

Abstract
Despite making up only a minor fraction of the total cellular glutathione, recent studies indicate that the mitochondrial glutathione pool is essential for cell survival. Selective depletion of mitochondrial glutathione is sufficient to sensitize cells to mitochondrial oxidative stress (MOS)1 and intrinsic apoptosis. Glutathione is synthesized exclusively in the cytoplasm and must be actively transported into mitochondria. Therefore, regulation of mitochondrial glutathione transport is a key factor in maintaining the antioxidant status of mitochondria. Bcl-2 is resident in the outer mitochondrial membrane where it acts as a central regulator of the intrinsic apoptotic cascade. In addition, Bcl-2 displays an antioxidant-like function that has been linked experimentally to the regulation of cellular glutathione content. We have previously demonstrated a novel interaction between recombinant Bcl-2 and reduced glutathione (GSH) which was antagonized by either Bcl-2 homology-3 domain (BH3) mimetics or a BH3-only protein, recombinant Bim. These previous findings prompted us to investigate if this novel Bcl-2/GSH interaction might play a role in regulating mitochondrial glutathione transport. Incubation of primary cultures of cerebellar granule neurons (CGNs) with the BH3 mimetic, HA14-1, induced MOS and caused specific depletion of the mitochondrial glutathione pool. Bcl-2 was co-immunoprecipitated with GSH following chemical cross-linking in CGNs and this Bcl-2/GSH interaction was antagonized by pre-incubation with HA14-1. Moreover, both HA14-1 and recombinant Bim inhibited GSH transport into isolated rat brain mitochondria. To further investigate a possible link between Bcl-2 function and mitochondrial glutathione transport, we next examined if Bcl-2 associated with the 2-oxoglutarate carrier (OGC), an inner mitochondrial membrane protein known to transport glutathione in liver and kidney. Following co-transfection of CHO cells, Bcl-2 was co-immunoprecipitated with OGC and this novel interaction was significantly enhanced by glutathione monoethylester (GSH-MEE). Similarly, recombinant Bcl-2 interacted with recombinant OGC in the presence of GSH. Bcl-2 and OGC co-transfection in CHO cells significantly increased the mitochondrial glutathione pool. Finally, the ability of Bcl-2 to protect CHO cells from apoptosis induced by hydrogen peroxide was significantly attenuated by the OGC inhibitor phenylsuccinate. These data suggest that GSH binding by Bcl-2 enhances its affinity for the OGC. Bcl-2 and OGC appear to act in a coordinated manner to increase the mitochondrial glutathione pool and enhance resistance of cells to oxidative stress. We conclude that regulation of mitochondrial glutathione transport is a principal mechanism by which Bcl-2 suppresses MOS.
Keywords: Reduced Glutathione (GSH), Mitochondrial Oxidative Stress (MOS), Bcl-2, 2-Oxoglutarate Carrier (OGC)
INTRODUCTION
Mitochondrial oxidative stress (MOS) plays a key role in the pathology underlying several neurodegenerative diseases, including amyotrophic lateral sclerosis, Parkinson’s disease, and Alzheimer’s disease [1]. Accordingly, elucidating the pathways that regulate the mitochondrial oxidant/antioxidant balance is essential to develop novel therapeutics for neurodegeneration. Glutathione is an endogenous tri-peptide antioxidant, and a key player in averting MOS and evading apoptosis [reviewed in 2]. It has been previously shown that selective depletion of mitochondrial glutathione sensitizes cells to oxidative or nitrosative stress [3, 4]. Moreover, glutathione depletion can induce apoptosis directly through opening of the mitochondrial permeability transition pore [5]. In addition, gamma-glutamylcysteine synthetase (γ–GCS) knockout mice, in which glutathione synthesis is inhibited and glutathione is depleted, display significant apoptotic cell death in multiple tissues [6]. These findings demonstrate that maintenance of cellular glutathione and in particular, the mitochondrial glutathione pool is crucial for cell survival [7, 8].
Glutathione synthesis occurs exclusively in the cytosol due to the fact that the enzymes required for its synthesis are absent within mitochondria [9]. Furthermore, glutathione has an overall negative charge at physiological pH and mitochondria exhibit a large negative membrane potential; consequently, glutathione transport into mitochondria cannot be explained by simple diffusion [9, 10]. Previously, two inner mitochondrial membrane (IMM) anion carriers were identified in kidney and liver as glutathione transporters, the 2-oxoglutarate carrier (OGC; Slc25a11) and the dicarboxylate carrier (DIC; Slc25a10) [11–14]. Overexpression of either OGC or DIC in a rat renal proximal tubular cell line (NRK-52E cells) significantly enhanced mitochondrial glutathione transport and protected these cells from chemically-induced apoptosis, such as that induced by tert-butyl hydroperoxide [15, 16].
In the context of the CNS, few studies have examined the mechanisms responsible for mitochondrial glutathione transport. In one study, glutathione transport into isolated rat brain mitochondria seemed to be influenced most by inhibitors of the tricarboxylate carrier rather than OGC or DIC [17]. However, another study showed that an inhibitor of DIC, butylmalonate, significantly decreased the glutathione content of isolated mouse brain mitochondria, suggesting that DIC may be the major glutathione transporter in mouse cerebral cortical mitochondria [18]. The authors of this study also showed that both OGC and DIC are expressed in cortical neurons and astrocytes. These studies suggest that multiple IMM anion transporters might be involved in mitochondrial glutathione transport in the CNS. Yet, essentially nothing is known about how the function of these IMM glutathione transporters is regulated.
Previous studies have shown that the anti-apoptotic protein, Bcl-2, displays an antioxidant-like effect in response to either exogenous oxidative stress or glutathione depletion [19, 20]. Overexpression of Bcl-2 leads to an increase in the cellular content of glutathione [21, 22]. In contrast, Bcl-2 knockout mice show reduced glutathione levels and glutathione peroxidase activity in brain tissue and demonstrate enhanced susceptibility to MOS-induced neuronal cell death [23]. Thus, the antioxidant-like function of Bcl-2 depends, in large part, on its potential to regulate the cellular glutathione status. In this context, we have previously shown that recombinant Bcl-2 is capable of directly binding to GSH in vitro, an interaction that is antagonized by the Bcl-2 homology-3 domain (BH3) mimetics, HA14-1 and compound 6, as well as the BH3-only protein, Bim [24]. Interestingly, several BH3-only proteins are known to induce a pro-oxidant state at mitochondria suggesting that disruption of this Bcl-2/GSH interaction might be an underlying factor in this effect [25, 26].
