

Abstract

Bicyclic γ-silyloxy-β-hydroxy-α-diazoketones in which the Cβ-Cγ bond is the ring fusion bond productively fragment when treated with tin(IV) chloride to provide medium-sized cyclic 2-alkynones. This method provides good to excellent yields of 10-membered, 11-membered and 12-membered alkynone products.
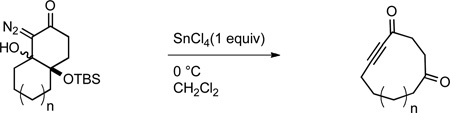
In view of the synthetic utility of ynones, and the fact that medium sized rings are an under-explored1 but important2 structural motif in biologically active compounds, medium sized cyclic 2-alkynones should be decidedly useful synthetic intermediates. However, these types of compounds have rarely been used in synthetic sequences;3–7 a fact that can undoubtedly be attributed to a lack of available methods to prepare this structural motif. While functional group manipulations on medium sized cycloalkynes8–12 or medium sized cycloalkenes13 have on occasion been used to prepare cyclic 2-alkynones, methods to directly prepare these compounds from precursors that do not contain a medium sized ring are limited to the aluminum trichloride promoted intramolecular cyclization of silylalkynes and acid chlorides developed by Utimoto and co-workers.14 Unfortunately, this strategy suffers from the common limitation of macrocyclization strategies, namely the requirement of high dilution conditions. With this in mind, we became interested to determine whether the Lewis acid-promoted fragmentation of cyclic γ-silyloxy-β-hydroxy-α-diazocarbonyls (e.g. 1 to 2, Figure 1) we recently discovered15,16 could be applied to the direct formation of medium-sized cyclic 2-alkynones. In this letter we wish to report that bicyclic γ-silyloxy-β-hydroxy-α-diazoketones in which the Cβ-Cγ bond is the ring fusion bond, productively fragment to provide medium sized cyclic 2-alkynones in good to excellent yield.17
Figure 1.

Fragmentation of cyclic γ-silyloxy-β-hydroxy-α-diazocarbonyls yields aldehyde tethered ynoates
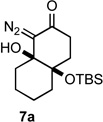
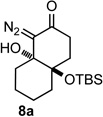
To test our hypothesis that bicyclic diazoketones could be precursors to medium sized cyclic 2-alkynones we prepared the diastereomeric bicyclo[4.4.0]decane diazo compounds 7a and 8a by the route shown in Scheme 1. d’Angelo-type18 conjugate addition provided ester 4a in 78% yield and subsequent hydrogenolysis provided the free acid 5a in 98% yield. The requisite diazo carbonyl 6a was then formed in 63% yield over two steps from acid 5a via the corresponding mixed anhydride. It was interesting to note the facility by which the pendant diazo ketone (6a) underwent intramolecular aldol-type addition to provide bicycles 7a and 8a; this intramolecular reaction was achieved with the mild base DBU over the course of 12 h. Intermolecular aldol-type additions of diazo carbonyl compounds to ketones require the generation of a lithiated diazo species, which is most often formed by deprotonation of the corresponding diazo carbonyl with either n-butyl lithium or LDA.19,20 Due to their greater electrophilicity, aldehydes do react with diazo carbonyl compounds in the presence of DBU,21 and in our case the proximity of the reacting partners likely makes the aldoltype addition more favorable. Potassium hydroxide in methanol induced the ring closure22 more efficiently and provided bicycles 7a and 8a as a separable mixture of diastereomers in 60% and 30% yield respectively after only 15 minutes.23
Scheme 1.

Preparation of bicyclic fragmentation substrates
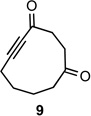
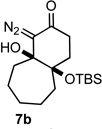
With the requisite fragmentation precursors in hand, we turned our attention to the fragmentation reaction. Treating the cis-fused bicyclo[4.4.0]decane diazo 7a with 1 molar equivalent of SnCl4 at 0 °C resulted in observable gas evolution and provided essentially pure cyclodec-5-yne-1,4-dione (9, Table 1) in near quantitative yield as determined by 1H and 13C NMR analysis. Purification resulted in some compound loss and isolation by chromatography returned cyclic ynone 9 in 80% yield. The trans-fused diastereomer 8a also productively fragmented to give alkyne 9 in 71% yield.
