

Abstract
Activation of NAD(P)H oxidase has been reported to produce superoxide (O2 ·-) extracellularly as an autocrine/paracrine regulator or intracellularly as a signaling messenger in a variety of mammalian cells. However, it remains unknown how the activity of NAD(P)H oxidase is regulated in arterial myocytes. Recently, CD38-associated ADP-ribosylcyclase has been reported to use NAD(P)H oxidase product, NAD+ or NADP+ to produce cyclic ADP-ribose (cADPR) or nicotinic acid adenine dinucleotide phosphate (NAADP), which mediates intracellular Ca2+ signaling. The present study was designed to test a hypothesis that CD38/cADPR pathway as a downstream event exerts feedback regulatory action on the NAD(P)H oxidase activity in production of extra- or intracellular O2 ·-in mouse coronary arterial myocytes (CAMs). By fluorescent microscopic imaging, we simultaneously monitored extra- and intracellular O2 ·-production in wild-type (CD38+/+) and CD38 knockout (CD38-/-) CAMs in response to oxotremorine (OXO), a muscarinic type 1 (M1) receptor agonist. It was found that CD38 deficiency prevented OXO-induced intracellular but not extracellular O2 ·-production in CAMs. Consistently, the OXO-induced intracellular O2 ·-production was markedly inhibited by CD38 shRNA or CD38 inhibitor nicotinamide in CD38+/+ CAMs. Further, Nox4 siRNA inhibited OXO-induced intracellular but not extracellular O2 ·- production, whereas Nox1 siRNA attenuated both intracellular and extracellular O2 ·-production in CD38+/+ CAMs. Direct delivery of exogenous cADPR into CAMs markedly elevated intracellular Ca2+ concentration and restored intracellular O2 ·-production in CD38-/- CAMs. Functionally, CD38 deficiency or Nox1 siRNA and Nox4 siRNA prevented OXO-induced contraction in isolated perfused coronary arteries in CD38 WT mice. These results provide direct evidence that CD38/cADPR pathway importantly controls Nox4-mediated intracellular O2 ·-production and that CD38-dependent intracellular O2 ·-production is augmented via an autocrine manner of CD38-independent Nox1-derived extracellular O2 ·-production in CAMs.
Keywords: ADP-ribose, Ca2+, redox signaling, artery, vasoconstriction, coronary circulation
There is a large body of evidence that NAD(P)H oxidase is a major source of O2 ·-in the vascular cells [1-3] and that NAD(P)H oxidase is an important redox signaling enzyme to produce O2 ·- under physiological condition to regulate vascular functions[4]. However, it remains poorly understood how the activity of vascular NAD(P)H oxidase is regulated in response to a variety of physiological and pathological stimuli. Since NAD(P)H oxidase and its isoforms have been reported to be expressed in cell membrane, cytosol and different organelles and it produces and releases O2 ·- in different cellular compartments[3, 5-8]. Therefore, the temporospatial regulation of NAD(P)H oxidase activity is rather complicate. In this regard, recent studies have suggested that in the vasculature NAD(P)H oxidase and its isofroms can be detected in cell plasma membrane and different subcellular compartments including the lamellipodial focal complexes, membrane ruffles, caveolae and lipid rafts, endosomes, sarcoplasmic reticulum, and nucleus[3, 5-8]. This distribution of NAD(P)H oxidase may determine the concentrations of O2 ·- and related reactive oxygen species (ROS) within and outside vascular cells. In a recent study, we indeed demonstrated that in coronary arterial smooth muscle cells membrane-bound NAD(P)H oxidase produced O2 ·-toward the outside of these cells to regulate cell functions[9]. In other studies, NAD(P)H oxidase in cytosol and cell organelles such as lysosomes and sarcoplasmic reticulum contributes to intracellular levels of O2 - and ROS[3, 10-11]. This intracellular O2 - and ROS may serve as second messenger to regulate cellular activities [12]. Upon different stimuli or agonists, NAD(P)H oxidase may be activated in different cellular compartments to produce O2 - thereby regulating cell function. How such compartmental production and action of O2 - are regulated remains a mystery.
Among different regulatory pathways, CD38 as a multifunctional enzyme use NAD+ or NADP+ as substrate to produce related signaling molecules such as cyclic ADP-ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP), the products of its ADP-ribosylcyclase activity that are potent intracellular Ca2+ mobilizers [13]. Our recent studies have demonstrated that this CD38-ADP-ribosylcyclase-mediated signaling pathway importantly contributes to the vasomotor response. Since both NAD+ and NADP+ as substrates for the ADP-ribosylcyclase of CD38 are the products of NAD(P)H oxidase, it is plausible that there may be important interplay between both CD38-mediated Ca2+ and NAD(P)H oxidase-mediated redox signaling. Such interplay of both signaling pathways was indeed found to be present in different cells or tissues. For example, it has been shown that production of reactive oxygen species (ROS) was markedly reduced in embryonic fibroblasts from CD38-/- mice, suggesting that ROS production may be dependent upon an intact CD38 signaling pathway [14]. On the other hand, O2 ·- and ROS were reported to enhance CD38-ADP-ribosylcyclase activity to enhance Ca2+ signaling [15]. In the present study, we attempted to test whether CD38-ADP-ribosylcyclase signaling pathway participates in the regulation of NAD(P)H oxidase-dependent O2 ·-production extra- and intracellularly in coronary arterial myocytes (CAMs) in response to M1 agonist, a classical activator of CD38-ADP-ribosylcyclase in these cells[16]. We first examined whether intracellular O2 ·-production induced by the M1 receptor agonist OXO is altered in CAMs from CD38-/- mice. Then, we tested the role of NAD(P)H oxidase in CD38-associated intracellular and extracellular O2 ·-production in CAMs. We further determined whether induction of intracellular Ca2+ release by exogenous supplementation of cADPR could restore intracellular O2 ·-production in CD38-/- CAMs. Finally, we examined the role of CD38/cADPR-associated O2 ·-production in vasoconstrictor action of OXO in coronary arteries.
Materials and methods
Mice
Both CD38-/- and wild-type mice were purchased from the Jackson Laboratory. Male and female mice, 8wk old, were used in all experiments. All experimental protocols were reviewed and approved by the Animal Care Committee of Virginia Commonwealth University.
Isolation and culture of mouse CAMs
CAMs were isolated from CD38+/+ or CD38-/- mice as previously described [17]. In brief, mice were deeply anesthetized with intraperitoneal injection of pentobarbital sodium (25 mg/kg). The heart was excised with an intact aortic arch and immersed in a petri dish filled with ice-cold Krebs-Henseleit (KH) solution (20 mM HEPES, 128 mM NaCl, 2.5 mM KCl, 2.7 mM CaCl2, 1 mM MgCl2, 16 mM glucose, pH 7.4). A 25-gauge needle filled with Hanks’ buffered saline solution (HBSS) (in mM: 5.0 KCl, 0.3 KH2PO4, 138 NaCl, 4.0 NaHCO3, 0.3 Na2HPO4·7H2O, 5.6 D-glucose, and 10.0 HEPES, with 2% antibiotics) was inserted into the aortic lumen opening while the whole heart remained in the ice-cold buffer solution. The opening of the needle was inserted deep into the heart close to the aortic valve. The needle was tied in place with the needle tip as close to the base of the heart as possible. The infusion pump was started with a 20-ml syringe containing warm HBSS through an intravenous extension set at a rate of 0.1 ml/min for 15 min. HBSS was replaced with warm enzyme solution (1 mg/ml collagenase type I, 0.5 mg/ml soybean trypsin inhibitor, 3% BSA, and 2% antibiotic-antimycotic), which was flushed through the heart at a rate of 0.1 ml/min. Perfusion fluid was collected at 30-, 60-, and 90-min intervals. At 90 min, the heart was cut with scissors, and the apex was opened to flush out the cells that collected inside the ventricle. The fluid was centrifuged at 1,000 rpm for 10 min, the cell-rich pellets were mixed with the one of the media described below, and the cells were plated on 2% gelatin-coated six-well plates and incubated in 5% CO2-95% O2 at 37°C. Advanced DMEM with 10% FBS, 10% mouse serum, and 2% antibiotics was used for isolated smooth muscle cells. The medium was replaced three days after cell isolation and then once or twice each week until the cells grew to confluence. Mouse CAMs were identified according to their morphology, immunohistological staining, Western blot analysis of marker proteins, and flow cytometric characteristics (Fig 1). In these experiments, smooth muscle α-actin (α-SMA) and Dil-Ac-LDL (Biomedical Technology) were used as markers of smooth muscle cells and vascular endothelial cells.