Collectively, these findings prompted us to hypothesize that Bcl-2 might be a key regulator of the mitochondrial glutathione pool. Here, we show that Bcl-2 interacts with GSH in intact primary cerebellar granule neurons (CGNs). As we have shown previously using recombinant proteins, this Bcl-2/GSH interaction is disrupted by the BH3 mimetic, HA14-1. Consistent with a central role for Bcl-2 in maintenance of the mitochondrial glutathione pool, both HA14-1 and Bim inhibited mitochondrial GSH transport. Most significantly, in co-transfected CHO cells, Bcl-2 co-immunoprecipitates with OGC and this novel interaction is markedly enhanced by glutathione monoethylester (GSH-MEE). Moreover, Bcl-2 and OGC co-expression significantly increases the mitochondrial glutathione pool. Finally, we show that the ability of Bcl-2 to protect cells from apoptosis induced by hydrogen peroxide depends on OGC activity. We conclude that Bcl-2 is a novel interacting partner for OGC and a central regulator of the mitochondrial glutathione pool. This newly discovered property of Bcl-2 suggests a molecular mechanism by which Bcl-2 protects cells from oxidative injury.
MATERIALS AND METHODS
Materials
Ethacrynic Acid, Triton X-100, DTT, anti-tubulin antibody, DAPI, and phenylsuccinic acid were received from Sigma Aldrich (St. Louis, MO). HA14-1 was purchased from Alexis Biochemicals (Enzo Life Sciences, Plymouth Meeting, PA). Glutathione assay kit was obtained from Oxford Biomedical (Rochester Hills, MI). Mitochondrial Cytosolic fractionation kit was purchased from Biovision (Mountain View, CA). MDA assay kit was obtained from OxisResearch (Percipio Bioscience, Foster City, CA). Anti-Cox-IV was purchased from Cell Signalling (Beverly, MA). Glutathione monoethylester and rotenone were received from Calbiochem (San Diego, CA). Anti-V5 antibody was purchased from Abcam (Cambridge, MA). Recombinant BimL was obtained from R&D systems (Minneapolis, MN). The anti-GSH antibody and protein A/G beads were received from Santa Cruz Biotechnology (Santa Cruz, CA). Disuccinimyidyl suberate (DSS) was purchased from Pierce Chemical (Rockford, IL). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA). Anti-Bcl-2 antibody was from BD Pharmigen (Franklin Lakes, NJ). Mouse TrueBlot® ULTRA Anti-Mouse Ig HRP secondary antibody was obtained from eBioscience (San Diego, CA). GST tagged Slc25a11 (GST-OGC) recombinant protein and Slc25a11 (OGC) antibody were purchased from Novus Biologicals (Littleton, CO). Recombinant Bcl-2 protein was obtained from Calbiochem (Darmstadt, Germany). Optimem media was purchased from Gibco (Carlsbad, CA). Maxiprep Kit was obtained from Qiagen (Valencia, CA). Anti-active caspase-3 was purchased from Promega (Madison, WI). Fitc secondary antibody for immunohistochemistry was from Jackson ImmunoResearch Inc (West Grove, PA). ECL, Percoll, and secondary antibodies for immunoblotting were purchased from GE Life Sciences (Piscataway, NJ). V5-OGC plasmid was a generous gift from Dr. Lash, Wayne State University (Detriot, MI). The Bcl-2 plasmid was a generous gift from Dr. Hardwick, Johns Hopkins University (Baltimore, MD).
Cerebellar Granule Neuron (CGN) Culture
CGNs were isolated from P7 Sprague Dawley rat pups and cultured as previously described [24]. All animal manipulations were performed in accordance with and under approval of the University of Denver Institutional Animal Care and Use Committee.
Immunohistochemistry
CGNs or CHOs were transfected and/or treated as described under the Figure Legends section and fixed with 4% paraformaldehyde. Next, cells were blocked and permeabilized in PBS, pH 7.4 with 5% BSA and 0.2% Triton X-100 for 1 h, followed by incubation with the primary antibody overnight at 4°C diluted in 0.2% Triton X-100 and 2% BSA in PBS. After which, the primary antibody was removed and cells were washed 5X with PBS over 30 min. Next, the cells were incubated with secondary antibody and DAPI for 1 h at room temperature, diluted in 0.2% Triton X-100 and 2% BSA in PBS. Cells were washed 5X with PBS over 30 min, and placed in anti-quench. Images were captured using a Zeiss Axioplan 2 fluorescence microscope equipped with a Cooke Sensicam CCD camera and Slidebook Image analysis software (Intelligent Imaging Innovations, Inc Denver, CO).
CHO cell culture
K1-CHO (Chinese Hamster Ovary) cells were plated on 35-mm diameter plastic dishes in Ham’s F12 media containing 10% fetal bovine serum, 2 mM L-glutamine and (100 U/mL/100 μg/mL) penicillin/streptomycin. Cells were cultured overnight at 37°C in 10% CO2. The following day cells were prepared for transfection or treatment, at which point cultures were 60–80% confluent.
Plasmid Preparation
DsRed2, Bcl-2, and V5-OGC plasmids were transformed using 50 ng of plasmid in JM109 Escherichia coli (E. Coli) cells, and grown on LB agar plates containing 35 μg/mL kanamycin sulfate or 100 μg/mL ampilicillin sodium salt at 37°C overnight. Starter cultures were grown in LB broth with 35 μg/mL kanamycin sulfate or 100 μg/mL ampilicillin sodium salt for 8 h at 37°C, and diluted 1:250 into overnight cultures, for plasmid purification using the Qiagen Maxi Prep Kit (Valencia, CA). DNA concentrations were determined using a ThermoScientific NanoDrop 2000.
Transfection
DsRed2 (Con), Bcl-2, or V5-OGC were used at a concentration of 5 μg/mL. Plasmids were transfected using a standard Lipofectamine 2000 protocol. CHO cells cultures were incubated with the plasmid/Lipofectamine 2000 mixture in Opti-MEM, for 6 h at 37°C and 10% CO2. Transfection was removed and cell cultures were placed in 1 mL of Ham’s F12 media and incubated overnight at 37°C and 10% CO2. Indicated treatments were administered 24 h post-transfection.