Table 1.
Fragmentation of fused bicyclic substrates
 | ||
|---|---|---|
| substrate | product | yield |
 |
 |
80% |
 |
9 | 71% |
 |
 |
99% |
 |
10 | 93% |
 |
 |
93% |
 |
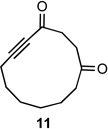
11 | 80% |
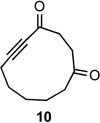
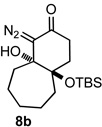
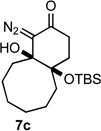
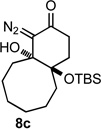
Applying the same sequence of reactions shown in Scheme 1 to the corresponding 2-silyloxycycloheptanone (3b) and 2-silyloxycyclooctanone (3c) provided the diazo bicyclo[5.4.0]undecane and bicyclo[6.4.0]dodecane systems (7b, 8b, and 7c, 8c; Table 1) respectively. While the intramolecular cyclization of diazo compound 6b occurred in high yield (92%) to give the expected diastereomeric aldol-products 7b and 8b, the cyclization of diazo compound 6c did not go to completion. Under the reaction conditions this latter system existed as an equilibrium mixture of the starting material 6c and the two diastereomeric products (7c and 8c) in a 4.6 to 1.2 to 1 ratio. Separating these compounds was trivial and the isolated bicyclic products were stable once removed from the basic reaction conditions. However, re-subjecting bicycle 7c to the cyclization reaction conditions provided the same equilibrium mixture of starting material and diastereomeric products, which highlights the reversibility of this aldol-type addition reaction. Attempts to shift the equilibrium toward the ring closed products by changing the solvent or base, or by trapping the bicyclic products as the silyl protected tertiary alcohols, failed. It seems likely that transannular interactions in the bicyclo[6.4.0]dodecane system make the ring closed form less favorable at equilibrium.
Both the cis (7b) and trans (8b) diastereomers of the bicyclo[5.4.0]undecane system productively fragmented to provide cycloundec-5-yne-1,4-dione (10) in 99% and 93% yield respectively after purification. The larger bicyclo[6.4.0]dodecane systems (7c and 8c) were also competent fragmentation substrates and provided cyclododec-5-yne-1,4-dione (11) in 93% and 80% yield respectively after purification.
In order to extend the utility of this transformation we examined the possibility of varying the size of the diazo containing ring, which would allow a variety of different medium sized rings to be formed from a common 5- or 6-membered ring precursor. Our synthetic routes to the bicyclo[4.3.0]nonane systems 15 and 16 and bicyclo[5.4.0]undecane systems 20a and 21a, in which the diazo functional group resides on a 5-membered and 7-membered ring respectively, are shown in Schemes 2 and 3. The known24 2-siloxy-2cyclohexenone (12) was converted in two steps to α-silyloxy ketone 13 by Grignard addition followed by base catalyzed silyl migration.25 The olefin was oxidatively cleaved to the carboxylic acid, which was subsequently converted to diazo ketone 14 under standard conditions. Diazo compound 14 smoothly cyclized to diastereomeric bicycles 15 and 16 in 43 and 45% yield respectively. Fragmentation of 15 and 16 provided complex mixtures which appeared to contain ynone 17 in up to 40% yield, but the desired product was isolated in at most 3% yield. Ynone 17 proved to be unstable and upon standing decomposed to an intractable mixture of products. It is not clear at this point if the low yield obtained of ynone 17 is due to an unproductive fragmentation or the inherent instability of 17.
Scheme 2.

Preparation of bicyclo[4.3.0]nonane fragmentation precursor
Scheme 3.

