2 hours. Data are expressed as means ± SEM from three independent experiments. All values are significantly different relative to the corresponding untreated controls and to the corresponding values after pre-treatment with sulfasalazine (p< .05).
JS-K Inhibits Tumor Growth in a U87 Xenograft Model
Subcutaneous tumor growth in a U87 xenograft model was significantly attenuated by treatment with JS-K (p< .0001) (Fig. 7). No deaths of unknown cause unrelated to tumor growth or sacrifice at the end of the observation period were observed. Macroscopic photos (Fig. 8 A+A*), T2-weighted MRI images of tumors from the control and the JS-K group (Fig. 8 B+B*) and H&E sections (Fig. 8 C+C*) showed reduced tumor size and increased signs of tumor necrosis in the JS-K group. The JS-K-treated tumor (9A*) appeared less vascularized than the untreated control (Fig. 9A). Visual scoring of representative immunohistochemical sections from both groups showed larger necrotic tumor portions and areas with less Ki-67 staining (Fig. 9 A+A*). Vessel density was not significantly reduced after JS-K treatment (Fig. 9 D vs. D*).
Figure 7.
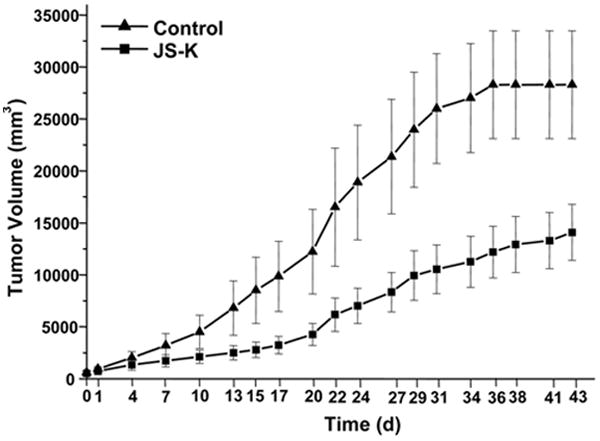
JS-K significantly reduced growth of U87 xenografts in vivo. Nude rats (rnu/nu, n=14) were inoculated subcutaneously bilaterally into the flank with 106 U87 cells. When tumors reached a volume of 8000-9000 mm3, JS-K (6 mg/kg) or vehicle was administered subcutaneously into the neck daily for 7 days. Tumor growth was expressed as the difference in tumor volume in mm3 compared to the tumor volume at day 0 set to 0. Tumor volumes are represented as means ± SEM for each group over time (day 0 to day 43).
Fig. 8.
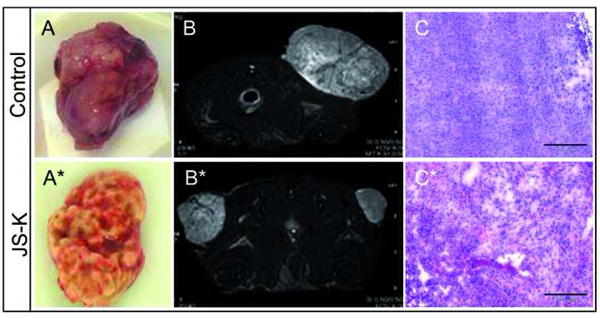
JS-K delays tumor growth compared to untreated controls.
Representative excised tumors are smaller und show less tumor angiogenesis in the JS-K treated tumor specimen (8A: untreated control; 8A*: JS-K-treated tumor). Coronal T2 weighted MRI images taken on day 29 after treatment show the difference in tumor size and vascularization between untreated (8B) and JS-K treated (8B*) animals. H&E staining demonstrate larger areas of necrosis after JS-K (8C*) compared to the controls (8C).
Fig. 9.