Fig. 1.
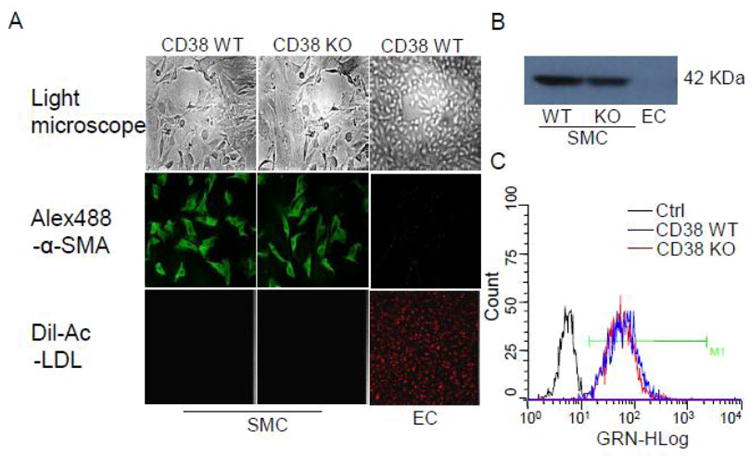
Coronary artery smooth muscle cell characterization. (A) The top panel showed cell morphology of CD38 WT and KO CAMs, and CAECs under light microscope. The middle panel showed that immunocytochemistry observes CAMs and CAECs, which were stained with α-SMA first antibody followed with Alex-488 conjugated second antibody. The low panel showed that Dil-Ac-LDL labels CAMs and CAECs. (B) Western blot assay for α-SMA protein expression in CAMs and CAECs. (C) Flowcytometry assay for the purity of CAMs which were stained with α-SMA antibodies followed with Alex-488 conjugated second antibody. Black peaks represent control of CD38 WT and KO CAMs without staining with α-SMA first antibody. Blue and Red peak represents positive stain of α-SMA of CD38 WT and OK CAMs. M1, cutoff point for estimating percentage of positive stain.
Simultaneous monitoring of O2 ·-production inside and outside CAM
Intracellular and extracellular O2 ·-production were simultaneously monitored by fluorescence microscopy as we previously described [9]. O2 ·-oxidizes dihydroethidium (DHE) to form membrane-impermeable ethidium, which binds to DNA and forms a strong red fluorescence. In this regard, intracellular O2 ·-production is detected when O2 ·-oxidizes intracellular DHE to ethidium which binds to nuclear DNA, whereas extracellular O2 ·-oxidizes DHE in the solution to ethidium which binds to Matrigel (BD Biosciences)-trapped extracellular DNA. In these experiments, freshly isolated CAMs (102 cells/well) were seeded into a 16-well chamber slide with a transparent glass bottom (LAB-TEK) and incubated overnight. On the day of the experiment, the CAMs were washed twice with Hanks’ buffer, and 40μl of salmon testes DNA solution (7.5 mg/ml; Sigma) were mixed with 40 μl of Matrigel solution (at 4°C) and then carefully loaded onto the top of the CAMs. After 5 min at room temperature, the gel was polymerized and exogenous DNA was immobilized around these cells. The CAMs with Matrigel were then overlayed with Hanks’ buffer containing 250 μM DHE. After a 60 min loading of DHE into the CAMs, the chamber slide was mounted on the stage of a fluorescent microscope, which we routinely used for high-speed wavelength-switching imaging acquisition and recording. For both intracellular and extracellular DHE oxidizing signals, a ratio of ethidium-DNA to DHE signals was recorded at an excitation of 480 nm and an emission of 610 nm. The DHE fluorescence signal was detected at an excitation of 380 nm and an emission of 445 nm. The ratio of ethidium-DNA to DHE fluorescent signals was recorded to represent O2 ·-levels. The ratiometric assay increases the sensitivity for O2 ·-detection and avoids an artifact in assays from cell volume changes or contractions during agonist stimulation. For each experiment, 8–10 CAMs were monitored simultaneously and an average value was used for statistical analysis. OXO (80 μM) was added to activate its receptor and consequently stimulate O2 ·-production. Nicotinamide (6 mM) was pre-incubated with the cells for 30 min, or CD38 shRNA was transferred into wild-type CAMs, and then the O2 ·-producing response to OXO was determined.
Confocal microscopic detection of extracellular O2 - production
To further determine extracellular O2 -., CAMs were prepared as described above for ethidium-DNA trapping imaging [9]. In brief, 0.5 ml of Matrigel was mixed with 25 μl of OxyBURST H2HFF Green BSA (10 μg/ml), a BSA-conjugated molecular probe and directly reacts with H2O2, and then used to evenly coat the 35 mm×10-mm cell culture dish. Freshly isolated CAM suspension (60 μl, 3×106/ml ) was loaded on the top of the ice-cold Matrigel matrix, and then the culture dish was gently tapped three or four times to distribute cells across the surface of the gel. After polymerizing the gel at room temperature for 5 min, 2 ml of Krebs-Ringer phosphate buffer, which contains PBS, pH 7.4, with 1.0 mM Ca2+, 1.5 mM Mg2+, and 5.5 mM glucose, were added. Confocal fluorescent microscopic images were acquired by an Olympus Fluoview system (version 4.2, FV300), consisting of an Olympus BX61WI inverted microscope with an Olympus Lumplan F1×60, 0.9 numerical aperture, water-immersion objective. A single z-section was taken or 0.1μm sections were obtained through the cell with excitation and emission wavelengths of 488 and 530 nm, respectively for OxyBURST. Real-time microscopic fluorescence images were acquired every 1 or 2 min under control conditions and after different treatments. This OxyBURST H2HFF Green BSA detects H2O2, which mirrors O2 ·-production outside the cells because BSA cannot enter the cells [18].