Preparation and fragmentation of bicyclo[5.4.0]undecane ring system
Diazo compounds 20a and 21a were prepared from ketone 13 by cross metathesis26,27 to incorporate the requisite side chain functionality and subsequent simultaneous reduction of the alkene and hydrogenolysis of the benzyl ester to provide acid 18a. Incorporation of the diazo functionality under standard conditions proceeded as expected to provide diazo ketone 19a. Subsequent intramolecular aldol-type cyclization to form the 7-membered ring failed in the presence of DBU and KOH in methanol, but diazo compound 19a did provide the expected cis- and trans-fused bicycles 20a and 21a in 44% and 14% yield respectively upon treatment with LDA. Fragmentation of bicycle 20a with SnCl4 returned cycloundec-6-yne-1,5-dione (22) in 67% yield. Ynone 22 differs from ynone 10 by the relative positions of the carbonyls. Attempts to apply this same sequence to the preparation of the bicyclo[6.4.0]dodecane system (20b and 21b), in which the diazo resides on an 8-member ring, were also made. Although the linear diazo precursor 19b was formed without incident, all attempts to induce cyclization failed to yield any of the desired bicyclic products. We suspect that in this case the unfavorable equilibrium noted above for the closure of diazo compound 6c is compounded by the fact that 8-member rings are inherently more difficult to form,28 which prevents a productive cyclization from occurring.
The preliminary results presented here demonstrate that bicyclic γ-silyloxy-β-hydroxy-α-diazoketones are competent fragmentation substrates that can provide access to 10-, 11- and 12-membered cyclo 2-alkynone products in good to excellent yields. It is not clear if the low yield obtained for the 9-membered cyclo 2-alkynone (17) is an inherent limitation of this method to form 9-membered ynones, or if this low yield is due to the instability of 17 under the reaction conditions. Further studies of this medium-sized ring forming reaction and applications of this method toward the synthesis of medium-ring containing natural products are underway and will be disclosed in future publications.
Supplementary Material
Acknowledgment
We thank Dr. John Greaves (University of California, Irvine), Dr. Jonathan Karty (Indiana University) and Mr. Bruce O’Rourke (University of Vermont) for obtaining mass spectral data and Dr. Bruce Deker (University of Vermont) for assistance with NMR characterization. This work was financially supported by the NIH (National Institute of General Medical Sciences Award Number R01GM092870). The National Science Foundation supported this work through instrumentation grant CHE-0821501.
Footnotes
Supporting Information Available Experimental details and spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Greve B, Imming P, Laufer S. Angew. Chem. Int. Ed. Engl. 1996;35:1221. [Google Scholar]
- 2.Yet L. Chem. Rev. 2000;100:2963. doi: 10.1021/cr990407q. [DOI] [PubMed] [Google Scholar]
- 3.Garcia J, Ariza X, Bach J, Berenguer R, Farras J, Fontes M, Lopez M, Ortiz J. J. Org. Chem. 2004;69:5307. doi: 10.1021/jo0495687. [DOI] [PubMed] [Google Scholar]
- 4.Covey DF, Parikh VD. J. Org. Chem. 1982;47:5315. [Google Scholar]
- 5.Saimoto H, Shinoda M, Matsubara S, Oshima K, Hiyama T, Nozaki H. Bull. Chem. Soc. Jpn. 1983;56:3088. [Google Scholar]
- 6.Marshall JA, Rothenberger SD. Tetrahedron Lett. 1986;27:4845. [Google Scholar]
- 7.Utimoto K, Kato S, Tanaka M, Hoshino Y, Fujikura S, Nozaki H. Heterocycles. 1982;18:149. [Google Scholar]