Mitochondrial/Cytosolic Fractionation
Cells were treated as indicated in the “Results” or “Figure Legend” sections after which the media was aspirated and cells were washed 1X in ice cold PBS, pH 7.4. 200 μL of cytosolic buffer (provided in the kit, diluted 1:5 in ddH2O, with added protease inhibitor cocktail and 1mM DTT as per the manufacturer’s recommendations) was added to the cells and allowed to incubate on ice for 20 min. Cells were scraped and harvested, then homogenized with 40 passes of a dounce homogenizer. Samples were spun down at 720 rcf for 10 min at 4°C. The supernatant from each sample was transferred to a new tube labeled mitochondrial fraction and spun at 10,000 rcf for 30 min at 4°C. The supernantant was then transferred to a new tube labeled cytosolic fraction and the pellet in the mitochondrial fraction tube was resuspended in 150 μL mitochondrial buffer (provided in the kit, with added protease inhibitor cocktail and 1mM DTT as per the manufacturer’s recommendations). Samples were measured for glutathione content as described below.
Glutathione Assay
Total Glutathione (GSH+GSSG) was measured using an assay kit (DTNB) from Oxford Biomedical, following the manufacturer’s protocol. All glutathione measurements were normalized to protein concentration.
MDA Assay
MDA was measured using an MDA assay kit from OxisResearch, following the manufacturer’s protocol. All MDA measurements were normalized to protein concentration.
Immunoblot analysis
Immunoblot analysis was completed as previously described in [24].
GSH Transport Assay
GSH transport into isolated mitochondria was measured as previously published [24]. 20 μL of rat brain mitochondria were added to 230 μL of GSH transport buffer (5 mM HEPES, pH 7.2, 220 mM mannitol, 70 mM sucrose, 0.1 mM EDTA, 0.1% BSA (fatty acid-free), 5 mM succinate, and 1 mM potassium phosphate) at room temperature. The isolated mitochondria were then preincubated for 20 min with either 0.5 μL Me2SO (vehicle, Con), 20 μM HA14-1 (HA14), or 2 μM recombinant Bim (Bim). A trace amount (0.5 μCi) of radiolabeled [3H]GSH in buffer containing unlabeled (cold) GSH at a final concentration of 50 μM was then added to the isolated mitochondria, vortexed and incubated for 15 sec at room temperature. 1 mL volume of icecold transport buffer was then added to each tube, and the samples were pelleted and washed 2X with 500 μL ice-cold buffer, and then 100 μL of 1N NaOH was added to dissolve the final pellets. [3H]GSH uptake into the isolated mitochondria was counted by liquid scintillation. The data were calculated as percentages of [3H]GSH uptake relative to Con.
Isolation and treatment of rat brain mitochondria
Mitochondria were isolated as previously described in [24]. Mitochondria were treated as follows; For 4 h GSH-MEE Co-incubation, mitochondria were incubated with 2 mM GSH-MEE in combination with either vehicle (Con), 20 μM HA14-1, or 10 μM rotenone for 4 h 37°C, 300 rpm. For the 2 h GSH-MEE pre-load/2 h wash out, mitochondria were pre-incubated with 2 mM GSH-MEE for 2 h, 37°C, 300 rpm, washed 2X with mitochondrial isolation buffer, and then inbuated with either vehicle (Con), 20 μM HA14-1, or 10 μM rotenone for 2 h, 37°C, 300 rpm. All described treatments were completed in mitochondrial isolation buffer; 0.64 M sucrose, 2mM EDTA, and 20mM Tris-HCl, pH 7.4. The isolated mitochondria were then washed 3X in mitochondrial isolation buffer and assayed for glutathione content (Described above).
Immunoprecipitation
CHO cells transfected with Bcl-2 and V5-OGC (5 μg each) were treated with either vehicle or 2 mM GSH-MEE for 4 h, 37°C, 10% CO2 and lysed with 0.1% Triton X-100/Wahl buffer as described previosly (24), containing either 1 mM DTT or 2 mM GSH-MEE. Lysates were then immunoprecipitated using a monoclonal V5 antibody in 0.1% Triton X-100/Wahl buffer containing either 1 mM DTT or 2 mM GSH-MEE overnight, 4°C, mixing by inversion. Next, 50 μL or protein A/G agarose beads were incubated with the samples for 4 h, 4°C, mixing by inversion. Immnue complexes were washed 3X and resolved by SDS-PAGE as previously described (24). Whole cell lysates contained 200 μg of protein. Immunoblot analysis was completed for Bcl-2 (using Mouse TrueBlot® ULTRA Anti-Mouse Ig HRP secondary antibody to eliminate the light chain) and V5. Primary CGNs were pre-incubated with 20 μM HA14-1 or vehicle (0.4%DMSO) for 2 h, 37°C, 10% CO2 and samples were cross-linked with 500 μM DSS for 30 min, 37°C, 10% CO2. CGNs were lysed in 1% Triton X-100/Wahl buffer with 50 mM glycine (to quench the cross-link), after which lysates were immunoprecipitated using a monoclonal GSH antibody and Protein A/G agarose beads as described above, and subjected to SDS-PAGE as described previously (24). Recombinant immunoprecipitation experiments were performed using 50 ng of both recombinant Bcl-2 and recombinant GST-OGC, incubated in 0.1% Triton/Wahl buffer with 10 μM GSH overnight in the presence of either a monoclonal Bcl-2 antibody or a monoclonal Slc25a11 (OGC) antibody. Samples were immunoprecipiated as described above and subjected to SDS-PAGE as described previously (24). Ttotal samples contained 50 ng of both Bcl-2 and GST-OGC recombinant proteins.
RESULTS
The BH3 mimetic, HA14-1, induces MOS and apoptosis of CGNs
Consistent with our previous observations [24, 27], incubation of CGNs with the BH3 mimetic, HA14-1, induced an increase in active caspase-3, degradation of the microtubule network, and nuclear fragmentation indicative of apoptosis (Figure 1A). Exposure of CGNs to HA14-1 significantly increased the concentration of malondialdehyde (MDA) in mitochondrial fractions, demonstrating an induction of MOS (Figure 1B).
Figure 1. The BH3 mimetic, HA14-1, induces mitochondrial oxidative stress and apoptosis of CGNs.