ESR detection of extracellular and intracellular O2 - level
For the detection of the total and extracellular O2 ·-production dependent on NAD(P)H oxidase, the proteins from CAMs extracted using sucrose buffer or gently collected cells were and resuspended with modified Krebs–Hepes buffer containing deferoximine (100 μmol/L; Sigma, St. Louis, MO, USA) and diethyldithiocarbamate (5 μmol/L; Sigma). 1×106 of intact cells or their homogenates were subsequently mixed with 1mM of the cell permeable O2 -.-specific spin trap, 1-hydroxy-3-methoxycarbonyl- 2,2,5,5- tetramethylpyrrolidine (CMH), and substrate NAD(P)H in the presence or absence of manganese-dependent superoxide dismutase (SOD) (200 U/ml; Sigma). The mixture was loaded into glass capillaries and immediately analyzed for O2 ·-production kinetically for 10 min using a Miniscope MS200 ESR spectrometer (Magnettech Ltd., Berlin, Germany) as we described [19]. The SOD-inhibitable fraction of the signal in intact cells or homogenates reflects the extracellular or total O2 ·- levels, respectively. The difference between the total and extracellular O2 -.level was used to reflect intracellular O2 ·-level. The ESR settings were as follows: biofield, 3350; field sweep, 60 G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 3 G; 4096 points of resolution; receiver gain, 20 for tissue and 50 for cells [20]. The results were expressed as fold changes relative to control.
Fluorescence measurement of [Ca2+]i and cADPR delivery in CAMs
Intracellular Ca2+ responses to OXO or cADPR were determined using a fluorescence image analysis system with the Ca2+ indicator, fura-2, as described previously [15, 21]. Ca2+-free HBSS including 1 mM EGTA was used for Ca2+ measurement to ensure the Ca2+ response was solely derived from intracellular Ca2+ store release rather than from extracellular Ca2+ influx. Administration of cADPR (200μM) into the cells was carried out by wrapping this cell-impermeable nucleotide in Optison (Perflutren protein-type A microspheres) and delivering by ultrasound treatment as detailed previously [22-24].
Isolated small artery for tension recording
Small ventricular septal arteries (~150μm inner diameter) were dissected [25] and then mounted in a Multi Myograph 610M (Danish Myo Technology, Aarhus, Denmark) for recording of isometric wall tension [26-27] after 30 min of equilibration in physiological salt solution (PSS; pH 7.4) containing (in mM) 119 NaCl, 4.7 KCl, 1.6 CaCl2, 1.17 MgSO4 1.18 NaH2PO4, 2.24 NaHCO3, 0.026 EDTA, and 5.5 glucose at 37°C bubbled with a gas mixture of 95% O2 and 5% CO2. After the basic tension was set, the dose effect of OXO alone (0, 20, 40, 60, 80 and 100μM) on the tension changes of wild type and CD38-/- septal arterial wall were measured with or without the presence of different inhibitors, including 8-Br-cADPR (30 μM), or Nox1 and Nox4 siRNA. Ultrasound microbubble technology has been used to transfect mouse coronary artery smooth muscle with Nox1 and Nox4 siRNA as we described previously [22-23, 28-29]. In brief, after isolation, coronary arteries were transferred to a water-jacketed perfusion chamber and cannulated with two glass micropipettes at their in situ length and with PSS buffer in the lumen until transfection. 20 μg siRNA was mixed in 100 μl Optison (Amersham) and kept for 30 seconds at 37°C. Then the RNA-Optisim solution was perfused within the lumen of arteries. The arteries were treated with ultrasound for 1 minutes through a 6-mm diameter probe in the chamber with an input frequency of 1MHz, an output intensity of 1.0-2.0 W/cm2 and a pulse duty ratio of 10-50%(Rich-Mar). After transfection, the arteries were removed from glass micropipettes and incubated in DMEM medium for 24-48 hours at 37°C to knockdown Nox1 and Nox4.
Statistics
Data are presented as means ± SE. Significant differences between and within multiple groups were examined using ANOVA for repeated measures, followed by Duncan’s multiple-range test. A Students’ t-test was used to detect significant differences between two groups. P<0.05 was considered statistically significant.
Results
Coronary artery smooth muscle cell characterization
As shown on the top of Fig. 1A, CAMs formed longitudinal bands of parallel cells and became thinner at a high density. Immunocytochemistry in the middle of Fig 1A showed that CD38 WT and KO CAMs were stained positively with anti-α-SMA antibody followed by Alex-488 conjugated second antibody. However, no staining was found in coronary arterial endothelial cells (CAECs). Similarly, Western blot analysis showed that α-SMA was detected only from CD38 WT and KO CAMs but not endothelial cells (Fig. 1B). Moreover, Dil-Ac-LDL, vascular endothelial cells marker, only labeled endothelial cells to produce red fluorescence on the bottom of of Fig 1A. To further ascertain the purity of CAMs, the percent of α-SMA positive CAMs was assessed with flowcytometry. The results showed that the purity was 97.6% for α-SMA-stained CD38 WT CAMs and 98.0% for CD38 KO CAMs (Fig. 1C).
Lack of intracellular, but not extracellular O2 ·-production in CAMs from CD38-/- mice
To determine whether CD38 has a role in agonist-induced O2 ·-production in CAMs, we simultaneously monitored fluorescence intensity of ethidium-DNA complex within CD38+/+ and CD38-/- CAMs and in Matrigel around these cells. Fig. 2A shows representative fluorescence images before and 50 min after a CAM received OXO at a concentration of 80 μM. The dynamic changes in ratios of intracellular and extracellular ethidium-DNA fluorescent signals to DHE fluorescent signals were acquired and summarized in Fig. 2B. We found that the OXO induced increases in extracellular O2 ·-levels (becoming red) in both CD38+/+ and CD38-/- CAMs (Fig. 2A). In addition, CD38 deficiency has no effect on dynamic responses of extracellular ethidium-DNA fluorescence in CAMs (Fig. 2B). In contrast, OXO-induced increases in O2 ·-levels inside cells were markedly attenuated in CD38-/- CAMs compared to those in CD38+/+ CAMs. Therefore, our findings suggest that CD38 is key for agonist-induced intracellular O2 ·-production in CAMs.
Fig. 2.
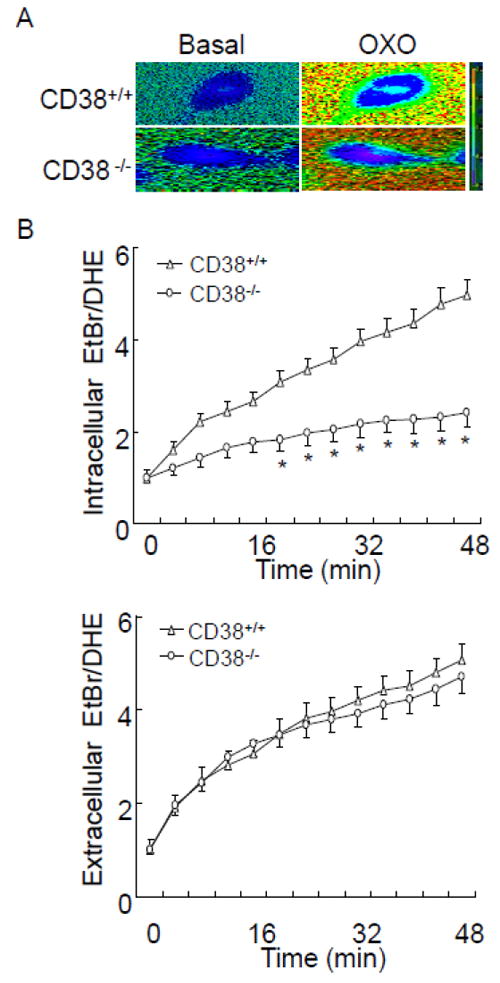
Fluorescent microscopic imaging analysis to simultaneously monitor the OXO-induced O2 ·-production inside and outside single coronary arterial myocyte (CAM) from CD38+/+ and CD38-/- mice. (A) Typical time-dependent increase in oxidized dihydroethidium (DHE) fluorescence ratio [ethidium bromide (EtBr)/DHE] image inside and outside a CAM. Red fluorescence indicates O2 ·-level higher than blue background. (B, C) Summarized digitized data showing the spatiotemporal pattern of O2 ·-increase inside and outside CD38+/+ and CD38-/- CAM during OXO (80μM) stimulation. (n=6, *P<0.05 vs. CD38+/+ CAM)
We also examined whether OXO induces similar extracellular ROS production between CD38+/+ and CD38-/- CAMs using OxyBURST Green, which is a BSA-conjugated probe and directly reacts with H2O2. Because O2 -., rapidly dismutate into H2O2, OxyBURST Green fluorescence intensity in extracellular Matrigel reflects extracellular O2 -.-derived ROS level. Fig. 3A shows representative confocal fluorescent images in CAMs. OXO stimulation induced similar increases in OxyBURST green fluorescence in CD38+/+ and CD38-/- CAMs. As summarized in Fig. 3B, CD38 deficiency has no effect on OXO-induced dynamic responses of OxyBURST green fluorescence in CAMs.
