

Abstract
Regulatory Foxp3+ T cells are a critical cell population that suppresses T cell activation in response to microbial and viral pathogens. We identify a cell-intrinsic mechanism by which effector CD4+ T cells overcome the suppressive effects of Treg cells in the context of three distinct infections, Toxoplasma gondii, Listeria monocytogenes, and vaccinia virus. The acute responses to the parasitic, bacterial, and viral pathogens resulted in a transient reduction in frequency and absolute number of Treg cells. The infection-induced partial loss of Treg cells was essential for the initiation of potent Th1 responses and for host protection against the pathogens. The observed disappearance of Treg cells was a result of insufficiency in IL-2 caused by the expansion of pathogen-specific CD4+ T cells with a limited capacity of IL-2 production. Exogenous IL-2 treatment during the parasitic, bacterial, and viral infections completely prevented the loss of Treg cells, but restoration of Treg cells resulted in a greatly enhanced susceptibility to the pathogens. These results demonstrate that the transient reduction in Treg cells induced by pathogens via IL-2 deprivation is essential for optimal T cell responses and for host resistance to microbial and viral pathogens.
Introduction
Foxp3+ T regulatory (Treg) cells exert pleiotropic immunoregulatory effects essential for immune homeostasis, the prevention of autoimmunity and the regulation of pathogen-induced inflammatory reactions (1). The transcription factor Foxp3 controls Treg cell development and function, and deficiency in the Foxp3 gene results in hyperactivation of CD4+ T cells, overproduction of proinflammatory cytokines, and massive multiorgan pathology (2-3). It was initially thought that the functions of Treg cells were limited to the control of immune responses to autoantigens, but later studies demonstrated that regulatory T cells also exert suppressive effects on the immune responses to tumors (4) and infectious antigens (5-6). These and other studies formally established that Treg cells have profound effects on immunity towards pathogens (7). A number of distinct suppressive mechanisms by which Treg cells target antigen-presenting cells, in particular dendritic cells, and T lymphocytes have been identified (8). The dominant suppressor functions of Treg cells present a serious obstacle in the establishment of robust protective immunity toward pathogens; several studies with bacterial, viral, or parasitic infections formally demonstrated that Treg cells restrict the efficiency and magnitude of T cell responses, which results in an increased pathogen burden (9-11). Some studies have also revealed that by limiting immune responses to an infectious agent, Tregs minimize tissue damage and potentiate a rapid pathogen clearance (12-13). Importantly, the potential reciprocal interactions between effector and Treg cells during the initiation of immune responses are not completely understood.
In this report, we establish a CD4+ T cell-intrinsic mechanism for overcoming the suppressive effects of Treg cells upon the induction of protective T cell responses in the context of three distinct infections, Toxoplasma gondii, Listeria monocytogenes, and vaccinia virus. We observed that the acute responses to the investigated infectious agents resulted in the dramatic loss of Treg cells. This infection-induced reduction of Treg cells was essential for the initiation of potent Th1 responses and for host protection against the parasitic, bacterial, and viral infections. Furthermore, we identified a mechanism responsible for the transient disappearance of Treg cells. We observed that the pathogen-induced expansion of effector T cells was associated with a limited amount of available IL-2. The lack of this growth factor, which is known to be essential for Treg cell development and survival (14), resulted in the loss of Treg cells during the acute response to the pathogens. The infection-induced Treg cell deficiency was completely prevented by treatment with exogenous IL-2 during the course of the experimental diseases. However, the restoration of Treg cells by IL-2 treatment had a negative impact on the induction of Th1 responses and resulted in greatly enhanced susceptibility to the microbial and viral pathogens. Taken together, our results establish that the pathogen-induced transient reduction in Treg numbers caused by IL-2 insufficiency is essential for optimal effector CD4+ T cell responses and host resistance to the parasitic, bacterial, and viral pathogens. Furthermore, because the observed loss of Treg cells was transient and incomplete, it enhanced the protective immune responses to pathogens, but it did not result in severe immunopathology. Our results also suggest that thymus-derived Treg cells, rather than those that develop through peripheral conversion, have a major role in Treg reconstitution following the acute response to microbial pathogens.
Materials and Methods
Animals
C57BL/6 mice were obtained from the University of Texas Southwestern (UTSW) Medical Center Mouse Breeding Core Facility. B6.Cg-Foxp3tm2Tch/J (Foxp3EGFP) mice were obtained from the Jackson Laboratory. All animals used were age- and sex-matched and maintained in the SPF barrier facility at the University of Texas Southwestern Medical Center at Dallas. All experiments were performed using protocols approved by the Institutional Animal Care and Use Committees of the UTSW Medical Center.
Parasitic, bacterial, and viral infections
Mice were infected intraperitoneally (IP) or orally with an average of 20 T. gondii (ME49 strain) cysts as previously described (15). For infection of mice with L. monocytogenes, log-phase cultures of L. monocytogenes (10403 serotype) were washed twice and diluted in PBS to the desired concentration. L. monocytogenes was injected in the lateral tail vein at 104 CFU per mouse. Vaccinia virus was injected in the lateral tail vein at a dosage of 106 PFU per mouse.
IL-2 treatment
Recombinant IL-2 and the anti-mouse IL-2 monoclonal antibody (Clone JES6-1A12) were purchased from eBioscience. The IL-2/anti IL-2 complex was prepared as described previously (16) with a few minor modifications: 50 μg anti-IL-2 antibody was mixed with 1.5 μg IL-2 in 200 μl PBS 15 minutes prior to intraperitoneal injection on days 3 and 5 post-infection with T. gondii, L. monocytogenes, or vaccinia virus.
Flow cytometry and ELISA
On days 3, 5, 7, 10, 14 and 15 post-infection the animals were necropsied, and their spleens and mesenteric lymph nodes were harvested for the analysis of Treg cells by flow cytometry. Cells were stained in ice-cold FACS buffer (PBS supplemented with 2 mM EDTA, 1% heat-inactivated FCS, and 0.02% sodium azide). Analyses of intracellular cytokine expression were performed on splenocytes restimulated with 0.5 μg/ml αCD3 (BD Biosciences) for 5 hr in the presence of GolgiPlug (Brefeldin A, BD Biosciences). After in vitro restimulation, cells were washed once in FACS buffer, stained with PerCP-Cy5.5-labeled anti-CD4 mAb or anti-CD8 mAb, and fixed for 40 min in BD Cytofix/Cytoperm. Cells were then stained using fluorochrome-conjugated antibodies according to the manufacturer's protocol (BD Biosciences). In short, after incubation in the permeabilization buffer supplemented with anti-FcγRII/III mAb (2.4G2; 5 μg/ml) at 4°C, cells were stained for 30 min with FITC-labeled anti-TNF, PE-labeled anti-IL-2, and APC-labeled anti-IFN-γ mAb (BD Biosciences), washed three times, and resuspended in PBS plus 1% FBS. Cell fluorescence was measured using a FACSCalibur or LSRII flow cytometer, and data were analyzed using FlowJo software (Tree Star; Ashland, OR).
Flow cytometric analysis of CD4+Foxp3+ regulatory T cells was performed using the mouse/rat Foxp3 Staining Set (eBioscience) as recommended by the manufacturer.
The ALDH-positive cells were detected using an ALDEFLUOR staining kit (StemCell Technologies). Splenocytes from naïve and infected mice were incubated in the dark for 35 minutes at 37°C in the ALDEFLUOR assay buffer containing the activated ALDEFLUOR substrate, with or without the ALDH inhibitor diethylaminobenzaldehyde (DEAB). Cells were subsequently stained using the CD11b-, CD11c-, and Gr1-specific antibodies (BD Biosciences) and were analyzed on a LSRII flow cytometer. Naïve GFP-(Foxp3-) CD4+ T cells were sort-purified from the spleen of the Foxp3EGFP (B6.Cg-Foxp3tm2Tch/J) mice. Sort-purified GFP-(Foxp3-)CD4+ T cells (105 cells/well) were co-cultured with ALDH+CD11b+CD11c- or CD11c+ cells (2×104 cells per well) in the presence of an αCD3 mAb (0.5 μg/ml) (BD Bioscience). In some experiments, ALDH+CD11b+CD11c- and CD11c+ cells were mixed together with the GFP-CD4+ T cells. TGF-β (3 ng/ml) and IL-2 (5 ng/ml) were purchased from eBioscience. After 3 days of culture, Foxp3 expression was measured by flow cytometry. The data were analyzed using FlowJo software (Tree Star).