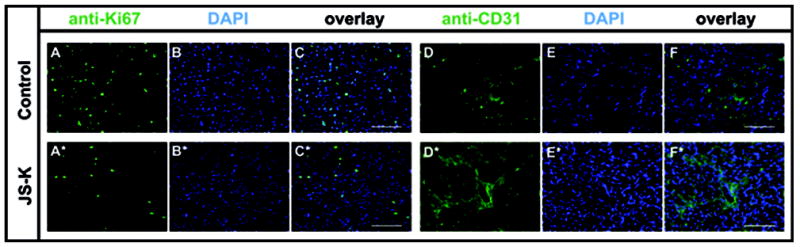
JS-K reduces proliferation in U87 flank tumors. U87 flank tumors treated with JS-K showed reduced expression of Ki-67 (9A*) compared to untreated controls (9A). No strong difference in CD31 expression was observed (Control: 9D, JS-K: 9D*). Frozen sections of excised tumor tissue were sliced in 10 μm sections and stained as described in the Methods section. Photomicrographs in columns 1 and 4 show Ki-67 or CD31 staining, respectively, columns 2 and 5 show nuclear staining with DAPI, and columns 3 and 6 show composites with overlay of both images (20x magnification, scale bar 100 μm).
DISCUSSION
In this study, we established for the first time the ability of the nitric oxide donor JS-K to induce cell death in human glioblastoma cells in vitro and in a U87 xenograft model in vivo. Cell death occurred in a dose-dependent fashion by various mechanisms including necrosis, caspase-dependent and caspase-independent cell death. The cytotoxic effect of JS-K was secondary to the release of copious amounts of NO after activation of JS-K by reaction with GSH under catalysis by GST.
GSTs were chosen as the enzymes to activate the NO-releasing prodrug JS-K because they are upregulated in many tumors, conferring multidrug resistance to tumor cells that have a survival advantage by enhanced detoxification of anticancer drugs37. The use of a GST-activated NO donor compound was considered a promising strategy to assess in a glioma model, as malignant gliomas display highly heterogeneous expression patterns and genetic polymorphisms of the GST genes27. Overexpression of the GST-π and GST-μ isoforms has been associated with drug resistance and poor outcome in malignant gliomas25, 28, 29. Grant et al. compared GST immunostaining in normal brain compared to brain tumors and showed weaker staining in neurons, astrocytes and oligodendrocytes and significant increases in intensity in glial tumors depending on their differentiation38. GST-α immunostaining was observed in endothelial cells and astrocytes in the vicinity of blood vessels39. Elevated expression of GST-α, and -π could be demonstrated in U87 cells and correlated with the strong antiproliferative effect of JS-K. These findings in immortalized U87 glioma cells could be reproduced in primary glioblastoma cells from surgical specimens, an effect that was higher than in astrocytes and fibroblasts.
The antiproliferative effect correlated well with the release of NO from JS-K in U87. Pre-treatment with sulfasalazine attenuated both NO production and the cytotoxic effect of JS-K to a considerable degree. This might be due to GST inhibition by sulfasalazine. A chemical interaction between the molecules was not observed (Joseph Saavedra, unpublished data). Reaction of JS-K with GSH to release NO seems to be a major prerequisite for induction of cell death. However, NO release after interaction of JS-K with glutathione and other thiols in the absence of GST has been described earlier and presumably accounts for the portion of the antiproliferative effect that was not blocked by GST inhibition18, 40. In multiple myeloma cells, pre-treatment with sulfasalazine blocked NO release from JS-K by 36%19. Sulfasalazine predominantly inhibits GST-α and GST-μ and to a lesser degree GST-π, which is the predominantly expressed isoform in gliomas41, 42. In addition to its capacity to block GSTs, sulfasalazine is a potent inhibitor of the transcription factor NFκB and may induce cell cycle arrest itself43. At high concentrations (1 mM), it also induces death signalling pathways and caspase-dependent apoptosis independent of NFκB in gliomas44. Structural analogs of JS-K that do not release NO (e.g., JS-43-126) have less biological activity than their NO-releasing counterparts45.