Fig. 3.
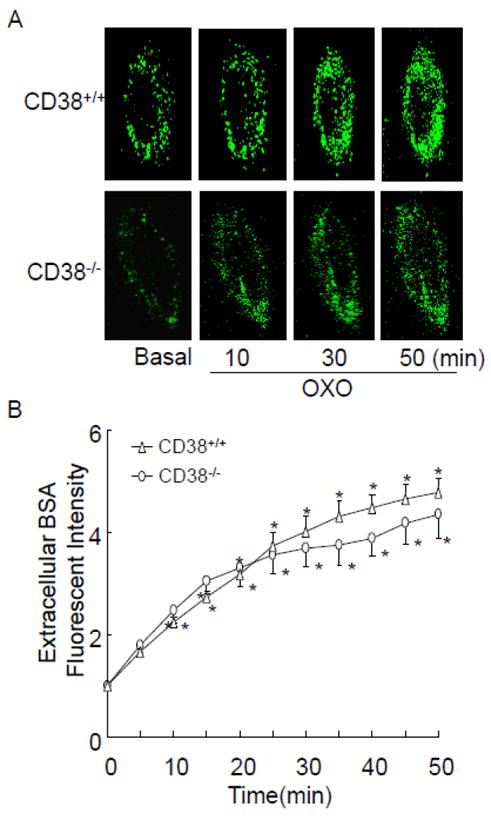
Confocal microscopic analysis to monitor O2 ·-outside CAM from CD38+/+ and CD38-/- mice. (A) Representative images showing as extracellular O2 ·-production as detected by OxyBURST H2HFF Green BSA trapped H2O2 outside a single CAM during OXO (80 μM) stimulation. (B) Summarized digitized data showing the time-dependent O2 ·-increases outside CD38+/+ and CD38-/- CAMs. (n=6, *P<0.05 vs. basal condition)
Inhibition of CD38/cADPR signaling pathway blocked intracellular O2·-production without effect on extracellular O2 ·- production in CAMs from CD38+/+ mice
CD38 is a key enzyme for production and metabolism of cADPR in vascular cells. We then examined whether CD38/cADPR is involved in OXO-induced intracellular O2 ·-production. As shown in Fig. 4, inhibiting CD38-mediated cADPR production with nicotinamide, or knockdown of CD38 via RNA interference, inhibited OXO-induced increases in intracellular but not extracellular O2 ·-production in CAMs.
Fig. 4.
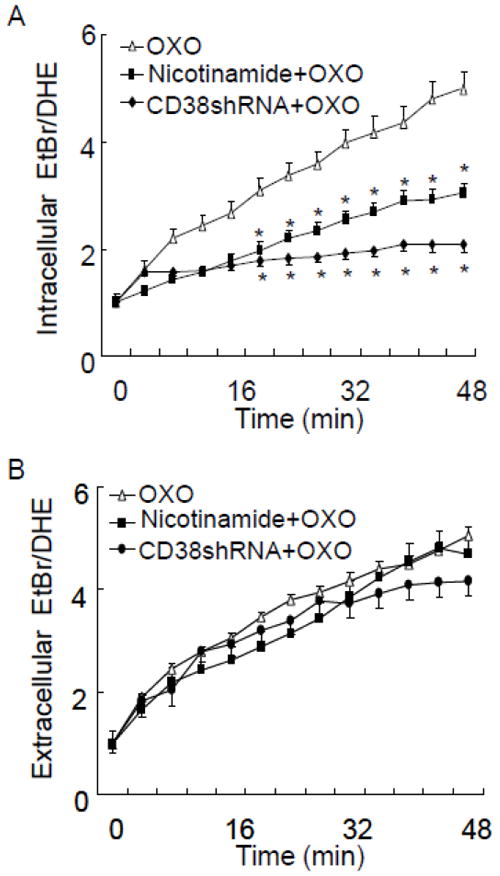
Effect of CD38 gene silencing or inhibition of ADP-ribosyl cyclase activity of CD38 on OXO-induced intracellular (A) and extracellular (B) O2 ·-production in CD38+/+ CAMs. CD38 gene was silenced by CD38 shRNA and CD38 cyclase activity was inhibited by nicotinamide (Nico, 6 mM). CD38+/+ CAM was treated with OXO (80μM) for 50 min in the presence or absence of CD38 shRNA or nicotinamide. (n=6, *P<0.05 vs. OXO treated CAM)
By using the ESR spectrometry, we further determined the role of CD38/cADPR in OXO-induced intracellular O2 ·-production in CAMs. Fig. 5A shows representative changes in SOD-inhibitable ESR spectrometric curve recorded under control conditions and after OXO stimulation. Summarized data showed that OXO significantly increased production of O2 ·-dependent on NAD(P)H oxidase outside and inside CD38+/+ CAMs. Inhibition of CD38/cADPR by CD38 inhibitor nicotinamide or cADPR antagonist, 8-Br-cADRP attenuated OXO-induced SOD-sensitive O2 ·-production inside but not outside CD38+/+ CAMs. Similarly, knockdown of CD38 by CD38shRNA or genectic deficiency of CD38 inhibited intracellular but not extracellular SOD-sensitive O2 ·-production in CAMs (Fig. 5B).
Fig. 5.