A. CGNs were treated for 24 h with either HA14-1 (HA14; 15 μM) or vehicle, control (Con, 0.3% DMSO). Top panels: Fitc, tubulin; DAPI, nuclei; Cy3, active caspase-3. Arrows indicate apoptotic cells with either condensed/fragmented nuclei, or immunoreactivity for active caspse-3. Bottom panels: decolorized DAPI images to show nuclear morphology. B. CGNs were treated with either vehicle (Con) or HA14-1 (HA14; 20 μM) for 4 h and then fractionated into cytosolic and mitochondrial fractions. Mitochondrial fractions were then measured for MDA concentration, or lipid peroxidation levels. Data are shown as μ moles of MDA present per μg of protein and analyzed using a Student’s T-test, ***p<0.001, n=4.
HA14-1 selectively depletes the mitochondrial glutathione pool in CGNs
We have previously shown that CGN apoptosis induced by HA14-1 is prevented by glutathione [24, 27]. Therefore, we measured mitochondrial and cytosolic pools of total glutathione in CGNs incubated in the absence and presence of HA14-1. CGNs treated with HA14-1 for 4 h displayed selective depletion of total mitochondrial glutathione to approximately 50% of the control level (Figure 2A). In contrast, CGNs incubated with ethacrynic acid, a compound that forms nonfunctional adducts with GSH, showed significant depletion of both the mitochondrial and cytosolic pools of glutathione (Figure 2A). Cox-IV blots were performed to demonstrate pure fractionation of mitochondria and cytoplasm (Figure 2B). The fraction of total cellular glutathione made up by mitochondrial (~10%) versus cytosolic (~90%) pools were consistent with previous studies (Figure 2C). In addition, control levels of total mitochondrial glutathione were an average of 6.78 +/− 0.33 nmoles/mg of protein (n=4) and these results are consistent with previous studies [28–30].
Figure 2. HA14-1 selectively depletes mitochondrial glutathione in CGNs.

A. CGNs treated with either vehicle (Con), HA14-1 (HA14; 20 μM), or ethacrynic acid (EA; 100 μM) for 4 h and then separated into mitochondrial and cytosolic fractions. Total glutathione content of each fraction was measured using a DTNB colorimetric assay. Data are shown as percent of control glutathione (GSH+GSSG) and analyzed using one-way ANOVA with a post-hoc Tukey’s analysis. *p<0.05, **p<0.01 vs Con, n=4. Control levels of total mitochondrial glutathione were an average of 6.78 +/− 0.33 nmoles per mg of protein. B. Immunoblot shown is for Cox-IV to demonstrate the purity of the mitochondrial and cytosolic fractions in (A). C. Graph showing the representative pools of cellular glutathione between mitochondrial and cytosolic fractions, n=4.
Bcl-2 co-immunoprecipitates with GSH following chemical cross-linking in CGNs; an interaction antagonized by the BH3 mimetic, HA14-1
We have previously shown that recombinant Bcl-2 directly interacts with GSH in vitro [24]. To determine if Bcl-2 interacts with GSH in intact cells we utilized a chemical cross-linking approach and measured the co-immunoprecipitation of Bcl-2 with GSH from CGNs. Cultured CGNs were cross-linked using DSS (a cell permeable, homobifunctional cross-linker possessing two amine-reactive N-hydroxysuccinimide esters), and upon quenching the cross-link with excess glycine, Bcl-2 was co-immunoprecipitated with GSH using a GSH antibody (Figure 3). In CGNs pre-incubated with DMSO vehicle and then cross-linked and immunoprecipitated with a GSH antibody, both monomeric and dimeric forms of Bcl-2 were co-immunoprecipitated with GSH. However, pre-treatment of the CGNs, prior to cross-linking, with HA14-1 significantly decreased the amount of both the monomeric and dimeric forms of Bcl-2 co-immunoprecipitated with GSH. These data strongly suggest that Bcl-2 and GSH interact in intact CGNs, and this interaction can be antagonized by the BH3 mimetic, HA14-1. Futhermore, we completed similar experiments in isolated rat brain mitochondria and found that HA14-1 also antagnoized the interaction between Bcl-2 and GSH (data not shown). These findings are consistent with our previously published data in which we showed that recombinant Bcl-2 and GSH interact in a direct manner. Moreover, this interaction shows specificity between Bcl-2 and GSH, since other Bcl-2 family proteins, such as Bcl-xL and Bcl-w, did not interact directly with GSH [24].
Figure 3. Bcl-2 co-immunoprecipitates with GSH following chemical cross-linking in CGNs, an interaction antagonized by the BH3 mimetic, HA14-1.
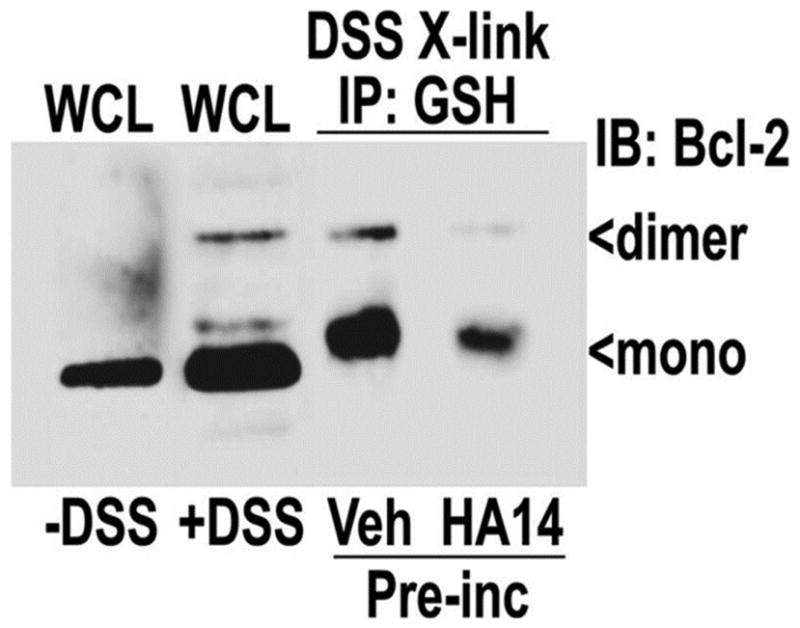
CGNs were pre-incubated with either vehicle (0.4% DMSO) or 20 μM HA14-1 for 2 h, after which they were cross-linked (x-link) with DSS, which was quenched upon lysing (see materials and methods). Lysates were immunoprecipitated (IP) with a monoclonal antibody to GSH. Immune complexes were resolved by SDS-PAGE and immunoblotted (IB) for Bcl-2. Mono=monomeric, dimer=dimeric, WCL=whole cell lysate.