The selectivity of the antiproliferative effect of JS-K has been tested earlier in leukemia, multiple myeloma and renal cancer cell lines. The IC50s for the normal cells were often an order of magnitude higher than those for the respective tumor cell lines19, 46. Testing the antiproliferative effect of JS-K using the NCI 51-Cell-Line Screen, IC50s between 10 and 100 μM were observed in solid tumors, which is in line with our findings of an IC50 of 15 μM for the glioma cell lines23. Nitric oxide modulates intracellular targets that affect cell growth through multiple mechanisms and with different kinetics. The most profound effect on cell viability of U87 cells was observed not immediately after the 24 h incubation but after 48 h, indicating that cell death pathways are activated that may proceed slower. On the other hand, a trend towards regrowth of surviving cells could be observed after 72 h, indicating that a portion of the tumor cells might be resistant to JS-K.
It has been described earlier that NO might have dual pro- and antitumor action, depending on the local concentration and duration of NO exposure, and cellular sensitivity to NO. Low NO concentrations can promote tumor growth, whereas high NO levels from activated iNOS or exogenous NO donors have cytotoxic effects3, 47. This phenomenon was also observed in our study.
Brune et al. described the complex relationship between NO and ROS (reactive oxygen species) and RNS (reactive nitrogen species) formation in conveying pro- or anti-apoptotic signals depending on concentration and cellular context48. While low NO concentrations evoked cell protection by inducing caspase inhibition, cGMP formation and expression of anti-apoptotic proteins, high NO doses caused p53 stabilization and caspase activation, resulting in cell death by necrosis and apoptosis. NO has been shown to induce cytochrome c release from mitochondria and activate the intrinsic apoptotic pathway49. The same effect was observed for JS-K in HL-60 cells inducing cytochrome c release and caspase-dependent apoptosis by mainly activating the intrinsic apoptosis pathway24.
Activation of the apoptosis pathway via caspase-3 activation was also observed in our experiments. JS-K simultaneously induced cell death and caspase-3 activation in U87 cells. However, inhibition of apoptosis by Z-VAD-FMK or QVD-OPH only partially abrogated the cytotoxic effect of JS-K.
In our study, inhibition of the cGMP pathway by pre-treatment with ODQ only moderately reduced the growth inhibitory effect of JS-K in U87 glioma cells. Many effects of NO are mediated by its ability to activate soluble guanylyl cyclase. The increase in intracellular cGMP levels regulates protein kinase G activity, protein phosphorylation, and many other biological processes50. However, it has been shown earlier that the cytotoxic and inflammatory effects of NO might be independent of cGMP and may be the result of the involvement of different death pathways in JS-K-induced cell death. While the vascular effects of NO donor therapy on the permeability of the blood-brain barrier clearly depend on activation of the cGMP pathway, other death signalling pathways appear to be more important in inducing cell death by JS-K10. Mujoo et al. showed in several human cancer models that tumor cell proliferation is mediated by both cGMP-dependent and independent mechanisms51. Caspase-3 activation after JS-K was not impaired but was even slightly enhanced. ODQ has been shown to activate caspase-3 in prostate cancer cells and may induce cell death independent from cGMP levels33.
Inhibition of the MAPK pathway using the MEK1+2 inhibitor U0126 partially prevents cell death by JS-K in U87 glioma cells without affecting caspase-3 activation, indicating that this pathway is involved in the growth-inhibitory effect of JS-K. It has already been observed that MEK inhibition antagonizes the growth inhibitory effect of JS-K in hepatoma in vitro and in vivo, as JS-K exerts its action to some degree by activation of the MAPK pathway22, 23. Involvement of the MAPK pathway induces cell death by dephosphorylation of ERK, resulting in caspase-independent cell death.
It has been shown in other tumor models that the potent antiproliferative effect of NO released from JS-K or related NO-donating compounds results from their capacity to alter protein function directly through posttranslational modification (nitration or nitrosylation) or indirectly through interactions with oxygen, superoxide, thiols, or heavy metals, the products of which can ultimately lead to S-glutathionylation19, 20, 22, 52. This interaction with metal ions, thiol groups, and other free radicals can mutate DNA and inhibit several key enzymes involved in energy metabolism and stress response pathways that involve p38 and JNK, eventually leading to cell death53. JS-K also induces apoptosis by inhibition of ubiquitinylation of proteins, increase of p53 levels and cleavage of DNA repair enzymes such as poly [ADP-ribose] polymerase 1 (PARP)54. Recent gene expression profiling data in HL-60 leukemia cells show induction of concentration and time-dependent changes in the expression of genes related to apoptosis, cell cycle control, invasion and acute phase proteins after JS-K treatment55.