- 8.Netland KA, Gundersen LL, Rise F. Synth. Commun. 2000;30:1767. [Google Scholar]
- 9.Sydnes LK, Bakstad E. Acta Chem. Scand. 1997;51:1132. [Google Scholar]
- 10.Gleiter R, Merger M. Synthesis. 1995:969. [Google Scholar]
- 11.Pfaltz A, Bulic B, Luecking U. Synlett. 2006:1031. [Google Scholar]
- 12.Bodenmann B, Keese R. Tetrahedron Lett. 1993;34:1467. [Google Scholar]
- 13.Eaton PE, Stubbs CE. J. Am. Chem. Soc. 1967;89:5722. [Google Scholar]
- 14.Utimoto K, Tanaka M, Kitai M, Nozaki H. Tetrahedron Lett. 1978:2301. [Google Scholar]
- 15.Bayir A, Draghici C, Brewer M. J. Org. Chem. 2010;75:296. doi: 10.1021/jo902405f. [DOI] [PubMed] [Google Scholar]
- 16.Draghici C, Brewer M. J. Am. Chem. Soc. 2008;130:3766. doi: 10.1021/ja801004d. [DOI] [PubMed] [Google Scholar]
- 17.The Eschenmoser-Tanabe fragmentation has been used to generate cyclic alkynes: Reese CB, Sanders HP. Synthesis. 1981:276. Tanabe M, Crowe DF, Dehn RL. Tetrahedron Lett. 1967:3943. Tanabe M, Crowe DF, Dehn RL, Detre G. Tetrahedron Lett. 1967:3739. Eschenmoser A, Felix D, Ohloff G. Helv. Chim. Acta. 1967;50:708. doi: 10.1002/hlca.19670500235. Schreiber J, Felix D, Eschenmoser A, Winter M, Gautschi F, Schulte-Elte KH, Sundt E, Ohloff G, Kalovoda J, Kaufmann H, Wieland P, Anner G. Helv. Chim. Acta. 1967;50(7):2101. Lange GL, Hall T-W. J. Org. Chem. 1974;39:3819. Gordon DM, Danishefsky SJ, Schulte GK. J. Org. Chem. 1992;57(26):7052. Dudley and Tummatorn have recently extended the vinylogous acyl triflate fragmentation strategy to the preparation of cycloalkynes: Tummatorn J, Dudley GB. Org. Lett. 2011;13(6):1572. doi: 10.1021/ol2003308.
- 18.d’Angelo J, Desmaële D, Dumas F, Guingant A. Tetrahedron: Asymmetry. 1992;3(4):459. [Google Scholar]
- 19.Schöllkopf U, Frasnelli H. Angew. Chem. Int. Ed. Engl. 1970;9:301. [Google Scholar]
- 20.Pellicciari R, Natalini B, Sadeghpour BM, Marinozzi M, Snyder JP, Williamson BL, Kuethe JT, Padwa A. J. Am. Chem. Soc. 1996;118:1. [Google Scholar]
- 21.Jiang N, Wang J. Tetrahedron Lett. 2002;43:1285. [Google Scholar]
- 22.Burkoth TL. Tetrahedron Lett. 1969;10:5049. [Google Scholar]
- 23.The relative stereochemistry of diastereomer 7a and 8a were easily distinguishable by NMR. The conformational flexibility of cis-fused bicyclic 7a led to broad signals in the 1H and 13C NMR spectra, which sharpened upon warming. Trans-fused diastereomer 8a, on the other hand, had sharp signals in the 1H and 13C NMR spectra at room temperature. The structures of the other diastereomers were assigned by analogy based on the following consistent observations: a) the hydroxyl proton appeared further downfield in each bicyclic structure assigned as cis-fused, presumably due to intramolecular hydrogen bonding with the adjacent silyl ether; b) the methyl groups of the silyl ether showed a greater difference in chemical shift for the bicycles assigned as transfused compared to the cis-fused diastereomers.
- 24.Kadow JF, Saulnier MG, Tun MM, Langley DR, Vyas DM. Tetrahedron Lett. 1989;30:3499. [Google Scholar]
- 25.Gulías M, Rodríguez JR, Castedo L, Mascareñas JL. Org. Lett. 2003;5:1975. doi: 10.1021/ol0345632. [DOI] [PubMed] [Google Scholar]
- 26.BouzBouz S, Simmons R, Cossy J. Org. Lett. 2004;6(20):3465. doi: 10.1021/ol049079t. [DOI] [PubMed] [Google Scholar]
- 27.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168. [Google Scholar]
- 28.Illuminati G, Mandolini L. Acc. Chem. Res. 1981;14:95. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.