HA14-1 depletes glutathione from isolated mitochondria. HA14-1 and Bim inhibit mitochondrial GSH transport in vitro
We examined the effects of HA14-1 on glutathione pools in isolated rat brain mitochondria. Incubation of isolated mitochondria with HA14-1 induced a significant depletion of total glutathione (GSH+GSSG) when mitochondria were co-incubated in the presence of GSH-MEE (Figure 4A, n=3, **p<0.01 vs. Con) for 4h. HA14-1 has previously been shown to have some uncoupling and inhibitory effects on mitochondrial respiration at high concentrations [31]. To exclude the possibility that HA14-1 (used at 20 μM) was depleting glutathione in isolated rat brain mitochondria by inhibiting mitochondrial respiration, and in turn inducing oxidative stress, we compared its effects to those of the complex I inhibitor, rotenone, which we used at a concentration significantly higher than its previously shown IC50 value for complex I. [32,33]. While HA14-1 (20 μM) significantly depleted total mitochondrial glutathione content, rotenone (10 μM) had no such effect (Figure 4A). In another series of experiments, we pre-loaded isolated mitochondria with 2 mM GSH-MEE for 2 h and then washed the mitochondria prior to adding HA14-1 or rotenone for an additional 2 h. Following the 2 h pre-incubation with GSH-MEE, mitochondrial glutathione content was increased by approximately 250% over non-loaded controls (data not shown). Incubation with HA14-1 induced a marked reduction of total glutathione in the pre-loaded mitochondria, suggesting that the Bcl-2 inhibitor actually stimulated glutathione efflux (Figure 4B, n=6, *p<0.05 vs. Con). In contrast, incubation with rotenone had no effect on mitochondria pre-loaded with GSH-MEE. We compared the amount of total glutathione in freshly isolated mitochondria versus the Con samples in the treatment paradigms of Figures 4A and 4B; no differences in the levels of total glutathione were observed (an average of 17.84 +/− 1.69 nmoles/mg, n=3, data not shown). Finally, we assessed the effects of HA14-1 or recombinant Bim on the transport of [3H]GSH into isolated mitochondria. Both Bcl-2 inhibitors, HA14-1 (20μM) or Bim (2μM), significantly inhibited GSH uptake into mitochondria (Figure 4C, #p<0.01 vs Con and *p<0.05 vs Con, n=3). These data show that HA14-1 and Bim suppress mitochondrial GSH transport and that HA14-1 is capable of inducing efflux of glutathione from isolated mitochondria.
Figure 4. HA14-1 depletes glutathione from isolated rat brain mitochondria, in addition HA14-1 and Bim inhibit mitochondrial GSH transport.

A. Isolated rat brain mitochondria were incubated in mitochondrial buffer with a combination of 2 mM GSH-MEE and either vehicle (Con), HA14-1 (20 μM), or rotenone (10 μM) for 4 h. Samples were then washed 3X in mitochondrial buffer and assyed for glutathione (GSH+GSSG) content. Data are shown as percent of control glutathione (GSH+GSSG) and analyzed using a one-way ANOVA with a post-hoc Tukey’s analysis. ** p<0.01 vs Con, n=3. B. Isolated rat brain mitochondria were incubated in mitochondrial buffer with GSH-MEE (2 mM) for 2 h at 37°C, washed 2X, and then incubated in mitochondrial buffer with either Con, HA14-1 (HA14; 20 μM), or rotenone (10 μM) for 2h at 37°C. After which samples were washed 3X with mitochondrial buffer and assayed for glutathione (GSH+GSSG) content using a DTNB colorimetric assay, data are normalized to protein concentration. Two controls are shown because some of the experiments performed with HA14-1 were carried out separately from several of the experiments performed with rotenone. Data are represented as percent of the respective control glutathione (GSH/GSSG) and analyzed using a Student’s T-test. *p<0.05 vs Con, n=6. Control total mitochondrial glutathione levels for the isolated rat brain mitochondria experiments were an average of 17.84 +/− 1.69 nmoles/mg of protein. C. Isolated rat brain mitochondria were incubated with either vehicle (Con; 0.4%DMSO), HA14-1 (HA14; 20 μM) or Bim (2 μM) for 20 min and then 0.5μ Ci of [3H]GSH for 15 sec at room temperature. [3H]GSH was measured as decribed under Materials and Methods. Data are respresented as percent of Con (CPM values) and analyzed using a one-way ANOVA with post-hoc Tukey’s anaylsis. **p<0.01 vs Con, *p<0.05 vs Con, n=3 experiments completed in triplicate.