IL-2 and IFN-γ in culture supernatants were measured by ELISA using commercially available kits (R&D Systems for IFN-γ and eBiosciences for IL-2).
Reverse-Transcription Polymerase Chain Reaction
RNA was isolated from splenocytes, sort or magnetic beads (Miltenyi Biotec) purified CD4+, CD8+, CD4+ Foxp3EGFP cells with The PureLink™ RNA Mini Kit (Invitrogen). cDNA was prepared using the SuperScript III kit (Invitrogen). Optimized primers targeting each gene (foxp3: 5-ttcatgcatcagctctccac-3, 5-ctggacacccattccagact-3; il2: 5-atgcagctcgcatcctgt-3, 5-ggagctcctgtaggtccatc-3; ifng: 5-actggcaaaaggatggtgac-3, 5-tgagctcattgaatgcttgg-3; cd25: 5-agaacaccaccgatttctgg-3, 5-agctggccactgctacctta-3; and hprt (5gcccttgactataatgag tacttcagg-3 5-ttcaacttgcgctcatcttagg-3) were designed using the Primer 3 software. cDNA was amplified with Power SYBR Green master mix (Applied Biosystems). The MyiQ Real-Time PCR Detection System (BioRad) was used to obtain Ct values. The relative expression of each sample was determined after normalization to HPRT using the the ddCt Method.
Statistical analysis
All data were analyzed with Prism (Version 5; GraphPad). Data were considered statistically significant for P values less than 0.05 using a two-tailed t-test.
Results
Acute responses to parasitic, bacterial and viral infections result in the transient loss of Treg cells
Treg cells are involved in the potent regulation of the immune responses to a variety of microbial pathogens. For this reason, we investigated the interactions between the induction of protective T cells and that of regulatory Foxp3+ CD4+ T cells in the context of infection with three distinct pathogens, the parasite T. gondii, the Gram-positive bacteria L. monocytogenes, and vaccinia virus.
T. gondii is a protozoan parasite that stimulates potent and rapid Th1-biased CD4+ T cell immune responses (17). A protective immune response to this pathogen requires a delicate balance between proinflammatory effector mechanisms, primarily regulated by the TLR-dependent activation of MyD88, and concomitant induction of an anti-inflammatory program (17-18). Lack of either of these mechanisms results in high susceptibility to this parasite, as is evident from the rapid mortality observed in T. gondii-infected MyD88-/- and IL-10-/- mice (19-21). Because Treg cells have an important role in the limitation of T cell responses, we investigated the reciprocal interactions between IFN-γ+ CD4+ T cells and regulatory Foxp3+ CD4+ T cells during the acute response to T. gondii. We observed that systemic infection with the parasite resulted in a dramatic reduction in the frequency of Foxp3+ CD4+ T cells by days 5-7 post-infection (Figures 1A, D). As is evident from Figures 1A and 1D, the observed loss of Treg cells during T. gondii infection was transient, and Treg cells recovered by the end of the acute response to the parasite. In addition, the remaining Treg cells expressed reduced levels of Foxp3, when compared to naïve controls (Figure 1A).
Figure 1.

The acute response to T. gondii results in the transient loss of Treg cells.
WT mice (five animals per group) were infected intraperitoneally with an average of 20 T. gondii strain ME49 cysts per mouse, and (A) the frequency of Treg cells defined as CD4+Foxp3+ cells was analyzed in the spleen of the infected and control (d0) mice at the indicated number of days post-infection. (B) Foxp3 expression levels were analyzed in splenocytes (left graph), isolated CD4+ T cells (central graph), or sort-purified CD4+Foxp3+ T cells (right) isolated from naïve or T. gondii infected mice (day 7 post infection) by real-time PCR. All the data were normalized to the expression level seen in sort-purified CD4+CD44-Foxp3- naïve T cells. The data shown are the mean ± SD. (C) WT mice were infected orally with an average of 20 T. gondii strain ME49 cysts per mouse, and the frequency of Treg cells was analyzed in the spleen of the infected mice at the indicated number of days post-infection. (D) Average frequency of Foxp3+ cells in the spleens of mice infected intraperitoneally (filled circles) or orally (open circles). (E) Absolute quantification of Treg cells in the spleens of mice infected intraperitoneally (filled circles) or orally (open circles) with the parasite. The data shown are representative of five independent experiments. (F). Absolute quantification of CD4+ and CD4+IFN-γ+ T cells in the spleens of naïve (d0) and T. gondii infected mice (d7). The data shown are representative of at least six independent experiments. * P< 0.05; ** P< 0.01.
The relative loss of Foxp3+CD4+ T cells in the T. gondii infected mice was also observed during the analysis of Foxp3 mRNA expression in splenocytes and isolated CD4+ T cells (Figure 1B). We observed a substantial reduction in the amount of Foxp3 mRNA seen in splenocytes and purified CD4+ T cells isolated from T. gondii infected mice when compared to their naïve controls (Figure 1B). In addition, a reduction of Foxp3 mRNA levels in the Treg cells themselves was observed in the T. gondii infected mice (Figure 1B). The later observation was consistent with the flow-cytometry data demonstrating that infection with the parasite resulted not only in the loss of Treg cells, but also in reduced levels of Foxp3 seen in the infected mice, Figure 1A.
To examine the possibility that the observed loss of Treg cells was an artifact of the experimental intraperitoneal infection with the parasite, we next used a natural (oral) route of T. gondii infection. Using the oral route of infection, we observed a similar progressive and transient partial depletion of Treg cells (Figures 1C and 1D). Thus, infection with the protozoan parasite resulted in the transient reduction in frequency of Treg cells during the acute response to the pathogen independently of the route of infection (Figures 1A-D).
Because the Treg cell loss closely coincided with the peak of the CD4+ T cell response against T. gondii, the observed decrease in the frequency of CD4+Foxp3+ Treg cells measured by flow-cytometric or Foxp3 signals detected by PCR approaches could be explained by the expansion of pathogen-specific effector CD4+ T cells. In order to address this possibility, we performed an absolute quantification of CD4+Foxp3+ and CD4+IFN-γ+ T cells during the course of the parasitic infection. The quantification of Treg cells in mice infected intraperitoneally or orally with the parasite revealed that Treg cells (Figure 1E) were almost undetectable at the peak of the CD4+ T cell response against T. gondii (Figure 1F), formally demonstrating that T, gondii infection results in both absolute and relative loss of Treg cells. In addition, we also observed that the disappearance of Treg cells was not limited to the spleen but was also seen in all tissues examined (data not shown).
We next investigated whether the observed transient depletion of Treg cells is unique to the infection with the protozoan parasite T. gondii or whether it is a common feature of acute immune responses to microbial and viral pathogens. As a model bacterial pathogen we selected L. monocytogenes, a Gram-positive bacterium that is responsible for severe disease in immunocompromised humans. Host resistance to this pathogen depends on an intact innate immune system and requires potent IFN-γ production by T cells (22). Similar to our observations after infection with T. gondii, mice challenged with L. monocytogenes demonstrated a dramatic reduction of Treg cells, and this reduction had similar kinetics to those observed during the parasitic infection (Figure 2A). Similar to the T. gondii-induced Treg cell loss, the depletion of Treg cells triggered by infection with L. monocytogenes was transient, and the Treg cell frequency returned to the levels typically seen in naïve animals within 10-14 days post-infection (Figure 2A).
Figure 2.