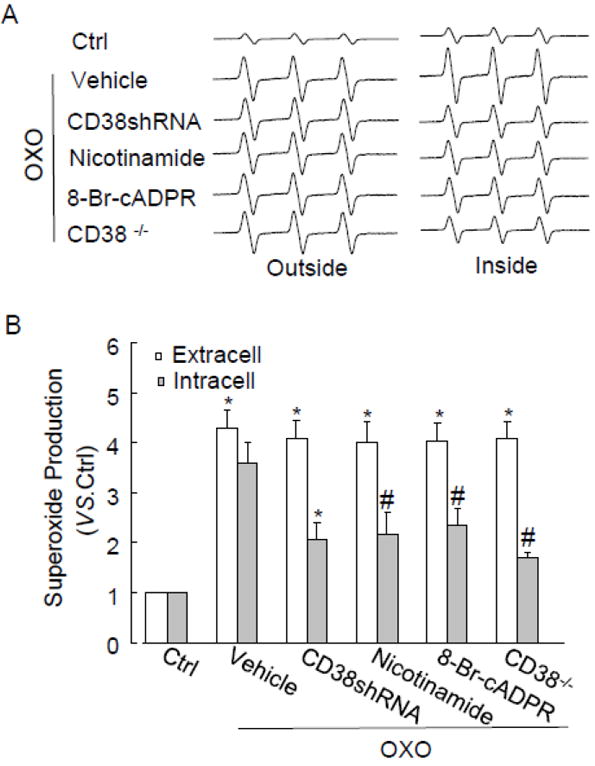
ESR spectrometric analysis of O2 ·-production in CAMs stimulated by OXO. (A) Representative ESR spectrographs of O2 ·-trapped by 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine. (B) Summarized data showing the effects of CD38 shRNA, nicotinamide (Nico, 6 mM), 8-Br-cADPR (30 μM) or CD38 deficiency on OXO-induced O2 -. production inside or outside CD38+/+ CAMs (n=6, *P<0.05 vs. unstimulated samples (control); #P<0.05 vs. samples treated with OXO alone)
Regulation of NAD(P)H oxidase isoform activity by CD38-cADPR-mediated signaling in CAMs
We previously reported that Nox4 only mediates intracellular O2 ·-production, whereas Nox1 is involved in both extracellular and intracellular O2 ·-production in bovine CAMs [9]. To further examine the mechanism of CD38-associated intracellular O2 ·-production, we determined ethidium/DHE fluorescence in mouse CAMs transfected with Nox4 siRNA or Nox1 siRNA. Nox1 and Nox4 siRNA have been demonstrated to inhibit the protein expression of Nox1 and Nox4 by 66.5% and 77.4%, respectively (Fig. 6). It was found that Nox4 siRNA or Nox1 siRNA significantly inhibited intracellular O2 ·-production in CD38+/+ CAMs (Fig. 7A), whereas intracellular O2 ·-production was not further decreased by Nox4 siRNA or Nox1 siRNA in CD38-/- CAMs (Fig. 7C). Moreover, Nox1 siRNA but not Nox4 siRNA blocked extracellular O2 ·-production in both CD38+/+ and CD38-/- CAMs (Fig. 7B and D).
Fig. 6.
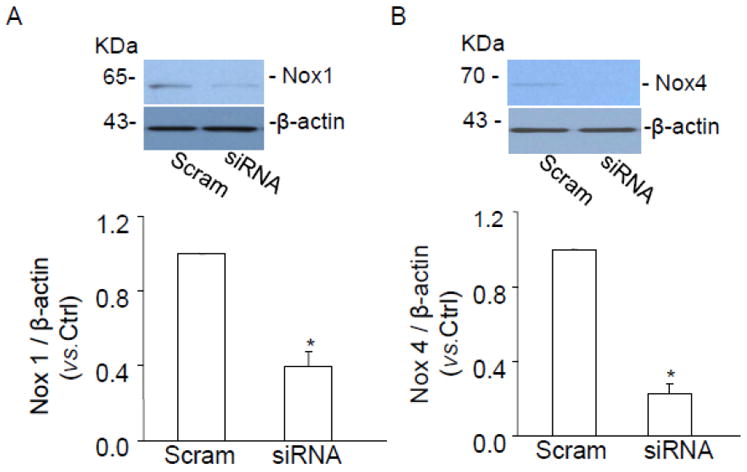
Western blot gel document presents the level of Nox1 (A), Nox4 (B) and β-actin from mouse CAMs before and after Nox1 and Nox4 siRNA. Summarized results shows normalized intensity ratio of Nox1 and Nox4 to β-actin. Nox1 and Nox4 protein expression was significantly inhibited when Nox1 and Nox4 siRNA was transfected into mouse CAMs. (n=3, *P<0.05 vs. scramble RNA transfected CAMs).
Fig. 7.
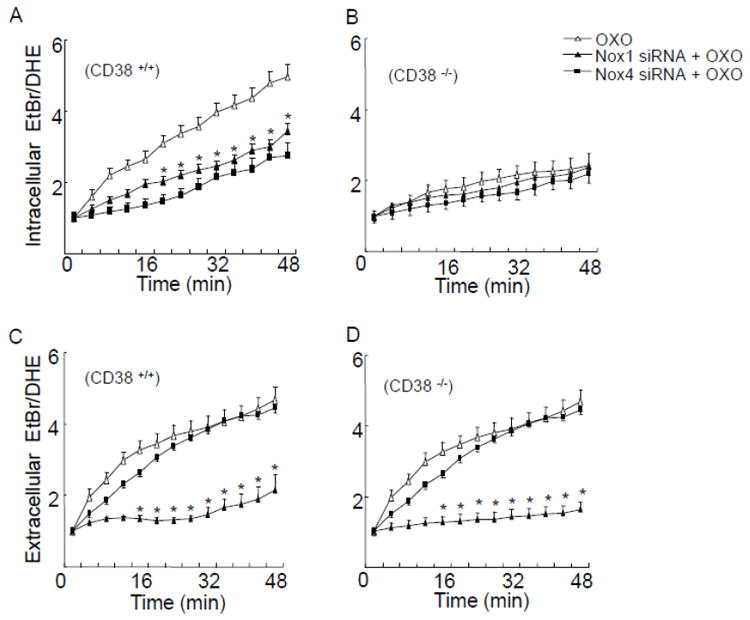
Effects of NAD(P)H oxidase inhibition on OXO-induced O2 ·-production. (A, B) Transfected Nox1 and Nox4 siRNA suppressed O2 ·-production inside CD38+/+ CAM, however, Nox1 and Nox4 siRNA had no significant effects on O2 -. production inside CD38-/- CAM. (C, D) O2 ·-production outside CD38+/+ and CD38-/- cells was inhibited by pretreatment with Nox1 siRNA, whereas extracellular O2 ·-production was not affected by Nox4 siRNA. (n=6, *P<0.05 vs. CAM treated with OXO alone).
Exogenous cADPR restored intracellular O2·-production without effect on extracellular O2·-production in CD38-/- CAMs
To confirm that failure in cADPR/Ca2+ response during OXO stimulation contributes to the defect in intracellular O2 ·-production in CD38-/- CAMs, we determined whether exogenous supplementation of cADPR restores Ca2+ response and O2 ·-production. As shown in Fig. 8A and B, exogenous cADPR triggered similar increases in intracellular Ca2+ concentration in CD38+/+ (from 184 nM to 321 nM) and CD38-/- CAMs (from 175 nM to 325 nM). Moreover, exogenous cADPR induced similar intracellular but not extracellular O2 ·-production in CD38+/+ and CD38-/- CAMs.
Fig. 8.