Bcl-2 co-immunoprecipitates with OGC and this interaction is enhanced by GSH-MEE
HA14-1, a BH3 mimetic that binds Bcl-2 within the hydrophobic groove, selectively depletes mitochondrial glutathione and antagonizes the interaction between Bcl-2 and GSH in CGNs, and inhibits GSH uptake/stimulates glutathione efflux in isolated brain mitochondria. These data suggest a possible role for Bcl-2 in regulating mitochondrial glutathione transport. It has been previously shown that the IMM anion transporters OGC and DIC are responsible for glutathione transport across the inner mitochondrial membrane into the matrix in liver and kidney [11, 12]. Therefore, we next examined the potential of Bcl-2 to interact with the OGC in a transient co-transfection system. CHO cells overexpressing Bcl-2 and a V5-tagged OGC were lysed in 0.1% Triton X-100 with 1 mM DTT, and immunoprecipitated with or without a V5 antibody. Bcl-2 co-immunoprecipitated with V5-OGC upon addition of the V5 antibody but did not precipitate in the presence of protein A/G agarose beads alone (Figure 5A). Next, we determined the effects of GSH-MEE on the interaction of Bcl-2 with OGC. CHO cells overexpressing Bcl-2 and V5-OGC were treated with 2 mM GSH-MEE for 4 h, and cells were lysed and immunoprecipitated in buffer also containing 2 mM GSH-MEE. In control conditions, CHO cells overexpressing Bcl-2 and V5-OGC were untreated, lysed, and immunoprecipitated in buffer containing 1 mM DTT. The relative amount of Bcl-2 co-immunoprecipitated with V5-OGC was appreciably increased in the presence of GSH-MEE versus DTT (Figure 5B). Densitometric analysis using normalization of the Bcl-2 band intensity to the V5-OGC band intensity revealed a significant increase (approximately five-fold) in Bcl-2 co-immunoprecipitation with V5-OGC in the presence of GSH-MEE versus DTT (Figure 5C, n=9, p<0.05 vs. DTT). GSH-MEE enhanced the interaction between Bcl-2 and OGC irrespective of the time in which it was added into the experiment, such as treating the cells only or simply adding GSH-MEE to the immunoprecipitation buffer (data not shown). Moreover, in a cell free system, recombinant Bcl-2 also interacted directly with recombinant GST-tagged OGC in the presence of GSH (Figure 5D). Both recombinant Bcl-2 and recombinant GST-OGC co-immunoprecipitated with a Bcl-2 antibody and a Slc25a11 (OGC) antibody (Figure 5D). Finally, upon the addition of ethacrynic acid to the immunoprecipitation buffer (which already contained GSH therefore ethacrynic acid was used to deplete the GSH), the interaction between recombinant OGC and recombinant Bcl-2 was antagonized (Figure 5E). These results indicate that Bcl-2 is an interacting partner for OGC and this novel protein-protein interaction is enhanced by GSH.
Figure 5. Bcl-2 co-immunoprecipitates with V5-OGC from co-transfected CHO cells, GSH-MEE enhances the interaction.

A. CHO cells were co-transfected with Bcl-2 and V5-OGC (5μg each) lysed in Wahl buffer containing 0.1% TritonX-100 and 1 mM DTT, and immunoprecipitated (IP) with a monoclonal V5 antibody. Immune complexes were then resolved by SDS-PAGE, immunoblotted for V5 and Bcl-2. WCL= Whole cell lysate, IP =immunoprecipitate, −V5 antibody=Protein A/G agarose alone was used in the IP. B. Same as in (A), except at 24 h post-transfection cells were treated with 2 mM GSH-MEE for 4 h (and GSH-MEE was added to the IP buffer). Alternatively, 1 mM DTT was added to the IP buffer. C. Densitometry comparing pixel density of co-immunoprecipitated Bcl-2 with V5-OGC in the presence of either 1 mM DTT (Con) or 2 mM GSH-MEE. Data are normalized to the amount of V5 immunoprecipitated and are expressed as a percent of Bcl-2 immunoprecipitated in the presence of DTT (Con). Data was analyzed using a Student’s T-test, *p<0.05, n=9. D. Recombinant Bcl-2 and recombinant GST-OGC were co-immunoprecipitated with either anti-Bcl-2 (Bcl-2 IP) or anti-Slc25a11 (OGC IP) in 0.1% Triton X-100/Wahl buffer with 11.1 μM GSH. Immune complexes were resolved by SDS-PAGE, immunoblotted for Bcl-2 and Slc25a11 (OGC). E. Recombinant Bcl-2 and recombinant GST-OGC were co-immunoprecipiated with anti-Bcl-2 in either 0.1% Triton X-100/Wahl buffer with 10 μM GSH (−EA) or 0.1% Triton X-100/Wahl buffer with 10 μM GSH and 100 μM ethacrynic acid (+EA). Immune complexes were resolved by SDS-PAGE, immunblotted for Bcl-2 and Slc25a11 (OGC).
Co-expression of Bcl-2 and OGC increases mitochondrial glutathione
We next examined the effects of Bcl-2 and OGC expression on mitochondrial glutathione content. Here, we utilized CHO cells expressing DsRed-2 (Con), Bcl-2, V5-OGC, or a combination of Bcl-2 and V5-OGC. At 24 h post-transfection, cells were lysed and mitochondrial and cytosolic factions were obtained by differential centrifugation and measured for total glutathione content. Control, Bcl-2, and OGC expressing CHO cells showed similar mitochondrial glutathione content (Figure 6A). However, with the co-expression of both Bcl-2 and OGC, total mitochondrial glutathione was significantly increased compared to Bcl-2 expression alone (Figure 6A, n=3, *p<0.05 vs. Bcl-2/OGC). Immunoblot analysis confirmed the expression of Bcl-2 and OGC in the appropriate mitochondrial fractions (Figure 6B), and a Cox-IV blot demonstrated the relative purity of the mitochondrial fractions (Figure 6C). Control levels of total mitochondrial glutathione were an average of 12.69 +/− 1.81 nmoles/mg of protein. These data suggest that Bcl-2 and OGC work in a concerted manner to increase the mitochondrial glutathione pool.
Figure 6. Co-transfection with Bcl-2 and V5-OGC significantly increases mitochondrial glutathione content.

A. CHO cells were transfected with control (DsRed), Bcl-2, V5-OGC, or a combination of Bcl-2 and V5-OGC (5μg each). After 24 h, cells were fractionated and mitochondrial glutathione (GSH+GSSG) content was measured using DTNB, and normalized to protein concentration. Data are expressed as percent of control glutathione and analyzed using a one-way ANOVA with a post-hoc Tukey’s analysis *p<0.05 vs Bcl-2/OGC, n=3. Control levels of total mitochondrial GSH were at an average of 12.69 +/− 1.81 nmoles/mg of protein. B. Immunoblots of mitochondrial fractions indicating expression of Bcl-2, V5-OGC, or a combination of both in transfected CHO cells. C. Immunoblot for Cox-IV indicating purity of mitochondrial and cytosolic fractions.