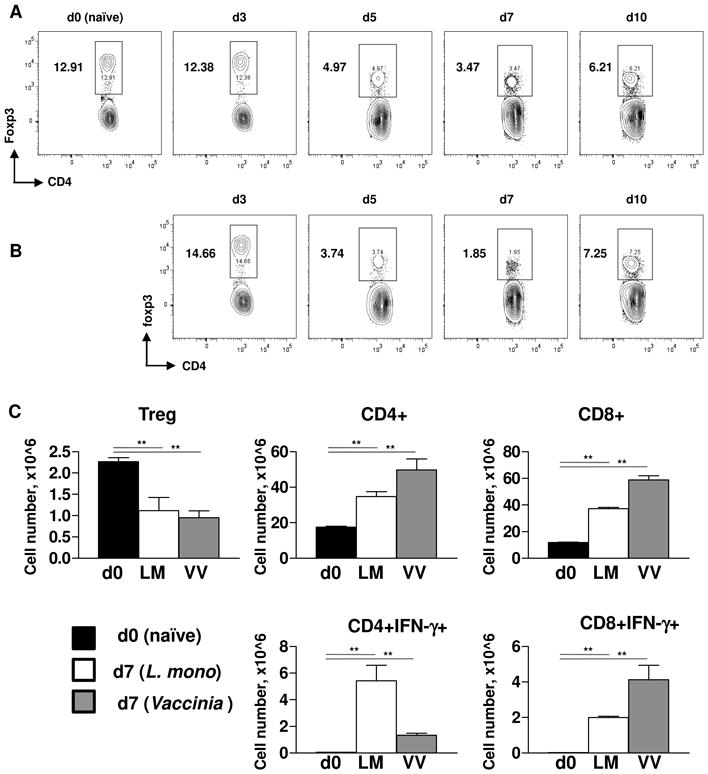
Acute responses to L. monocytogenes and vaccinia virus caused loss of Treg cells.
WT mice (five animals per group) were infected intravenously with (A) L. monocytogenes (104 CFU per mouse) or (B) vaccinia virus (106 PFU per mouse), and the frequency of splenic Treg cells was analyzed at the indicated number of days post-infection. (C) Absolute quantification of Treg cells (CD4+Foxp3+), CD4+, CD8+, CD4+IFN-γ+, and CD8+IFN-γ+ cells was performed in naïve (black bars), L. monocytogenes (open bars), or vaccinia virus (grey bars) infected mice on day 7 post infection. The data shown are the mean ± SD. For identification of IFN-γ+ cells splenocytes were restimulated with 0.5 μg/ml αCD3 for 5 hr in the presence of GolgiPlug. The data shown are representative of four independent experiments. ** P< 0.01.
Furthermore, we also observed that infection with vaccinia virus also resulted in the transient and systemic depletion of Treg cells (Figure 2B). The kinetics of the Treg cell disappearance and recovery closely resembled those observed in T. gondii or L. monocytogenes-infected mice (Figures 1 and 2). In all of the experimental infections (with the viral, bacterial, or parasitic pathogens), we observed that Treg cell depletion peaked by day 7 post-infection. Intriguingly, the loss of Treg cells was transient, and their frequency was largely restored within two weeks of the infection (Figures 1-2).
Quantifications of Treg cells in L. monocytogenes and vaccinia virus infected mice formally established that both infection resulted in absolute reduction of Treg cells (Figure 2C). Because both L. monocytogenes and vaccinia virus result in the expansion of effector CD4+ and CD8+ T cells, (Figure 2C), both infections caused a profound Treg cell insufficiency during the acute responses to these pathogens.
Activation of Toll-like receptors is not required for Treg cell disappearance
It has been previously established that the innate recognition of pathogens by TLRs can overcome the suppressive effects of Treg cells. The activation of TLRs on antigen-presenting cells results in the production of soluble factors, including IL-6, which render effector CD4+ T cells resistant to the suppressive effects of Treg cells (23). TLR activation on Treg cells can result in the elimination of the immunosuppressive effects of this cell population (24-26). TLR activation in effector T cells modulates CD4+ and CD8+ T cell responses (27-29). During T. gondii infection, we previously established that TLR11 is a major innate immune sensor for the parasite that coordinates CD4+ T cell responses to the pathogen (18). We thus investigated whether TLR11 activation was required for the depletion of Tregs during acute toxoplasmosis. We observed that similar to their WT counterparts, mice deficient in TLR11 demonstrated a progressive loss of Treg cells during both the systemic and mucosal responses to the parasite (Figure S1). The only differences between WT and TLR11-deficient mice were the slightly delayed kinetics and reduced magnitude of the disappearance of Tregs in TLR11-/- mice. Furthermore, the analysis of T. gondii-infected TLR2-, TLR4-, TLR2×TLR4-, and TLR9-deficient animals revealed that none of the examined TLRs were responsible for the loss of Treg cells during parasitic infection (data not shown). Taken together, our results suggest that TLR activation is dispensable for the infection-driven loss of Treg cells.
Pathogen-specific CD4+ T cells produce limited amounts of IL-2
We next determined the mechanism that was responsible for the disappearance of Treg cells during the course of parasitic, bacterial, and viral infections. We observed that the loss of Treg cells was independent of their proliferation status. During T. gondii infection, only a small fraction of Foxp3+ T cells was also positive for the proliferation marker Ki67 in infected WT mice, while Treg cells actively divided in mice infected with L. monocytogenes or vaccinia virus (Figure S2 and data not shown). Nevertheless, all three infections resulted in a similar loss of Foxp3+ T cells. We thus hypothesized that the infection-driven depletion of Treg cells was a result of a lack of a survival factor(s) essential for Treg cell maintenance.
Among various growth factors, IL-2 has an important role in the regulation of T cell function (30). While IL-2 is essential for T cell proliferation in vitro, a central function for IL-2 in vivo is the regulation of Treg cell development and survival (14). The lack of IL-2 or of the components of the IL-2 receptor results in a dramatic reduction in Treg cells in the thymus and in the periphery (14). We thus analyzed the effects of the parasitic, bacterial, and viral infections on the production of IL-2 by pathogen-specific CD4+ T cells.
WT animals were infected with T. gondii using an intraperitoneal or oral route, and intracellular staining for IFN-γ, TNF, and IL-2 was used to identify fully activated T. gondii-specific T cells. As previously reported (15, 17), T. gondii infection resulted in the rapid induction of CD4+ T cells producing high amounts of IFN-γ and TNF independently of the route of infection (Figures 3A and S3). In striking contrast, T. gondii-specific CD4+ T cells were largely deficient in the production of IL-2 (Figures 3B-3C, S3A-S3C). Real-time PCR based gene expression analysis also revealed a reverse correlation between expression of IL-2 and IFN-γ genes. We observed that T. gondii infections resulted not only in induction of IFN-γ Figure 3D), but also in the dramatic loss of IL-2 signals measured in purified T cells or bulk splenocytes isolated from infected mice (Figure 3E). These observations were also confirmed by an ELISA assay that demonstrated a reduced amount of IL-2 detected in splenocytes from the infected mice (Figure 3F and S3D). Furthermore, T. gondii infection resulted in the upregulation of CD25, a high affinity subunit of IL-2R, on the activated T cells (Figures 3G and S3E). These results suggest that the acquisition of IFN-γ secreting ability during T. gondii infection was associated with increased consumption of IL-2 coupled with reduced production of this cytokine.
Figure 3.

T. gondii-specific Th1 T cells produce limited amounts of IL-2
WT animals (five mice per group) were either left untreated (A) or infected with T. gondii intraperitoneally (B) or orally (C), and seven days later, the ability of splenic CD4+ T cells to produce IFN-γ, TNF, and IL-2 was analyzed. Analyses of intracellular cytokine expression were performed on splenocytes restimulated with 0.5 μg/ml αCD3 for 5 hr in the presence of GolgiPlug. (D) Expression levels of IFN-γ and (E) IL-2 were analyzed by real-time PCR in splenocytes, CD4+, CD8+, and CD4+Foxp3+ T cells isolated from naïve or T. gondii infected mice (day 7 post infection). (F) IL-2 in cell culture supernatant was measured by ELISA on splenocytes isolated from naïve (d0) or T. gondii infected mice at the indicated time points after restimulation with 0.01 ug/ml αCD3 for 48 hr. (G) Expression levels of CD25 were analyzed by real-time PCR in naïve or T. gondii infected mice (day 7) on the same samples as shown in (D and E). The data shown are representative of five independent experiments. * P< 0.05; ** P< 0.01, *** P< 0.001.