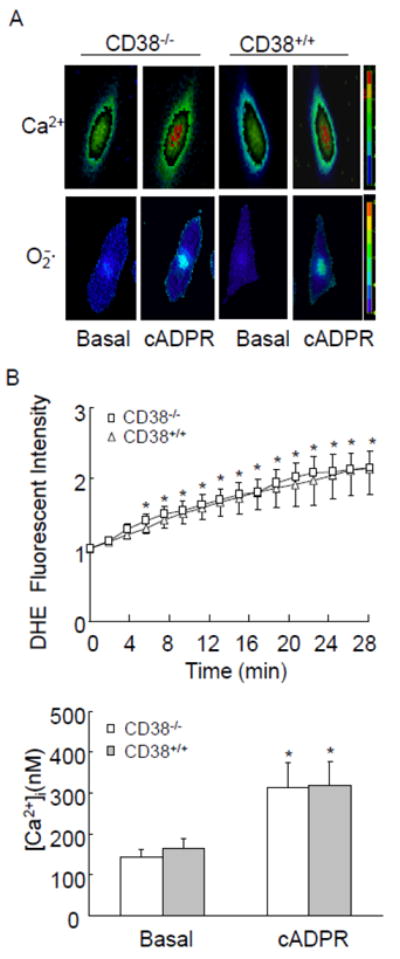
Effects of exogenous cADPR on Ca2+ release and O2 ·-production in CAMs. (A) Typical fluorescent images of CD38+/+ and CD38-/- CAMs stained by DHE or Flur-2 before (Basal) and after cADPR (200 μM) introduction. (B, C) summarized data showing that exogenous cADPR-induced increases in intracellular O2 ·-and Ca2+ concentration inside CD38+/+ and CD38-/- CAMs. (n=6, * P<0.05 compared with basal levels)
Vasoconstrictor response of coronary arteries to M1 receptor activation in CD38+/+ and -/- mice
To examine the functional relevance of CD38-cADPR signaling-mediated regulation of O2 ·-production, we examined the vasoconstrictor response of isolated perfused coronary arteries from both CD38+/+ and CD38-/- mice to OXO, a typical M1 receptor agonist. As shown in Fig. 9A, OXO dose-dependently induced vasoconstriction in coronary arteries from CD38+/+ mice with a maximum response of 56.9 ± 12.4% at the dose of 100 μM. This OXO-induced vasoconstriction response was significantly reduced in CD38-/- arteries with a maximum response of 14.5 ± 2.9%. Blockade of cADPR signaling using cADPR antagonist 8-Br-cADPR or inhibition of Nox1 or Nox4 by their siRNA significantly attenuated OXO-induced vasoconstriction in CD38+/+ arteries (Fig. 9B). Moreover, Nox1 or Nox4 siRNA had no effect on OXO-induced residual vasoconstrictor response in CD38-/- arteries (Fig. 9C).
Fig. 9.
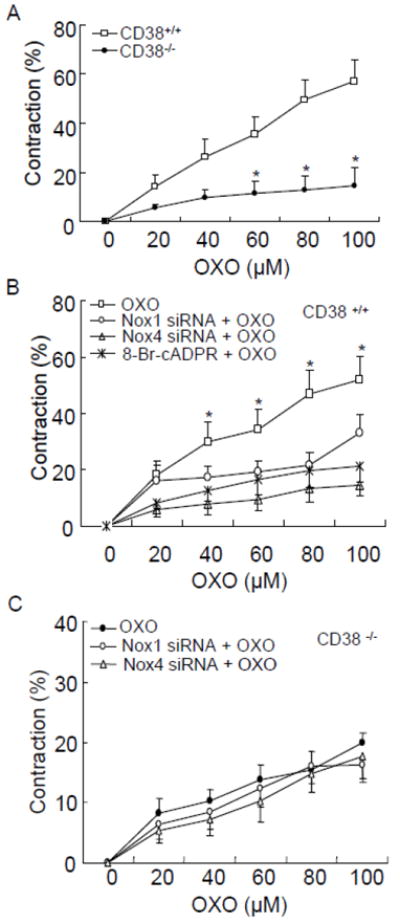
Effects of CD38 deficiency (A), NAD(P)H oxidase inhibition or cADPR/Ca2+ blockade (B, C) on OXO-induced vasoconstriction in coronary arteries. cADPR antagonist, 8-Br-cADPR (30 μM), or Nox1 and Nox4 siRNA were used. (n=3, *P<0.05 compared with CD38+/+ artery)
Discussion
The present study has demonstrated that CD38/cADPR pathway is involved in OXO-induced intracellular but not extracellular O2 ·-production in CAMs. CD38-regulated intracellular O2 ·-production is dependent on intracellular cADPR/Ca2+ signaling and NAD(P)H oxidase activity. Further, this CD38-mediated signaling can be augmented by membrane-bound NAD(P)H oxidase-derived extracellular O2 -.. These findings reveal that extracellular O2 - enhances CD38/cADPR-regulated intracellular O2 - production in CAMs upon M1 receptor activation.
Using fluorescent imaging with several ROS measurement methods, we first compared the spatiotemporal pattern of O2 - production in CD38+/+ and CD38-/- CAMs by using a sensitive and dynamic measurement of O2 - production both inside and outside cells. This method was developed in our laboratory by modifying a high-speed wavelength-switching fluorescent microscopic imaging system that we used for simultaneous monitoring of Ca2+ and nitric oxide [30]. With the help of Matrigel to trap DNA around CAMs, we were able to simultaneously measure extracellular and intracellular DHE-oxidizing signals, which represent O2 - levels. In the present study, we used this assay and found that the OXO-induced M1-receptor activation stimulated O2 ·-production both outside and inside CD38+/+ CAMs with a spatiotemporal pattern that more rapid extracellular increase in O2 - levels was followed by a slow increase in intracellular O2 - levels, which was similar to those previously observed in bovine CAMs [9]. More importantly, the present study showed that OXO-induced increases in intracellular O2 ·-levels were abolished in CD38-/- CAMs or in CD38+/+ CAMs transfected with CD38 shRNA. In contrast, no difference in OXO-induced extracellular O2 ·-productions was found between CD38+/+ and CD38-/- CAMs. These findings indicate that CD38 is essentially involved in OXO-induced intracellular O2 ·-production in CAMs. Consistent with our findings, a recent study in mouse embryonic fibroblast has shown that production of ROS in CD38-/- cells were markedly reduced compared with CD38+/+ cells during hypoxia/reoxygenation [14]. Based on these observations, we believe that M1 receptor activation results in at least two distinct pathways for O2 ·-production in CAMs, namely, CD38-dependent intracellular O2 ·-production, and CD38-independent extracellular O2 ·-production.
To further determine the contribution of CD38/cADPR pathway to intracellular O2 ·-production, we examined whether OXO-induced intracellular O2 ·-increases can be blocked in wild type CAMs by two widely used cADPR signaling inhibitors - nicotinamide (an inhibitor for ADP-ribosyl cyclase activity of CD38 which is responsible for synthesis of cADPR) and 8-Br-cADPR (a cADPR antagonist). Fluorescent imaging analysis and ESR studies demonstrated that, in the presence of nicotinamide, OXO-induced O2 ·-production inside CD38+/+ CAMs was significantly reduced. In addition, blockade of cADPR pathway by 8-Br-cADPR inhibited OXO-induced intracellular O2 ·-production as detected by ESR. These findings support the view that intracellular O2 ·-is associated with increases in ADP-ribosyl cyclase activity of CD38 and synthesis of cADPR in wild type CAMs during M1 receptor activation.