Bcl-2 protection from hydrogen peroxide toxicity is dependent on the function of OGC
Finally, we examined the consequences of increasing the mitochondrial glutathione pool via co-expression of Bcl-2 and OGC in transiently transfected CHO cells. In agreement with many previous studies [19, 34, 35], overexpression of Bcl-2 alone significantly protected CHO cells from hydrogen peroxide-induced apoptosis as assessed by measuring the percentage of adherent cells expressing active caspase-3 (Figure 7A, n=4, *p<0.05 vs Con+H2O2, **p<0.01 vs Con+H2O2). In a similar manner, expression of either OGC alone or OGC in combination with Bcl-2 also significantly protected cells from hydrogen peroxide. Interestingly, co-incubation of CHO cells with the OGC inhibitor, phenylsuccinate (PhS), at a concentration (500 μM) which was titrated down to a maximally non-cytotoxic level, did not significantly enhance the active caspase-3 staining either on its own or in combination with the hydrogen peroxide exposure (Figure 7B). Overexpression of OGC, in either the absence or presence of Bcl-2, maintained its protective effect against hydrogen peroxide even in the presence of this low dose of PhS (Figure 7C, n=4, *p<0.05 vs Con+PhS and H2O2, #p<0.01 vs Bcl-2+PhS and H2O2). These results are consistent with the low dose of PhS being sufficient to only inhibit the transport capabilities of the endogenous OGC within CHO cells. In marked contrast to the effects observed with OGC, Bcl-2 appeared to lose its protective effect against hydrogen peroxide when the endogenous levels of OGC within CHO cells were simultaneously inhibited by PhS. This point is demonstrated by the findings that Bcl-2 alone was ineffective at attenuating caspase-3 activation in CHO cells treated concomitantly with PhS and hydrogen peroxide (Figure 7C). These results suggest that Bcl-2 protects cells from hydrogen peroxide-induced apoptosis via an OGC-dependent mechanism (i.e., via an increase in mitochondrial glutathione transport resulting in an enhancement of the mitochondrial glutathione pool).
Figure 7. Bcl-2 protection against H2O2-induced apoptosis is OGC-dependent.

A. CHO cells transfected with DsRed, Bcl-2, V5-OGC, or a combination of Bcl-2 and V5-OGC were treated with 100 μM H2O2 for 24 h and stained for active caspase-3. Data are represented as mean +/− SEM. Data were analyzed using a one-way ANOVA with a post-hoc Tukey’s analysis. *p<0.05 vs Con+H2O2, **p<0.01 vs Con+H2O2, *** p<0.001 vs Con+H2O2, and #p<0.001 vs Con, n=4. B. CHO cells transfected as in (A) and treated with either vehicle, 100 μM H2O2 for 24 h alone or in combination with 500 μM phenylsuccinate (PhS), or 500 μM phenylsuccinate (PhS) alone for 24 h and stained for active caspase- 3. NS=not significant, data are represented as mean +/− SEM and analyzed using a Student’s T-test. C. CHO cells transfected as in (A), treated with a combination of 100 μM H2O2 and 500 μM phenylsuccinate (PhS) for 24 h and stained for active caspase- 3. Data are represented as mean +/− SEM. Data were analyzed using a one-way ANOVA with a post-hoc Tukey’s analysis. *p<0.05 vs Con+PhS and H2O2 and **p<0.01 vs Con+PhS and H2O2, n=4.
DISCUSSION
Bcl-2 is a key sentinel of the mitochondria that acts to suppress the intrinsic apoptotic cascade via its inhibitory interactions with pro-apoptotic family members [36]. Beyond its classical anti-apoptotic role, Bcl-2 is also known to have a critical antioxidant-like function [19]. This antioxidant-like property has been linked experimentally to the regulation of cellular glutathione content [reviewed in 37]. For example, overexpression of Bcl-2 increases cellular glutathione via an enhancement of glutathione synthesis and a diminution of cellular glutathione efflux [21, 38, 39]. Accordingly, overexpression of Bcl-2 protects cells from oxidative stress induced by glutathione depleting agents [20, 40, 41]. Furthermore, the protective effects of Bcl-2 against oxidative damage are significantly diminished by depriving cells of glutathione precursors, for instance through incubation in cysteine/methionine-deficient media [42]. Finally, Bcl-2 null mice show altered antioxidant enzyme activities in brain, such as decreased glutathione peroxidase activity, and demonstrate increased sensitivity to neural oxidative injury [23]. Many studies have provided evidence of a connection between cellular glutathione status and the antioxidant-like action of Bcl-2. However, the specific molecular mechanism(s) underlying this relationship between Bcl-2 and glutathione has not yet been elucidated.
In our previous work we demonstrated a novel interaction between recombinant Bcl-2 and GSH in a cell-free assay [24]. We showed that two dissimilarly structured BH3 mimetics predicted to bind in the hydrophobic surface groove (i.e., the BH3 groove) of Bcl-2, HA14-1 and compound 6, each antagonized this novel Bcl-2/GSH interaction, as did recombinant Bim [24]. From these previous data, we concluded that Bcl-2 is a de facto GSH-binding protein and we hypothesized that this newly discovered property of Bcl-2 might play a central role in its antioxidant-like function at mitochondria. Consistent with our hypothesis, we report here that HA14-1 induces a specific depletion of the mitochondrial glutathione pool in CGNs and this BH3 mimetic inhibits the Bcl-2/GSH interaction in intact CGNs. In addition, HA14-1 and Bim inhibit the mitochondrial uptake of [3H]GSH and HA14-1 induces glutathione efflux in isolated rat brain mitochondria. Importantly none of these results are observed in isolated mitochondria incubated with the complex I inhibitor, rotenone, demonstrating that the effects of HA14-1 on mitochondrial glutathione transport are independent of any inhibitory actions on the mitochondrial respiratory machinery.
To further establish a link between Bcl-2 function and mitochondrial glutathione transport, we used a transient co-transfection system in CHO cells to demonstrate a novel interaction between Bcl-2 and the IMM glutathione transporter, OGC. Significantly, this unique protein-protein interaction is markedly enhanced by GSH-MEE. We also show the interaction between OGC and Bcl-2 is direct through the use of recombinant proteins in an immunoprecipitation and this interaction is dependent on glutathione because the addition of ethacrynic acid (which binds to GSH and forms non-functional adducts) significantly abolished the ability of OGC and Bcl-2 to bind. Additional experiments using co-transfected CHO cells reveal that Bcl-2 and OGC work in conjunction to significantly increase the mitochondrial glutathione pool. Finally, we demonstrate that the ability of Bcl-2 to protect cells from oxidative stress is largely dependent on an intact transporter function of OGC because phenylsuccinate (an OGC inhibitor) abolishes the protective effect of Bcl-2 against hydrogen peroxide-induced apoptosis in CHO cells.