Intracellular staining was next used to detect IFN-γ and IL-2 proteins during the course of L. monocytogenes and vaccinia virus infections. Similar to infection with the parasite, infection with the bacterial or viral pathogen also resulted in a high frequency of CD4+IFN-γ+ T cells (Figure 4A-B). At the same time, only a small fraction of activated CD4+ T cells produced high levels of IL-2 (Figure 4A-B). Real-time PCR (Figure 4C) and ELISA (Figure 4D) methods also confirmed the flow-cytometric observation regarding the loss of IL-2 expression and acquisition of IFN-γ production during the acute responses to the pathogens.
Figure 4.
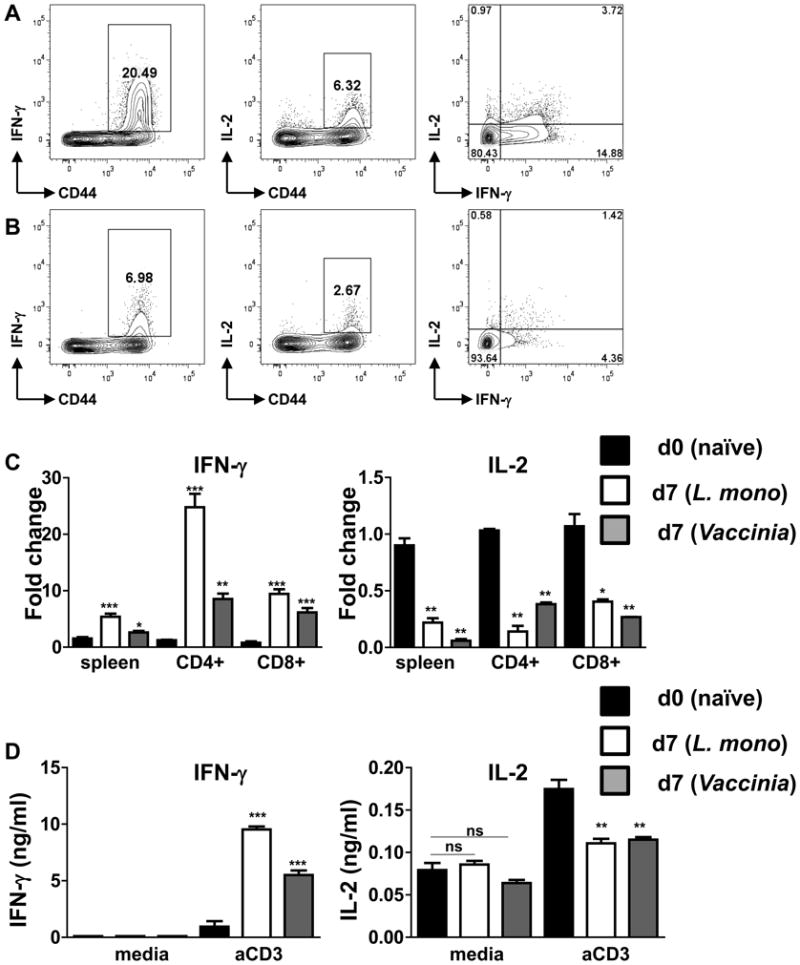
L. monocytogenes- and vaccinia virus-specific CD4+ T cells produce limited amounts of IL-2.
WT mice (five animals per group) were infected with (A) L. monocytogenes (104 CFU per mouse) or (B) vaccinia virus (106 PFU per mouse), and the ability of CD4+ T cells to secrete IFN-γ and IL-2 was analyzed by flow cytometry after restimulation with 0.5 ug/ml αCD3 for 5 hr in the presence of GolgiPlug. (C) Expression levels of IFN-γ (left panel) and IL-2 (right panel) were analyzed by real-time PCR in naïve (black bars), L. monocytogenes (open bars) and vaccinia virus (grey bars) infected mice on day 7 post infection. (D) IFN-γ and IL-2 were measured by ELISA in unstimulated (media) or 0.01 ug/ml αCD3 restimulated splenocytes isolated from naïve (black bars), L. monocytogenes (open bars) and vaccinia virus (grey bars) infected mice. The data shown are representative of four independent experiments. * P< 0.05; ** P< 0.01, *** P< 0.001.
Taken together, our results suggest that the effector phenotype during acute responses to T. gondii, L. monocytogenes, and vaccinia virus is associated with a limited ability to produce IL-2.
Rescue of Treg cells by IL-2 negatively regulates the host resistance to microbial and viral infections
We next investigated whether the limited production of IL-2, coupled with the expansion of pathogen-specific T cells, is responsible for the infection-induced loss of Treg cells. This hypothesis is based on previous experiments which revealed high sensitivity of Treg cells to the decreased availability of IL-2, as well as our observation regarding limited amounts of IL-2 produced by pathogen-specific CD4+ Tcells (Figures 3 and 4). To directly examine this possibility, we determined whether exogenous treatment with IL-2 would prevent the disappearance of Treg cells caused by the microbial and viral infections.
Animals infected with T. gondii, L. monocytogenes, or vaccinia virus were additionally treated with an IL-2/anti-IL-2 complex, which is known to increase the biological activity of the cytokine (16). As is evident in Figure 5, IL-2 supplementation during the course of all of these infections prevented the disappearance of Treg cells. IL-2 treatment completely prevented the loss of Treg cells triggered by T. gondii infection (irrespective of the route of infection) in the spleen (Figures 5A-5C) and in all of the examined tissues (data not shown). Similarly, IL-2 treatment of L. monocytogenes- or vaccinia virus-infected mice prevented the loss of Treg cells (Figures 5D-F). These results suggested that insufficiency of IL-2 is a major cause of the loss of Treg cells triggered by the parasitic, bacterial, and viral pathogens.
Figure 5.
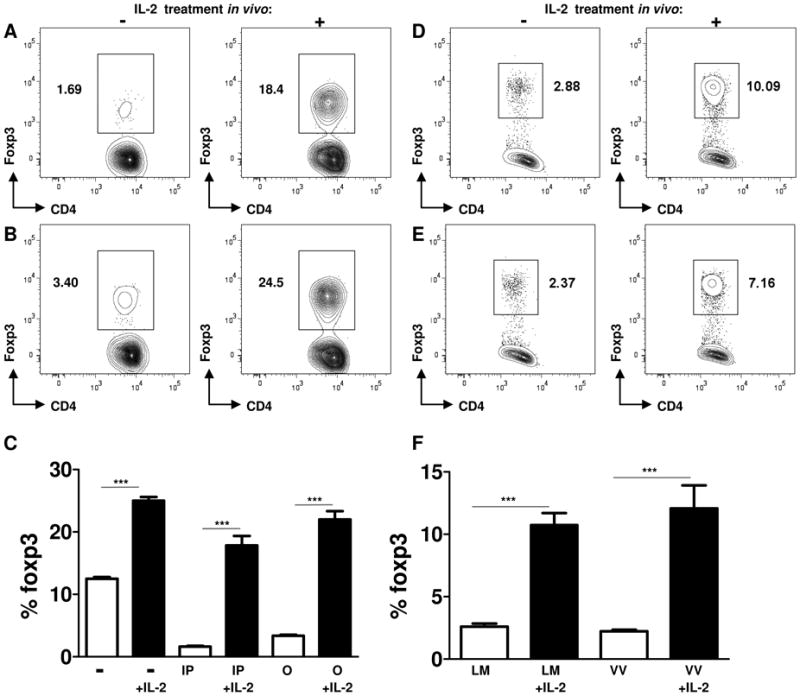
Exogenous IL-2 treatment during parasitic, bacterial, and viral infections prevented the loss of Treg cells
WT mice (n=5) were infected intraperitoneally (A) or orally (B) with T. gondii and additionally treated with IL-2 (1.5 μg of IL-2 plus 50 μg of anti-IL-2 antibody per mouse). The Treg cell frequency was analyzed by flow cytometry on day 7 post-infection. (C) Average frequency of Foxp3+ cells in the spleens of control (−), intraperitoneally (IP) or orally (O) infected mice treated with IL-2 plus anti-IL-2 antibody (+IL-2). The data shown are the mean ± SD. Animals were infected with (D) L. monocytogenes (104 CFU per mouse) or (E) vaccinia virus (106 PFU per mouse) and where indicated, were treated with exogenous IL-2 as described above. Treg cells were analyzed in IL-2 treated (+) and untreated (−) mice on day 7 post-infection. (F) The data shown are the mean frequency of Treg cells ± SD in L. monocytogenes (LM) or vaccinia virus (VV) infected mice. The data shown are representative of three independent experiments. *** P< 0.001.