The results of the inhibition experiments and the time course of cell death and apoptosis in U87 strongly suggest that, similar to findings in other tumor models, JS-K exerts its antiproliferative action in gliomas by several mechanisms including direct induction of DNA double strand breaks, interaction with proteins, e.g. DNA repair enzymes by promoting their phosphorylation, arylation, glutathionylation, or modulation of transcription factors, ultimately leading to induction of caspase-dependent and caspase-independent apoptosis.
The potent growth inhibitory effect of JS-K in gliomas could be reproduced in a xenograft model showing significant delay and reduction of tumor growth after JS-K treatment. JS-K treatment was started when the flank tumors already had a considerable size and was given by subcutaneous injection distant from the tumor location daily for one week. Histological workup showed large areas of necrosis and reduced cell proliferation and possibly a mild effect on vessel density. Comparable antitumor effects were observed in other xenograft models when JS-K was administered i.v. over several weeks, leading to a long-lasting tumor control alone or in combination with chemotherapeutic drugs without toxicity 18-23. JS-K doses up to 15 μmol/kg have been tested in rats and no vascular side effects or organ toxicity were observed (Paul Shami, personal communication).
The molecular weight of 384 D and presence of a diazeniumdiolate group in the molecule may not make JS-K or its analogs ideal candidates for penetration of the blood-brain barrier and use in intracranial gliomas. However, NO donor therapy may be especially promising for the treatment of brain tumors based on the dual role of NO as vasodilator and anti-neoplastic agent. NO released from NO donor drugs exerts its antitumor action not only by direct cytotoxic effects but also by opening the blood-tumor barrier, by increasing the uptake of therapeutic compounds, by chemo- or radiosensitization and by other mechanisms. Earlier studies have shown that uptake of radiolabelled tracers and carboplatin (MW 500 D) is significantly increased by selective opening of the blood-tumor barrier in C6 gliomas using the NO donor PROLI/NO, leading to long-term tumor control9. Therefore we hypothesize that spontaneous NO release from JS-K in the presence of glutathione or after JS-K-activation by GST expressed in endothelial cells and astrocytes in the vicinity of blood vessels may open the blood-brain barrier and thus increase the uptake of JS-K into the tumor. There, JS-K is then catalyzed to exert its tumor cell-killing effect by a variety of mechanisms. Local antitumor effects were observed in intracranial U87 xenografts after treatment with the GST-π-activated NO donor PABA/NO after delivery by s.c. injection or convection-enhanced delivery56.
CONCLUSION
This study for the first time shows the potent antiproliferative effect of the NO donor prodrug JS-K in gliomas in vitro and in vivo. Among other factors, this antitumor effect depends on the strong expression of GST in gliomas and the release of high levels of NO from JS-K, and it involves a variety of cell death pathways. Modifications of dosing and route and duration of delivery and assessment of the efficacy in an intracranial glioma model are still required to consolidate the role of this novel NO-releasing prodrug in glioma treatment.
Acknowledgments
We thank Jan Beckervordersandforth and Dr. Marie Follo for help with histology and immunocytochemistry and Dr. Wilfried Reichardt for MRI Imaging.
FINANCIAL SUPPORT: This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (JS) and by Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E.
Footnotes
CONFLICT OF INTEREST The authors declare that there is no conflict of interest with regard to the manuscript submitted for review.