NAD(P)H oxidase has been reported to be a major source of ROS in the vasculature [2]. The vascular NAD(P)H oxidase has characteristics of phagocyte NAD(P)H oxidase, which is composed of two transmembrane-bound catalytic proteins of gp91phox and p22phox, and three cytosolic subunits of p47phox, p67phox and p40phox. In addition to gp91phox named as Nox2, some other homologues of gp91phox such as Nox1, Nox4 and Nox5 were identified in the vascular cells such as endothelial and smooth muscle cells [1]. It has been shown that Nox2 localizes in plasma membranes as well as in intracellular compartments and activation of Nox2 causes O2 ·-production in response to a variety of agonists such as angiotensin II in vascular cells [9]. In addition to Nox2, recent studies have indicated that Nox4 is primarily responsible for intracellular O2 ·-production localized in different organelles of vascular smooth muscle cells including the SR, whereas Nox1 mainly produces extracellular O2 ·-[3, 9, 31]. In this regard, Nox1 has been shown to be enriched in membrane fraction and Nox4 is predominately found in the intracellular compartments such as the SR of vascular cells [3, 5]. In the present study, the use of Nox4 siRNA to silence this gene significantly attenuated OXO-induced intracellular O2 ·-production in CD38+/+ CAMs, but it did not have further effects in CD38-/- CAMs. These results suggest that CD38/cADPR-regulated intracellular O2 ·-production is primarily dependent on Nox4 activity inside CAMs. However, introduction of siRNA to silence Nox1 gene not only significantly attenuated OXO-induced intracellular O2 ·-production, but also extracellular O2 ·-in CD38+/+CAMs, suggesting that Nox1 may contribute to the production of both intra- and extracellular O2 -.. It has been well documented that the production of cADPR is increased by oxidants, which is dependent on a redox regulation of ADP ribosyl cyclase activity of CD38 possibly via enzyme dimerization causing enhancement of its activity [15, 32-33]. As we demonstrated in our previous study, extracellular O2 ·-serves as an autocrine to enhance CD38-dependent intracellular O2 ·-production in response to M1 receptor activation. This action of Nox1-dependent extracellular O2 ·-production may be associated with redox activation of ADP ribosyl cylase activity of CD38.
Another important finding of the present study was that delivery of exogenous cADPR into cells resulted in intracellular Ca2+ release and restored intracellular O2 ·-production in CD38-/- CAMs. This finding provides direct evidence that cADPR-induced intracellular Ca2+ mobilization is coupled with activation of intracellular NAD(P)H oxidase in CAMs. Previous studies have shown that Nox4 activity is sensitive to intracellular Ca2+ regulation associated with cADPR-induced Ca2+ release from ryanodine receptor, a Ca2+ channel on the SR membrane [3, 10, 34]. In the present study, Nox4 was also demonstrated to primarily contribute to OXO-induced intracellular O2 ·-production. Therefore, it is possible that CD38-derived cADPR production induces Ca2+ mobilization from the SR and thereby results in local activation of Nox4 on the SR. Indeed, silencing Nox4 gene was also found to substantially attenuate intracellular O2 ·-production induced by direct delivery of cADPR into the CAMs from wild type mice. Moreover, exogenous cADPR did not increase extracellular O2 ·-levels in these CAMs, indicating that cADPR-Ca2+ signaling does not participate in the activation of Nox1 or Nox2 and consequent extracellular O2 ·-production in CAMs. This further confirms the findings obtained from CAMs with CD38 deficiency or blockade of cADPR signaling that OXO-induced extracellular O2 ·-production remains to occur. Thus, it is possible that Nox1 activation is an upstream event of cADPR signaling, but the Nox4 activation is a downstream event of cADPR signaling in CAMs. However, a recent study has shown that Nox1 mRNA level was markedly lower in cultured mouse embryonic fibroblasts from CD38-/- mice compared to wild-type cells during hypoxia/reoxygenation and that the transcription of Nox1 mRNA was up-regulated by calcium ionophore ionomycin in these wild-type cells [14]. It seems that CD38/cADPR-Ca2+ signaling also controls Nox1-derived extracellular O2 ·-production in mouse embryonic fibroblasts by alternation Nox1 expression. Such inconsistency may be due to difference in treatment of agonists or stimuli. The previous study exposed fibroblasts to hypoxia for 48 hours or to ionomycin for 24 hours. The present study administrated OXO for no more than 1 hour and therefore the results reveal a rapid extracellular and intracellular O2 ·-production response to M1-receptor activation (as early as in 5 minutes) in mouse CAMs. In addition, different cell types and species may make a different response in this Nox1 regulation during acute or chronic treatments.
Using isolated small coronary arterial preparations, we also examined the functional significance of Nox-derived extracellular O2 ·-in CD38/cADPR-mediated coronary arterial response to agonists. It was demonstrated that isolated small coronary arteries from CD38 deficient mice or from CD38+/+ mice with inhibition of cADPR signaling lacked OXO-induced vasoconstriction. This provides strong evidence that CD38/cADPR signaling is essential for the coronary arterial constrictor response to OXO. Nox1 or Nox4 siRNA can disrupt the assembly of Nox1 or Nox4 with cytosolic NAD(P)H oxidase subunits to form an integrated enzyme causing failure in NAD(P)H oxidase activation. The present study demonstrated that Nox1 or Nox4 siRNA substantially blocked OXO-induced vasoconstriction in CD38+/+ coronary arteries, suggesting that generation of both extracellular and intracellular O2 ·-is involved in OXO-induced vasoconstriction. However, in CD38-/- coronary arteries, Nox1 or Nox4 siRNA had no further effect on OXO-induced vasoconstriction. This suggests that CD38 deficiency or cADPR signaling blockade inhibits OXO-induced vasoconstriction by the similar mechanism to Nox1 or Nox4 siRNA, namely, inhibition of NAD(P)H oxidase. With respect to the mechanism by which Nox-derived ROS in mediating vasoconstriction, we previously demonstrated that extracellularly produced O2 .- activated cADPR production and thereby mobilized intracellular Ca2+ from the SR, which produced or enhanced coronary vasoconstriction [15, 21]. In addition, we also found that intracellular O2 .- increase may directly enhance ryanodine receptor activity on the SR and thereby increase Ca2+ release response, resulting in enhanced vasoconstriction [15, 21].
In summary, the present study demonstrated that during M1 receptor activation, CD38 is essential for intracellular O2 ·-production via cADPR/Ca2+-mediated activation of Nox4. Moreover,Nox1-derived extracellular O2 ·-enhances cADPR/Ca2+-mediated intracellular O2 ·-production in CAMs and vasoconstriction in coronary arteries. These data provide new insights into the regulation of NAD(P)H oxidase-mediated O2 ·-production by CD38-cADPR signaling pathway.
Highlights.
CD38/cADPR pathway controls Nox4-mediated intracellular O2 ·- production.
Nox1-derived extracellular O2 ·- production is independent on CD38/cADPR pathway.