We next investigated the effects of Treg cell reconstitution on the induction of host resistance to T. gondii, L. monocytogenes, and vaccinia virus. We observed that IL-2 treatment of T. gondii-infected mice dramatically reduced the ability of CD4+ T cells to acquire a Th1 phenotype, a hallmark of the immune response to this parasite (Figures 6A). Furthermore, the reconstitution of Treg cells resulted in increased mortality of the infected mice compared with WT animals without IL-2 treatment (Figure 6B). These results strongly suggest that the infection-triggered depletion of Treg cells is essential for establishing a protective Th1 response during the acute response against the parasite. We also observed that in WT mice that survived the acute phase of toxoplasmosis, a short treatment with IL-2 at the initial phase of the adaptive immune response to the parasite resulted in an enhanced brain cyst load during the chronic phase of the infection (Figure 6B).
Figure 6.
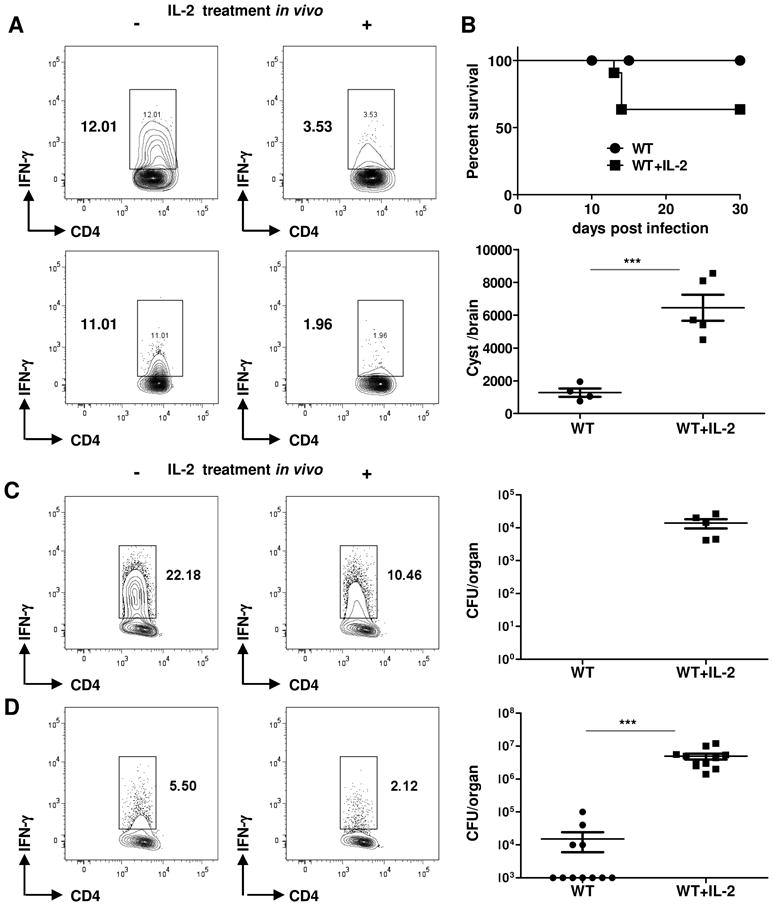
Exogenous IL-2 treatment during parasitic, bacterial, and viral infections results in enhanced susceptibility to the pathogen.
(A) T. gondii-infected animals (five mice per group) were treated with the IL-2/anti-IL-2 complex, and Th1 polarization was analyzed on day 7 post-infection. (B) The cumulative survival and the cyst burden on day 30 post-infection are shown for untreated and IL-2-treated mice. (C) L. monocytogenes- or (D) vaccinia virus-infected animals were additionally treated with IL-2 (+), and Th1 polarization and pathogen loads were analyzed on day 7 post-infection. The data shown are representative of five independent experiments for T. gondii and three independent experiments for L. monocytogenes and vaccinia virus infections. *** P< 0.001.
Similar to the results obtained with T. gondii-infected mice, we observed that in vivo IL-2 treatment impaired Th1 responses to L. monocytogenes and enhanced susceptibility to the pathogen (Figure 6C). While mock or untreated WT animals completely cleared L. monocytogenes by day 7 post-infection, animals that maintained the Treg cell population as a result of IL-2 treatment contained high numbers of bacteria (Figure 6C). IL-2 treatment and Treg cell reconstitution in vaccinia virus-infected animals also had a major negative impact on the clearance of the virus (Figure 6D).
Regeneration of Treg cells during microbial infection
Deficiency in IL-2 or Treg cells results in a fatal autoimmune disease affecting multiple organs (1, 14). In contrast, infection-induced loss of Treg cells was transient and even beneficial for the induction of pathogen-specific immune responses (Figure 6). These observations suggest that mechanisms are in place that regulate Treg cell recovery following acute responses to pathogens. Because Treg cells can either originate from the thymus or develop as a result of peripheral conversion from naïve T cells (1), we investigated the relative contribution of these mechanisms in the restoration of Treg cells.
The de novo induction of Treg cells in the periphery critically depends on the vitamin A metabolite retinoic acid, and cells expressing the aldehyde dehydrogenase (ALDH) enzymes are known to have a major role in the generation of inducible Treg cells (31). We used flow cytometry to analyze ALDH-expressing cells during the course of T. gondii infection. While only a small number of cells expressed ALDH under steady-state conditions, we observed a rapid accumulation of ALDH+ cells in the spleen of infected animals during the acute response to the parasite (Figures 7A and S4). The ALDH-positive cells also expressed CD11b and Gr1, but not CD11c, surface markers (Figure S4 and data not shown). Intriguingly, the highest numbers of ALDH+ cells were detected in the T. gondii-infected mice at the start of Treg cell reconstitution (Figures 7 and S4). To examine whether the infection-induced ALDH+ cells were responsible for the restoration of the Treg cell pool, we evaluated the capacity of CD11b+ALDH+ cells to generate Tregs in vitro from Foxp3-negative CD4+ T cells. Non-Treg naïve CD4+ T cells were isolated from Foxp3-GFP mice and were incubated with sorted ALDH+CD11b+CD11c- cells alone or with the addition of recombinant IL-2 and TGF-β (Figure 7B). For comparison, we also isolated CD11c+ DC from the same mice and analyzed the Treg cell conversion ability of these cells alone or in the combination with ALDH+ cells. We observed that ALDH+ cells alone or those co-cultured with DC failed to induce Treg cells in the absence of TGF-β. The addition of this cytokine to the culture of DC (CD11c+MHCII+ALDH-) or ALDH+CD11b+CD11c- cells dramatically enhanced the ability of both cell populations to induce Treg cells (Figure 7B). The addition of exogenous IL-2 resulted in a further increased generation of Treg cells. Nevertheless, under all tested conditions, ALDH+ cells were not superior to ALDH- DC in the conversion of Foxp3-negative CD4+ T cells into Foxp3-positive lymphocytes (Figure 7B). Furthermore, the efficient induction of Treg cells by ALDH+CD11b+ and CD11c+ cells was in part dependent on IL-2 (Figure 7B), arguing against the possibility that the peripheral induction of Treg cells could compensate for the loss of Treg cells that results from infection-induced IL-2 insufficiency.
Figure 7.