References
- 1.Lala PK, Orucevic A. Role of nitric oxide in tumor progression: lessons from experimental tumors. Cancer Metastasis Rev. 1998 Mar;17(1):91–106. doi: 10.1023/a:1005960822365. [DOI] [PubMed] [Google Scholar]
- 2.Lam-Himlin D, Espey MG, Perry G, Smith MA, Castellani RJ. Malignant glioma progression and nitric oxide. Neurochem Int. 2006 Dec;49(8):764–768. doi: 10.1016/j.neuint.2006.07.001. Epub 2006 Sep 2012. [DOI] [PubMed] [Google Scholar]
- 3.Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev. 2007 May;27(3):317–352. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 4.Ridnour LA, Isenberg JS, Espey MG, Thomas DD, Roberts DD, Wink DA. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc Natl Acad Sci U S A. 2005 Sep 13;102(37):13147–13152. doi: 10.1073/pnas.0502979102. Epub 12005 Sep 13142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wink DA, Mitchell JB. Nitric oxide and cancer: an introduction. Free Radic Biol Med. 2003 Apr 15;34(8):951–954. doi: 10.1016/s0891-5849(02)01362-x. [DOI] [PubMed] [Google Scholar]
- 6.Rutka JT, Weyerbrock A, Liang ML. Gliomas: quo vadis. Clin Neurosurg. 2006;53:58–63. [PubMed] [Google Scholar]
- 7.Matsukado K, Inamura T, Nakano S, Fukui M, Bartus RT, Black KL. Enhanced tumor uptake of carboplatin and survival in glioma-bearing rats by intracarotid infusion of bradykinin analog, RMP-7. Neurosurgery. 1996;39(1):125–133. doi: 10.1097/00006123-199607000-00025. [DOI] [PubMed] [Google Scholar]
- 8.Mitchell JB, DeGraff W, Kim S, et al. Redox generation of nitric oxide to radiosensitize hypoxic cells. International Journal of Radiation Oncology Biology Physics. 1998;42(4):795–798. doi: 10.1016/s0360-3016(98)00327-7. [DOI] [PubMed] [Google Scholar]
- 9.Weyerbrock A, Walbridge S, Pluta RM, Saavedra JE, Keefer LK, Oldfield EH. Selective opening of the blood-tumor barrier by a nitric oxide donor and long-term survival in rats with C6 gliomas. J Neurosurg. 2003;99(4):728–737. doi: 10.3171/jns.2003.99.4.0728. [DOI] [PubMed] [Google Scholar]
- 10.Weyerbrock A, Walbridge S, Saavedra JE, Keefer LK, Oldfield EH. Differential effects of nitric oxide on blood-brain barrier integrity and cerebral blood flow in intracerebral C6 gliomas. Neuro Oncol. 2011 Feb;13(2):203–211. doi: 10.1093/neuonc/noq161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wink DA, Cook JA, Christodoulou D, et al. Nitric oxide and some nitric oxide donor compounds enhance the cytotoxicity of cisplatin. Nitric Oxide-Biology and Chemistry. 1997;1(1):88–94. doi: 10.1006/niox.1996.0108. [DOI] [PubMed] [Google Scholar]
- 12.Keefer LK, Nims RW, Davies KM, Wink DA. “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 13.Griffin RJ, Makepeace CM, Hur WJ, Song CW. Radiosensitization of hypoxic tumor cells in vitro by nitric oxide. Int J Radiat Oncol Biol Phys. 1996 Sep 1;36(2):377–383. doi: 10.1016/s0360-3016(96)00329-x. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell JB, Cook JA, Krishna MC, et al. Radiation sensitisation by nitric oxide releasing agents. Br J Cancer Suppl. 1996 Jul;27:S181–184. [PMC free article] [PubMed] [Google Scholar]
- 15.Wink DA, Cook JA, Pacelli R, et al. The effect of various nitric oxide-donor agents on hydrogen peroxide-mediated toxicity: a direct correlation between nitric oxide formation and protection. Arch Biochem Biophys. 1996;331(2):241–248. doi: 10.1006/abbi.1996.0304. [DOI] [PubMed] [Google Scholar]