Nox1-derived O2 ·- augments CD38/cADPR signaling and consequent Nox4-dependent O2 ·-.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 2.Mohazzab KM, Kaminski PM, Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol. 1994;266:H2568–2572. doi: 10.1152/ajpheart.1994.266.6.H2568. [DOI] [PubMed] [Google Scholar]
- 3.Yi XY, Li VX, Zhang F, Yi F, Matson DR, Jiang MT, Li PL. Characteristics and actions of NAD(P)H oxidase on the sarcoplasmic reticulum of coronary artery smooth muscle. Am J Physiol Heart Circ Physiol. 2006;290:H1136–1144. doi: 10.1152/ajpheart.00296.2005. [DOI] [PubMed] [Google Scholar]
- 4.Griendling KK, Ushio-Fukai M. NADH/NADPH Oxidase and Vascular Function. Trends Cardiovasc Med. 1997;7:301–307. doi: 10.1016/S1050-1738(97)00088-1. [DOI] [PubMed] [Google Scholar]
- 5.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu RF, Xu YC, Ma Z, Nwariaku FE, Sarosi GA, Jr, Terada LS. Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol. 2005;171:893–904. doi: 10.1083/jcb.200507004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang AY, Yi F, Zhang G, Gulbins E, Li PL. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension. 2006;47:74–80. doi: 10.1161/10.1161/01.HYP.0000196727.53300.62. [DOI] [PubMed] [Google Scholar]
- 9.Zhang G, Zhang F, Muh R, Yi F, Chalupsky K, Cai H, Li PL. Autocrine/paracrine pattern of superoxide production through NAD(P)H oxidase in coronary arterial myocytes. Am J Physiol Heart Circ Physiol. 2007;292:H483–495. doi: 10.1152/ajpheart.00632.2006. [DOI] [PubMed] [Google Scholar]
- 10.Zhang F, Jin S, Yi F, Xia M, Dewey WL, Li PL. Local production of O2- by NAD(P)H oxidase in the sarcoplasmic reticulum of coronary arterial myocytes: cADPR-mediated Ca2+ regulation. Cell Signal. 2008;20:637–644. doi: 10.1016/j.cellsig.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin S, Yi F, Li PL. Contribution of lysosomal vesicles to the formation of lipid raft redox signaling platforms in endothelial cells. Antioxid Redox Signal. 2007;9:1417–1426. doi: 10.1089/ars.2007.1660. [DOI] [PubMed] [Google Scholar]
- 12.Das DK, Maulik N, Sato M, Ray PS. Reactive oxygen species function as second messenger during ischemic preconditioning of heart. Mol Cell Biochem. 1999;196:59–67. [PubMed] [Google Scholar]
- 13.Cancela JM. Specific Ca2+ signaling evoked by cholecystokinin and acetylcholine: the roles of NAADP, cADPR, and IP3. Annu Rev Physiol. 2001;63:99–117. doi: 10.1146/annurev.physiol.63.1.99. [DOI] [PubMed] [Google Scholar]
- 14.Ge Y, Jiang W, Gan L, Wang L, Sun C, Ni P, Liu Y, Wu S, Gu L, Zheng W, Lund FE, Xin HB. Mouse embryonic fibroblasts from CD38 knockout mice are resistant to oxidative stresses through inhibition of reactive oxygen species production and Ca(2+) overload. Biochem Biophys Res Commun. 2010;399:167–172. doi: 10.1016/j.bbrc.2010.07.040. [DOI] [PubMed] [Google Scholar]
- 15.Zhang AY, Yi F, Teggatz EG, Zou AP, Li PL. Enhanced production and action of cyclic ADP-ribose during oxidative stress in small bovine coronary arterial smooth muscle. Microvasc Res. 2004;67:159–167. doi: 10.1016/j.mvr.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Jia SJ, Jin S, Zhang F, Yi F, Dewey WL, Li PL. Formation and function of ceramide-enriched membrane platforms with CD38 during M1-receptor stimulation in bovine coronary arterial myocytes. Am J Physiol Heart Circ Physiol. 2008;295:H1743–1752. doi: 10.1152/ajpheart.00617.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teng B, Ansari HR, Oldenburg PJ, Schnermann J, Mustafa SJ. Isolation and characterization of coronary endothelial and smooth muscle cells from A1 adenosine receptor-knockout mice. Am J Physiol Heart Circ Physiol. 2006;290:H1713–1720. doi: 10.1152/ajpheart.00826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CS. Phorbol ester induces elevated oxidative activity and alkalization in a subset of lysosomes. BMC Cell Biol. 2002;3:21. doi: 10.1186/1471-2121-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin S, Zhang Y, Yi F, Li PL. Critical role of lipid raft redox signaling platforms in endostatin-induced coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2008;28:485–490. doi: 10.1161/ATVBAHA.107.159772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2005;102:9056–9061. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 22.Ohta S, Suzuki K, Tachibana K, Yamada G. Microbubble-enhanced sonoporation: efficient gene transduction technique for chick embryos. Genesis. 2003;37:91–101. doi: 10.1002/gene.10232. [DOI] [PubMed] [Google Scholar]
- 23.Taniyama Y, Tachibana K, Hiraoka K, Namba T, Yamasaki K, Hashiya N, Aoki M, Ogihara T, Yasufumi K, Morishita R. Local delivery of plasmid DNA into rat carotid artery using ultrasound. Circulation. 2002;105:1233–1239. doi: 10.1161/hc1002.105228. [DOI] [PubMed] [Google Scholar]
- 24.Zhang F, Zhang G, Zhang AY, Koeberl MJ, Wallander E, Li PL. Production of NAADP and its role in Ca2+ mobilization associated with lysosomes in coronary arterial myocytes. Am J Physiol Heart Circ Physiol. 2006;291:H274–282. doi: 10.1152/ajpheart.01064.2005. [DOI] [PubMed] [Google Scholar]
- 25.Lui AH, McManus BM, Laher I. Endothelial and myogenic regulation of coronary artery tone in the mouse. Eur J Pharmacol. 2000;410:25–31. doi: 10.1016/s0014-2999(00)00868-2. [DOI] [PubMed] [Google Scholar]
- 26.Brandin L, Bergstrom G, Manhem K, Gustafsson H. Oestrogen modulates vascular adrenergic reactivity of the spontaneously hypertensive rat. J Hypertens. 2003;21:1695–1702. doi: 10.1097/00004872-200309000-00019. [DOI] [PubMed] [Google Scholar]
- 27.Bund SJ, Lee RM. Arterial structural changes in hypertension: a consideration of methodology, terminology and functional consequence. J Vasc Res. 2003;40:547–557. doi: 10.1159/000075678. [DOI] [PubMed] [Google Scholar]
- 28.Han WQ, Xia M, Zhang C, Zhang F, Xu M, Li NJ, Li PL. SNARE-Mediated Rapid Lysosome Fusion in Membrane Raft Clustering and Dysfunction of Bovine Coronary Arterial Endothelium. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.00581.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li N, Chen L, Yi F, Xia M, Li PL. Salt-sensitive hypertension induced by decoy of transcription factor hypoxia-inducible factor-1alpha in the renal medulla. Circ Res. 2008;102:1101–1108. doi: 10.1161/CIRCRESAHA.107.169201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi FX, Zhang AY, Campbell WB, Zou AP, Van Breemen C, Li PL. Simultaneous in situ monitoring of intracellular Ca2+ and NO in endothelium of coronary arteries. Am J Physiol Heart Circ Physiol. 2002;283:H2725–2732. doi: 10.1152/ajpheart.00428.2002. [DOI] [PubMed] [Google Scholar]
- 31.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 32.Berruet L, Muller-Steffner H, Schuber F. Occurrence of bovine spleen CD38/NAD+glycohydrolase disulfide-linked dimers. Biochem Mol Biol Int. 1998;46:847–855. [PubMed] [Google Scholar]
- 33.Munshi C, Baumann C, Levitt D, Bloomfield VA, Lee HC. The homo-dimeric form of ADP-ribosyl cyclase in solution. Biochim Biophys Acta. 1998;1388:428–436. doi: 10.1016/s0167-4838(98)00204-0. [DOI] [PubMed] [Google Scholar]
- 34.Caborn DN, Coen M, Neef R, Hamilton D, Nyland J, Johnson DL. Quadrupled semitendinosus-gracilis autograft fixation in the femoral tunnel: a comparison between a metal and a bioabsorbable interference screw. Arthroscopy. 1998;14:241–245. doi: 10.1016/s0749-8063(98)70138-0. [DOI] [PubMed] [Google Scholar]