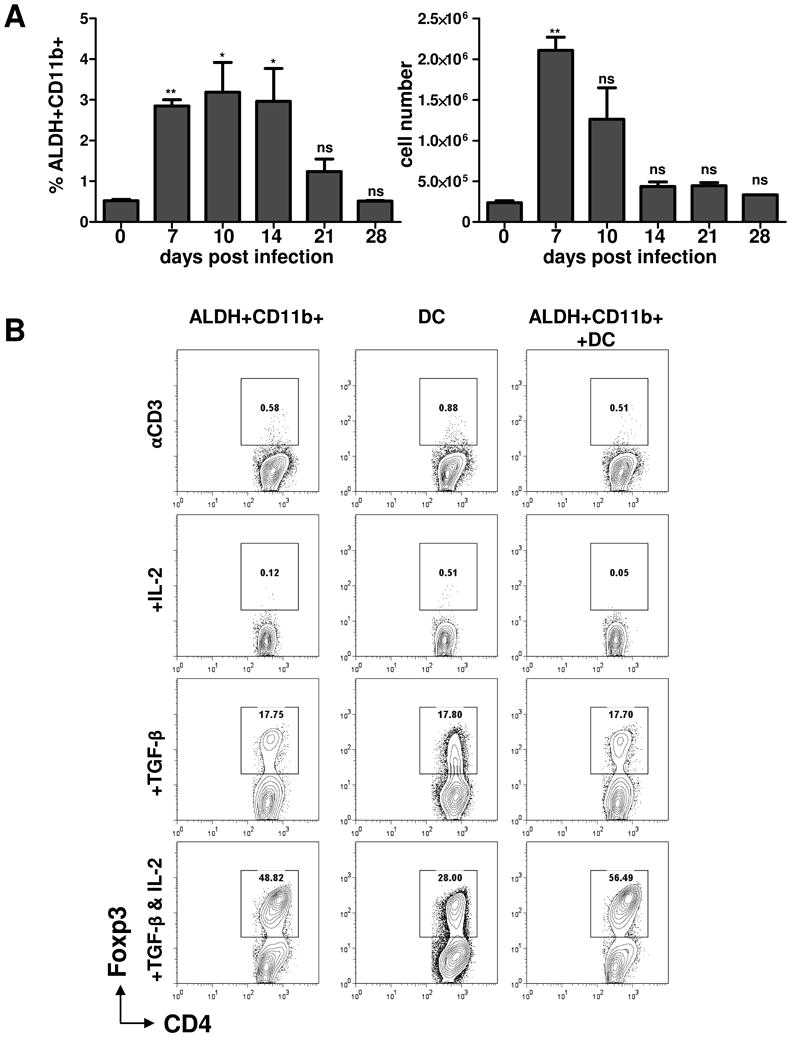
Peripheral Treg cell conversion by ALDH+CD11b+ and CD11c+ DC cells.
(A) The relative (left panel) and absolute (right panel) counts of ALDH+CD11b+ cells at the indicated time points after T. gondii infection. The appearance of ALDH+ cells in response to T. gondii infection was analyzed by flow cytometry on days 0, 7, 10, 14, 21, and 28 post-infection shown in Figure S4. (B) ALDH+CD11b+ and ALDH-CD11c+ cells were sort-purified from spleens of T. gondii-infected mice on day 7 post-infection and were mixed with sort-purified Foxp3GFP- CD4+ T cells in the presence of αCD3 alone or in combination with IL-2 and TGF-β. T cell Foxp3 expression was examined by flow cytometry after 3 days of culture. Plots are gated on CD4+ cells, and the percentages of Foxp3+ cells are shown. The data shown are representative of three experiments. * P< 0.1; ** P< 0.01.
We next analyzed whether an increased production of Treg cells in the thymus could have a role in the reconstitution of Treg cells after the acute response to parasitic infection. We first observed that while the frequency of thymic Treg cells was reduced in T. gondii-infected mice (Figures 8A, 8B), their loss was less prominent when compared with Treg cells in the periphery (Figure 1). Intriguingly, at the peak of peripheral Treg cell loss (day 7, Figure 1), we observed a relative expansion of thymic Foxp3+ cells that also expressed Ki67, a marker of recently divided cells (Figures 8A, 8C). Importantly, the thymic Treg cells rapidly recovered in both their absolute number and their frequency after the parasitic infection (Figures 8B, 8D), suggesting that thymus-derived Treg cells, rather than the peripheral induction of Foxp3+ CD4+ T cells, have a major role in Treg cell recovery following the acute response to the pathogen.
Figure 8.
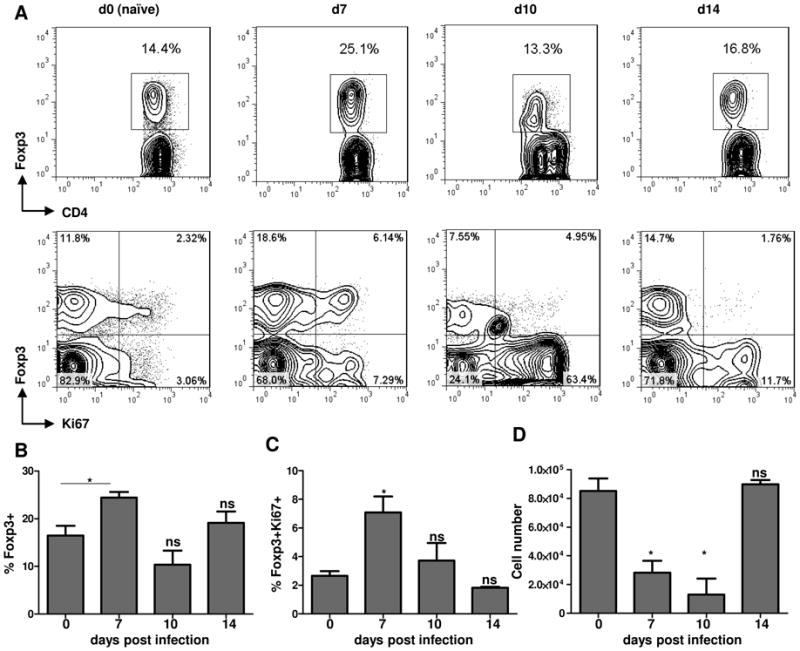
Thymic Treg cells are relatively resistant to infection-induced Treg cell loss.
(A) WT mice (five animals per group) were infected intraperitoneally with an average of 20 T. gondii strain ME49 cysts per mouse, and the frequency of Treg cells, among all CD+ T cells, was analyzed in the thymus of the infected and control mice at the indicated number of days post-infection. Analysis of thymic Treg cell proliferation was performed by intracellular staining for the nuclear antigen Ki67. Average frequency of (B) Foxp3+ cells and (C) Foxp3+Ki67+ CD4+ T cells in the thymus of T. gondii-infected mice. (D) Absolute quantification of Treg cells in the thymus of mice infected with the parasite. The data shown are representative of three independent experiments.
* P< 0.05.
Discussion
Treg cells expressing the transcription factor Foxp3 contribute to the dominant control of self-reactive T cells, thus contributing to the maintenance of immunologic self-tolerance (1, 8). Solid evidence from experimental animals and clinical observations have demonstrated that the expansion and accumulation of these immunosuppressive cells correlates with advanced tumor growth and predicts poor prognosis during infectious diseases. Treg cells not only prevent the induction of tumor- or pathogen-specific CD4+ and CD8+ T cell responses but also limit the efficacy of vaccinations and other therapeutic interventions (1).
The effect of Treg cells in the context of host-pathogen interactions has been particularly extensively studied during chronic infectious diseases. In a model of cutaneous leishmaniasis, Treg cells are responsible for the suppression of effector T cells and the establishment of a persistent parasitic infection (5). The removal of Treg cells during malaria infection limited the expansion of the parasite and prevented the host from developing cerebral malaria (6, 9). The immunosuppressive effects of Treg cells are not limited to parasitic diseases. The depletion of Treg cells greatly enhanced anti-viral responses during HIV and SIV infections (32-34). Similarly, failure to control hepatitis B and C viruses correlated with a high frequency of Treg cells (35). An important role for Treg cells in the progression of mycobacteria infection has also been demonstrated (10).