- 16.Konovalova NP, Goncharova SA, Volkova LM, Rajewskaya TA, Eremenko LT, Korolev AM. Nitric oxide donor increases the efficiency of cytostatic therapy and retards the development of drug resistance. Nitric Oxide. 2003 Feb;8(1):59–64. doi: 10.1016/s1089-8603(02)00142-8. [DOI] [PubMed] [Google Scholar]
- 17.Weyerbrock A, Baumer B, Papazoglou A. Growth inhibition and chemosensitization of exogenous nitric oxide released from NONOates in glioma cells in vitro. J Neurosurg. 2009 Jan;110(1):128–136. doi: 10.3171/2008.6.17607. [DOI] [PubMed] [Google Scholar]
- 18.Shami PJ, Saavedra JE, Wang LY, et al. JS-K, a Glutathione/Glutathione S-Transferase-activated Nitric Oxide Donor of the Diazeniumdiolate Class with Potent Antineoplastic Activity. Mol Cancer Ther. 2003;2(4):409–417. [PubMed] [Google Scholar]
- 19.Kiziltepe T, Hideshima T, Ishitsuka K, et al. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007 Jul 15;110(2):709–718. doi: 10.1182/blood-2006-10-052845. Epub 2007 Mar 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Li C, Qu W, et al. Nitric oxide prodrugs and metallochemotherapeutics: JS-K and CB-3-100 enhance arsenic and cisplatin cytolethality by increasing cellular accumulation. Mol Cancer Ther. 2004;3(6):709–714. [PubMed] [Google Scholar]
- 21.Maciag AE, Chakrapani H, Saavedra JE, et al. The nitric oxide prodrug JS-K is effective against non-small-cell lung cancer cells in vitro and in vivo: involvement of reactive oxygen species. J Pharmacol Exp Ther. 2011 Feb;336(2):313–320. doi: 10.1124/jpet.110.174904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren Z, Kar S, Wang Z, Wang M, Saavedra JE, Carr BI. JS-K, a novel non-ionic diazeniumdiolate derivative, inhibits Hep 3B hepatoma cell growth and induces c-Jun phosphorylation via multiple MAP kinase pathways. J Cell Physiol. 2003;197(3):426–434. doi: 10.1002/jcp.10380. [DOI] [PubMed] [Google Scholar]
- 23.Shami PJ, Saavedra JE, Bonifant CL, et al. Antitumor activity of JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] and related O2-aryl diazeniumdiolates in vitro and in vivo. J Med Chem. 2006 Jul 13;49(14):4356–4366. doi: 10.1021/jm060022h. [DOI] [PubMed] [Google Scholar]
- 24.Udupi V, Yu M, Malaviya S, Saavedra JE, Shami PJ. JS-K, a nitric oxide prodrug, induces cytochrome c release and caspase activation in HL-60 myeloid leukemia cells. Leuk Res. 2006 Oct;30(10):1279–1283. doi: 10.1016/j.leukres.2005.12.007. Epub 2006 Jan 1224. [DOI] [PubMed] [Google Scholar]
- 25.Ali-Osman F, Brunner JM, Kutluk TM, Hess K. Prognostic significance of glutathione S-transferase pi expression and subcellular localization in human gliomas. Clin Cancer Res. 1997 Dec;3(12 Pt 1):2253–2261. [PubMed] [Google Scholar]
- 26.Ezer R, Alonso M, Pereira E, et al. Identification of glutathione S-transferase (GST) polymorphisms in brain tumors and association with susceptibility to pediatric astrocytomas. J Neurooncol. 2002 Sep;59(2):123–134. doi: 10.1023/a:1019601305210. [DOI] [PubMed] [Google Scholar]
- 27.Juillerat-Jeanneret L, Bernasconi CC, Bricod C, et al. Heterogeneity of human glioblastoma: glutathione-S-transferase and methylguanine-methyltransferase. Cancer Invest. 2008 Jul;26(6):597–609. doi: 10.1080/07357900802072913. [DOI] [PubMed] [Google Scholar]
- 28.Kilburn L, Okcu MF, Wang T, et al. Glutathione S-transferase polymorphisms are associated with survival in anaplastic glioma patients. Cancer. 2010 May 1;116(9):2242–2249. doi: 10.1002/cncr.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nutt CL, Noble M, Chambers AF, Cairncross JG. Differential expression of drug resistance genes and chemosensitivity in glial cell lineages correlate with differential response of oligodendrogliomas and astrocytomas to chemotherapy. Cancer Res. 2000 Sep 1;60(17):4812–4818. [PubMed] [Google Scholar]
- 30.Shinoda J, Whittle IR. Nitric oxide and glioma: a target for novel therapy? Br J Neurosurg. 2001;15(3):213–220. doi: 10.1080/02688690120057628. [DOI] [PubMed] [Google Scholar]
- 31.Yang JJ, Yin JH, Yang DI. Nitric oxide donors attenuate clongenic potential in rat C6 glioma cells treated with alkylating chemotherapeutic agents. Neurosci Lett. 2007 May 11;418(1):106–110. doi: 10.1016/j.neulet.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 32.Saavedra JE, Srinivasan A, Bonifant CL, et al. The secondary amine/nitric oxide complex ion R(2)N[N(O)NO](-) as nucleophile and leaving group in S9N)Ar reactions. J Org Chem. 2001 May 4;66(9):3090–3098. doi: 10.1021/jo0016529. [DOI] [PubMed] [Google Scholar]
- 33.Haramis G, Zhou Z, Pyriochou A, Koutsilieris M, Roussos C, Papapetropoulos A. cGMP-independent anti-tumour actions of the inhibitor of soluble guanylyl cyclase, ODQ, in prostate cancer cell lines. Br J Pharmacol. 2008 Nov;155(6):804–813. doi: 10.1038/bjp.2008.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jadeski LC, Chakraborty C, Lala PK. Nitric oxide-mediated promotion of mammary tumour cell migration requires sequential activation of nitric oxide synthase, guanylate cyclase and mitogen-activated protein kinase. Int J Cancer. 2003 Sep 10;106(4):496–504. doi: 10.1002/ijc.11268. [DOI] [PubMed] [Google Scholar]
- 35.Zohrabian VM, Forzani B, Chau Z, Murali R, Jhanwar-Uniyal M. Rho/ROCK and MAPK signaling pathways are involved in glioblastoma cell migration and proliferation. Anticancer Res. 2009 Jan;29(1):119–123. [PubMed] [Google Scholar]
- 36.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic Biol Med. 2007 Oct 1;43(7):995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 37.Townsend DM, Findlay VL, Tew KD. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005;401:287–307. doi: 10.1016/S0076-6879(05)01019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grant R, Ironside JW. Glutathione S-transferases and cytochrome P450 detoxifying enzyme distribution in human cerebral glioma. J Neurooncol. 1995;25(1):1–7. doi: 10.1007/BF01054717. [DOI] [PubMed] [Google Scholar]
- 39.Johnson JA, el Barbary A, Kornguth SE, Brugge JF, Siegel FL. Glutathione S-transferase isoenzymes in rat brain neurons and glia. J Neurosci. 1993 May;13(5):2013–2023. doi: 10.1523/JNEUROSCI.13-05-02013.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar V, Hong SY, Maciag AE, et al. Stabilization of the nitric oxide (NO) prodrugs and anticancer leads, PABA/NO and Double JS-K, through incorporation into PEG-protected nanoparticles. Mol Pharm. 2010 Feb 1;7(1):291–298. doi: 10.1021/mp900245h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayeshi R, Chinyanga F, Chengedza S, Mukanganyama S. Inhibition of human glutathione transferases by multidrug resistance chemomodulators in vitro. J Enzyme Inhib Med Chem. 2006 Oct;21(5):581–587. doi: 10.1080/14756360600756105. [DOI] [PubMed] [Google Scholar]
- 42.Winter S, Strik H, Rieger J, Beck J, Meyermann R, Weller M. Glutathione S-transferase and drug sensitivity in malignant glioma. J Neurol Sci. 2000 Oct 1;179(S 1-2):115–121. doi: 10.1016/s0022-510x(00)00392-0. [DOI] [PubMed] [Google Scholar]