A role for Treg cells during the initiation of acute responses to pathogens is less clear. The profound inhibitory effects of Treg cells on the priming of pathogen- or model antigen-specific T cells have been well characterized (8). The experimental depletion of Treg cells resulted in greatly enhanced Th1 and Th2 responses and formally demonstrated a major role for Treg cells in the prevention of effector T cell activation (36, 37). What is not clear is how T cell responses are initiated in the presence of Treg cells in vivo. One mechanism of the modulation of the suppressive effects of Tregs depends on the activation of TLRs on DC and other antigen-presenting cells (APC) (38). Because TLRs are involved in the recognition of all known groups of pathogens, including viruses, bacteria, fungi, and protozoa, TLR-dependent inhibition of the suppressive effects of Treg cells can coordinate the immune responses to all groups of microorganisms. More recent studies revealed that in addition to APC, murine and human Treg cells express high levels of several TLRs, but the effects of TLR activation on Treg cells remain controversial. For example, TLR5 activation by flagellin increases the suppressive activity of Tregs (39). However, triggering of TLR8 on Treg cells results in the abrogation of their suppressive functions (24). The positive and negative effects of TLR2 activation on Treg cells depend on the nature of the TLR2 agonist (29). Similarly, TLR4 can both enhance and suppress Treg cell functions (40-41). In this report, we identified a distinct self-regulated mechanism that allows effector T cells to avoid the suppressive effects of Treg cells. Importantly, this mechanism does not require TLR-mediated recognition of the pathogen but depends on the tight regulation of IL-2 availability.
We observed that the acute immune response to three distinct pathogens, T. gondii, L. monocytogenes, and vaccinia virus, resulted in the transient and systemic partial loss of Treg cells. The disappearance of Treg cells did not require TLR activation but rather correlated with the degree of effector T cell activation and the inability of the activated CD4+ T cells to produce IL-2. We observed that the rapid expansion of pathogen-specific Th1 CD4+ T cells resulted in a transient insufficiency of IL-2. Because IL-2 has a dominant role in Treg cell development and in the regulation of the suppressive functions of Tregs (14), infection-induced insufficiency in IL-2 assures the loss of Treg cells during the initiation of pathogen-specific T cell responses. Our observations established that in contrary to the previous models in which deprivation of IL-2 was proposed to be a mechanism by which Treg cells could control effector T cells (42), limited amounts of IL-2 have more profound effects on Treg cells. Most importantly, we observed that the transient loss of Treg cells was essential for optimal host resistance to all of the tested pathogens, T. gondii, L. monocytogenes, and vaccinia virus. Prevention of the transient loss of Treg cells by treating the infected animals with IL-2 resulted in impaired pathogen-specific responses and, in the case of parasitic infection, was responsible for the high mortality of the infected mice. While Treg cell reconstitution did not result in mortality of vaccinia virus-infected mice, and only a small fraction of L. monocytogenes-infected animals died after Treg cell reconstitution, a dramatic elevation of the pathogen loads was observed in all of the experimental infections. This enhanced susceptibility to viral and microbial infection correlated with impaired IFN-γ production by effector T cells.
A recent report from Belkaid and colleagues revealed that the mucosal responses to T. gondii are characterized by the disappearance of Treg cells, which causes lethal intestinal and liver pathology (13). While we also observed that Treg cell reconstitution by IL-2 reduced the damage caused by the exaggerated immune responses to the parasite, several lines of evidence suggest that the main function for the transient loss of Treg cells is to enhance Th1 responses to pathogens. First, we established that T. gondii is not a unique pathogen in its ability to cause the loss of Treg cells. Mice infected with L. monocytogenes or vaccinia virus demonstrated a similar loss of Treg cells, but these bacterial and viral infections do not cause severe intestinal damage. Furthermore, we observed that systemic (intraperitoneal) infection with T. gondii also resulted in the IL-2 dependent disappearance of Treg cells, similar to that observed during the mucosal responses to the parasite. Because the intestinal pathology is triggered during mucosal, but not systemic, responses to the parasite, we conclude that while the infection-induced transient loss of Treg cells contributes to the pathological response, it is not sufficient for the initiation of the intestinal immunopathology. Indeed, we previously reported that TLR11-deficient animals are protected from T. gondii-induced ileitis (15), but TLR11 activation was not required for the infection-induced loss of Treg cells. These results suggest that the observed transient loss of Treg cells caused by microbial infection has a distinct biological function. We propose that the loss of Treg cells caused by the limited ability of the pathogen-specific CD4+ T cells to produce IL-2 is an important regulatory mechanism that is essential for host resistance to microbial infections.
Ablation of Treg cells is not always beneficial for the acute response to pathogens. In the context of mucosal herpes simplex virus infection, the ablation of Treg cells accelerates fatal infection associated with increased viral loads in the mucosa and central nervous system (12). This is because a complete lack of Treg cells results in uncoordinated responses to the virus, characterized by an exaggerated inflammatory reaction without the sufficient number of NK cells, DC, and T cells required for protection of the host against the herpes simplex virus infection. Similarly, the complete depletion of Tregs results in impaired RSV clearance in the lung, which is associated with a delay in the recruitment of RSV-specific CD8+ T cells (43). These studies clearly demonstrate that the complete and prolonged ablation of Treg cells can be detrimental not only because of fatal autoimmune reactions, but also because of uncoordinated pathogen-specific immune responses.
Our results revealed that T. gondii, L. monocytogenes and vaccinia virus caused a transient and incomplete infection-induced Treg cell depletion. The rapid recovery of Treg cells correlated with the enhanced replication of thymic Foxp3+CD4+ T cells, suggesting that thymus-derived Treg cells have a major role in the restoration of Treg cells in the periphery. In addition, we observed that thymic Treg cells were more resistant to the infection-induced IL-2 deprivation, and our results are consistent with previous publications that established that in the absence of an IL-2 signal, the thymic production of Foxp3+CD4+ T cells was only partially reduced, indicating that thymic Treg cells are less sensitive to IL-2 deficiency (44-45). In summary, we observed that parasitic, bacterial, and viral pathogens caused a prominent and transient loss of Treg cells. The infection-induced expansion of effector T cells was associated with a limited amount of IL-2 produced by activated CD4+ T cells, and the deficiency in this cytokine was responsible for the infection-triggered loss of Tregs. Our results revealed that the naturally occurring partial depletion of Treg cells is essential for establishing protective immunity to T. gondii, L. monocytogenes and vaccinia virus and suggest a mechanism that regulates adaptive immunity to pathogens that depends on the homeostatic IL-2-based interactions between effector and regulatory CD4+ T cells.
Supplementary Material
Acknowledgments
We thank Ellen Vitetta and Julie Mirpuri for carefully reading the manuscript.