- 43.Robe PA, Bentires-Alj M, Bonif M, et al. In vitro and in vivo activity of the nuclear factor-kappaB inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res. 2004 Aug 15;10(16):5595–5603. doi: 10.1158/1078-0432.CCR-03-0392. [DOI] [PubMed] [Google Scholar]
- 44.Chung WJ, Sontheimer H. Sulfasalazine inhibits the growth of primary brain tumors independent of nuclear factor-kappaB. J Neurochem. 2009 Jul;110(1):182–193. doi: 10.1111/j.1471-4159.2009.06129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simeone AM, McMurtry V, Nieves-Alicea R, et al. TIMP-2 mediates the anti-invasive effects of the nitric oxide-releasing prodrug JS-K in breast cancer cells. Breast Cancer Res. 2008;10(3):R44. doi: 10.1186/bcr2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chakrapani H, Kalathur RC, Maciag AE, et al. Synthesis, mechanistic studies, and antiproliferative activity of glutathione/glutathione S-transferase-activated nitric oxide prodrugs. Bioorg Med Chem. 2008 Nov 15;16(22):9764–9771. doi: 10.1016/j.bmc.2008.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jadeski LC, Chakraborty C, Lala PK. Role of nitric oxide in tumour progression with special reference to a murine breast cancer model. Can J Physiol Pharmacol. 2002 Feb;80(2):125–135. doi: 10.1139/y02-007. [DOI] [PubMed] [Google Scholar]
- 48.Brune B. The intimate relation between nitric oxide and superoxide in apoptosis and cell survival. Antioxid Redox Signal. 2005 Mar-Apr;7(3-4):497–507. doi: 10.1089/ars.2005.7.497. [DOI] [PubMed] [Google Scholar]
- 49.Yabuki M, Tsutsui K, Horton AA, Yoshioka T, Utsumi K. Caspase activation and cytochrome c release during HL-60 cell apoptosis induced by a nitric oxide donor. Free Radic Res. 2000 Jun;32(6):507–514. doi: 10.1080/10715760000300511. [DOI] [PubMed] [Google Scholar]
- 50.Koesling D. Modulators of soluble guanylyl cyclase. Naunyn Schmiedebergs Arch Pharmacol. 1998 Jul;358(1):123–126. doi: 10.1007/pl00005232. [DOI] [PubMed] [Google Scholar]
- 51.Mujoo K, Sharin VG, Martin E, et al. Role of soluble guanylyl cyclase-cyclic GMP signaling in tumor cell proliferation. Nitric Oxide. 2010 Jan 1;22(1):43–50. doi: 10.1016/j.niox.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Townsend DM, Findlay VJ, Fazilev F, et al. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol Pharmacol. 2006 Feb;69(2):501–508. doi: 10.1124/mol.105.018523. Epub 2005 Nov 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shami PJ, Maciag AE, Eddington JK, et al. JS-K, an arylating nitric oxide (NO) donor, has synergistic anti-leukemic activity with cytarabine (ARA-C) Leuk Res. 2009 Nov;33(11):1525–1529. doi: 10.1016/j.leukres.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kitagaki J, Yang Y, Saavedra JE, Colburn NH, Keefer LK, Perantoni AO. Nitric oxide prodrug JS-K inhibits ubiquitin E1 and kills tumor cells retaining wild-type p53. Oncogene. 2009 Jan 29;28(4):619–624. doi: 10.1038/onc.2008.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J, Malavya S, Wang X, et al. Gene expression profiling for nitric oxide prodrug JS-K to kill HL-60 myeloid leukemia cells. Genomics. 2009 Jul;94(1):32–38. doi: 10.1016/j.ygeno.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kogias E, Osterberg N, Baumer B, et al. Growth-inhibitory and chemosensitizing effects of the glutathione-S-transferase-pi activated nitric oxide donor PABA/NO in malignant gliomas. Int J Cancer. 2011 Mar 31; doi: 10.1002/ijc.26106. [DOI] [PMC free article] [PubMed] [Google Scholar]