This research was supported by NIH R01 AI085263
References
- 1.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nature Immunology. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 3.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 4.Nishikawa H, Jager E, Ritter G, Old LJ, Gnjatic S. CD4(+) CD25(+) regulatory T cells control the induction of antigen-specific CD4(+) helper T cell responses in cancer patients. Blood. 2005;106:1008–1011. doi: 10.1182/blood-2005-02-0607. [DOI] [PubMed] [Google Scholar]
- 5.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4(+)CD25(+) regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 6.Hisaeda H, Maekawa Y, Iwakawa D, Okada H, Himeno K, Kishihara K, Tsukumo S, Yasutomo K. Escape of malaria parasites from host immunity requires CD4(+)CD25(+) regulatory T cells. Nature Medicine. 2004;10:29–30. doi: 10.1038/nm975. [DOI] [PubMed] [Google Scholar]
- 7.Belkaid Y, Tarbell K. Regulatory T Cells in the Control of Host-Microorganism Interactions. Annu Rev Immunol. 2009;27:551–589. doi: 10.1146/annurev.immunol.021908.132723. [DOI] [PubMed] [Google Scholar]
- 8.Shevach EM. Mechanisms of Foxp3(+) T Regulatory Cell-Mediated Suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Amante FH, Stanley AC, Randall LM, Zhou YH, Haque A, McSweeney K, Waters AP, Janse CJ, Good MF, Hill GR, Engwerda CR. A role for natural regulatory T cells in the pathogenesis of experimental cerebral malaria. American Journal of Pathology. 2007;171:548–559. doi: 10.2353/ajpath.2007.061033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kursar M, Koch M, Mittruecker HW, Nouailles G, Bonhagen K, Kamradt T, Kaufmann SHE. Cutting edge: Regulatory T cells prevent efficient clearance of Mycobacterium tuberculosis. J Immunol. 2007;178:2661–2665. doi: 10.4049/jimmunol.178.5.2661. [DOI] [PubMed] [Google Scholar]
- 11.Scott-Browne JP, Shafiani S, Tucker-Heard G, Ishida-Tsubota K, Fontenot JD, Rudensky AY, Bevan MJ, Urdahl KB. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. Journal of Experimental Medicine. 2007;204:2159–2169. doi: 10.1084/jem.20062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science. 2008;320:1220–1224. doi: 10.1126/science.1155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O'Brien S, Blank R, Lamb E, Natarajan S, Kastenmayer R, Hunter C, Grigg ME, Belkaid Y. Decrease of Foxp3(+) Treg Cell Number and Acquisition of Effector Cell Phenotype during Lethal Infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nature Reviews Immunology. 2004;4:665–674. doi: 10.1038/nri1435. [DOI] [PubMed] [Google Scholar]
- 15.Benson A, Pifer R, Behrendt CL, Hooper LV, Yarovinsky F. Gut Commensal Bacteria Direct a Protective Immune Response against Toxoplasma gondii. Cell Host & Microbe. 2009;6:187–196. doi: 10.1016/j.chom.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- 17.Denkers EY, Gazzinelli RT. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clinical Microbiology Reviews. 1998;11:569. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yarovinsky F. Toll-like receptors and their role in host resistance to Toxoplasma gondii. Immunology Letters. 2008;119:17–21. doi: 10.1016/j.imlet.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, Medzhitov R, Sher A. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- 20.Gazzinelli RT, Wysocka M, Hieny S, SchartonKersten T, Cheever A, Kuhn R, Muller W, Trinchieri G, Sher A. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4(+) T cells and accompanied by overproduction of IL-12, IFN-gamma, and TNF-alpha. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 21.Wilson EH, Wille-Reece U, Dzersznski F, Hunter CA. A critical role for IL-10 in limiting inflammation during toxoplasmic encephalitis. J Neuroimmunol. 2005;165:63–74. doi: 10.1016/j.jneuroim.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 22.Pamer EG. Immune responses to Listeria monocytogenes. Nature Reviews Immunology. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 23.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4(+)CD25(+) T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 24.Peng GY, Guo Z, Kiniwa Y, Voo KS, Peng WY, Fu TH, Wang DY, Li YC, Wang HY, Wang RF. Toll-like, receptor 8-mediated reversal of CD4(+) regulatory T cell function. Science. 2005;309:1380–1384. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 25.Sutmuller RPM, den Brok M, Kramer M, Bennink EJ, Toonen LWJ, Kullberg BJ, Joosten LA, Akira S, Netea MG, Adema GJ. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang YP, Huang CT, Huang XP, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nature Immunology. 2004;5:508–515. doi: 10.1038/ni1059. [DOI] [PubMed] [Google Scholar]
- 27.LaRosa DF, Gelman AE, Rahman AH, Zhang JD, Turka LA, Walsh PT. CpG DNA inhibits CD4+CD25+ Treg suppression through direct MyD88-dependent costimulation of effector CD4+ T cells. Immunology Letters. 2007;108:183–188. doi: 10.1016/j.imlet.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gelman AE, Zhang JD, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4(+) T cell survival. J Immunol. 2004;172:6065–6073. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Current Opinion in Immunology. 2007;19:39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Yu AX, Malek TR. Selective availability of IL-2 is a major determinant controlling the production of CD4(+)CD25(+)Foxp3(+) T regulatory cells. J Immunol. 2006;177:5115–5121. doi: 10.4049/jimmunol.177.8.5115. [DOI] [PubMed] [Google Scholar]
- 31.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal T(H)17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 32.Andersson J, Boasso A, Nilsson J, Zhang R, Shire NJ, Lindback S, Shearer GM, Chougnet CA. Cutting edge: The prevalence of regulatory T cells lymphoid tissue is correlated with viral load HIV-infected patients. J Immunol. 2005;174:3143–3147. doi: 10.4049/jimmunol.174.6.3143. [DOI] [PubMed] [Google Scholar]
- 33.Karlsson I, Malleret B, Brochard P, Delache B, Calvo J, Le Grand R, Vaslin B. FoxP3(+) CD25(+) CD8(+) T-Cell induction during primary simian immunodeficiency virus infection in cnomolgus macaques correlates with low CD4(+) T-Cell activation and high viral load. Journal of Virology. 2007;81:13444–13455. doi: 10.1128/JVI.01466-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiss L, Donkova-Petrini V, Caccavelli L, Balbo M, Carbonneil C, Levy Y. Human immunodeficiency virus-driven expansion of CD4(+)CD25(+) regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood. 2004;104:3249–3256. doi: 10.1182/blood-2004-01-0365. [DOI] [PubMed] [Google Scholar]
- 35.Manigold T, Shin EC, Mizukoshi E, Mihalik K, Murthy KK, Rice CM, Piccirillo CA, Rehermann B. Foxp3(+)CD4(+)CD25(+) T cells control virus-specific memory T cells in chimpanzees that recovered from hepatitis C. Blood. 2006;107:4424–4432. doi: 10.1182/blood-2005-09-3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKee AS, Pearce EJ. CD25(+)CD4(+) cells contribute to Th2 polarization during helminth infection by suppressing Th1 response development. J Immunol. 2004;173:1224–1231. doi: 10.4049/jimmunol.173.2.1224. [DOI] [PubMed] [Google Scholar]
- 37.Taylor JJ, Mohrs M, Pearce EJ. Regulatory T cell responses develop in parallel to Th responses and control the magnitude and phenotype of the Th effector population. J Immunol. 2006;176:5839–5847. doi: 10.4049/jimmunol.176.10.5839. [DOI] [PubMed] [Google Scholar]
- 38.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nature Immunology. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 39.Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4(+) T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4(+)CD25(+) T regulatory cells. J Immunol. 2005;175:8051–8059. doi: 10.4049/jimmunol.175.12.8051. [DOI] [PubMed] [Google Scholar]
- 40.Abdollahi-Roodsaz S, Joosten LAB, Koenders MI, Devesa I, Roelofs MF, Radstake T, Heuvelmans-Jacobs M, Akira S, Nicklin MJH, Ribeiro-Dias F, Van den Berg WB. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118:205–216. doi: 10.1172/JCI32639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewkowicz P, Lewkowicz N, Sasiak A, Tchorzewski H. Lipopolysaccharide-activated CD4(+)CD25(+) T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol. 2006;177:7155–7163. doi: 10.4049/jimmunol.177.10.7155. [DOI] [PubMed] [Google Scholar]
- 42.Pandiyan P, Zheng LX, Ishihara S, Reed J, Lenardo MJ. CD4(+) CD25(+) Foxp3(+) regulatory T cells induce cytokine deprivation -mediated apoptosis of effector CD4(+) T cells. Nature Immunology. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 43.Fulton RB, Meyerholz DK, Varga SM. Foxp3(+) CD4 Regulatory T Cells Limit Pulmonary Immunopathology by Modulating the CD8 T Cell Response during Respiratory Syncytial Virus Infection. J Immunol. 2010;185:2382–2392. doi: 10.4049/jimmunol.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bayer AL, Yu AX, Malek TR. Function of the IL-2R for thymic and peripheral CD4(+)CD25(+) Foxp3(+) T regulatory cells. J Immunol. 2007;178:4062–4071. doi: 10.4049/jimmunol.178.7.4062. [DOI] [PubMed] [Google Scholar]
- 45.D'Cruz LM, Klein L. Development and function of agonist-induced CD25(+)Foxp3(+) regulatory T cells in the absence of interleukin 2 signaling. Nature Immunology. 2005;6:1152–1159. doi: 10.1038/ni1264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.