Based on our current and previously published data, we propose the following model to explain how Bcl-2 acts as a central regulator of the mitochondrial glutathione pool. Under normal circumstances, OGC functions as an IMM glutathione transporter that helps to maintain an adequate mitochondrial glutathione pool. We suggest that Bcl-2 localized to the outer mitochondrial membrane acts as a “sensor” of cytoplasmic glutathione content, and upon GSH-binding, a conformational change is induced within Bcl-2 that increases its affinity for OGC. The Bcl-2/OGC interaction in turn, enhances the mitochondrial glutathione transporter function of OGC to increase the mitochondrial glutathione pool. In this manner, an increase in cytoplasmic glutathione content, which may occur via either enhanced synthesis or reduced efflux [38, 39], may be transduced into the mitochondria to induce a corresponding elevation in mitochondrial glutathione content. However, when HA14-1 or Bim binds to Bcl-2 within the BH3 groove, this antagonizes (either directly or allosterically) the interaction between GSH and Bcl-2, and as a consequence lowers the affinity of Bcl-2 for OGC. Thus, BH3 mimetics and BH3-only proteins inhibit the capacity of Bcl-2 to regulate mitochondrial glutathione transport through OGC, leading to a selective depletion of mitochondrial glutathione, an increase in MOS, and activation of the intrinsic apoptotic cascade.
Our proposed model is supported by several previously published observations. First, in addition to our results showing that the BH3 mimetic, HA14-1, depletes mitochondrial glutathione in CGNs, other BH3 mimetics have been shown to have similar effects in different cell types. The highly selective Bcl-2 inhibitor, ABT-737, has been shown to deplete cellular glutathione in Jurkat cells and HeLa cells, although mitochondrial glutathione content was not explicitly measured in this study [43]. These findings, along with our data showing that Bim directly inhibits GSH uptake into isolated mitochondria, strongly suggest that inhibition of Bcl-2 is a common mechanism of depleting mitochondrial glutathione. Second, there is a precedent for Bcl-2 to interact with and modulate the activity of IMM transporters. For instance, Bcl-2 has been shown to enhance the ADP/ATP exchange activity of the IMM adenine nucleotide translocase in proteoliposomes, isolated mitochondria, and intact cells [44]. Interestingly, Bax displaces Bcl-2 from the translocase and acts to inhibit the ADP/ATP exchange activity. Third, a previous study demonstrated a positive correlation between an accumulation of Bcl-2 in the nuclear membrane and a redistribution of glutathione into the nucleus, adding further support to a central role for Bcl-2 in the regulation of glutathione transport that is not restricted to mitochondria [45]. Finally, the ability of BH3 mimetics and BH3-only proteins (e.g., Bim) to disrupt the Bcl-2/GSH interaction is entirely consistent with the pro-oxidant state induced at mitochondria by pro-apoptotic Bcl-2 family members [25, 46, 47].
Collectively, the results described above suggest that Bcl-2 plays a central role in the regulation of mitochondrial glutathione transport and maintenance of the mitochondrial glutathione pool. Moreover, this key antioxidant-like function of Bcl-2 likely involves direct GSH-binding and a GSH-stimulated interaction with the IMM GSH transporter, OGC. While our paper was in prepration, Gallo et al. found a similar interaction between the C. elegans homolog of OGC (MISC-1) and the Bcl-2 homolog (Ced-9), further providing evidence for an interaction between Bcl-2 and OGC [48].
Given the substantial roles of glutathione depletion and MOS in neurodegeneration, one could postulate how negatively impacting this novel function of Bcl-2 might contribute to the pathophysiology of various neurodegenerative diseases. For example, GSH depletion in the spinal cord of mice harboring a familial ALS mutation (G93A) in the SOD1 gene has been shown to correlate with motor neuron degeneration during disease progression [49]. In transgenic mice and rats, as well as human familial ALS patients, various mutant forms of SOD1 are apparently recruited to spinal cord mitochondria [50]. In general, these mutant forms of SOD1 are observed as misfolded aggregates that have been localized to essentially every compartment of the mitochondria; however, mutant SOD1 has also been shown to aggregate with Bcl-2 at mitochondria [51]. Most significantly, this interaction of mutant SOD1 with Bcl-2 has been shown to induce a conformational change in Bcl-2 that exposes its BH3 domain and converts Bcl-2 into a “toxic” protein at mitochondria [52]. Although the authors of this elegant study support a direct toxic function of the conformationally altered form of Bcl-2 at mitochondria, it is tempting to speculate that the interactions of Bcl-2 with mutant SOD1 may also interfere with the novel role that Bcl-2 plays in regulating mitochondrial glutathione transport. For instance, if only a fraction of Bcl-2 is actually altered in conformation by its interaction with mutant SOD1, then this “toxic” form of Bcl-2 may in fact act like a BH3-only protein and disrupt the ability of adjacent unaltered Bcl-2 molecules to bind GSH and stimulate OGC-dependent glutathione transport into the mitochondria. In fact, this model is consistent with findings in NSC34 motor neuronal cells stably expressing G93A mutant SOD1. These cells demonstrate a significant depletion of the mitochondrial glutathione pool with no effect on cytosolic glutathione content [53]. Moreover, these mutant SOD1-expressing cells demonstrate a more pronounced depletion of mitochondrial glutathione and display an enhanced sensitivity to apoptosis in response to ethacrynic acid when compared to non-transfected or wild type SOD1-expressing NSC34 cells [53, 54]. Thus, motor neuron toxicity induced by mutant SOD1 likely involves the interaction of this protein with Bcl-2 in the outer mitochondrial membrane. The precise consequences of this interaction, if any, on the capacity of Bcl-2 to regulate mitochondrial glutathione transport requires further study.
In conclusion, we identify the regulation of mitochondrial glutathione transport and maintenance of the mitochondrial glutathione pool as essential and novel functions of Bcl-2. We establish Bcl-2 as a direct GSH-binding protein and a novel interacting partner for the IMM glutathione transporter, OGC. Either BH3 mimetics or BH3-only proteins (e.g., Bim) interfere with the Bcl-2/GSH interaction and result in inhibition of mitochondrial glutathione transport. Bcl-2 and OGC appear to work in a concerted manner to significantly increase the mitochondrial glutathione pool. Accordingly, the ability of Bcl-2 to protect cells from oxidative stress is largely dependent on an intact transporter function of OGC. These findings suggest a molecular mechanism for the well established antioxidant-like function of Bcl-2, particularly at the level of the mitochondria. Finally, this novel function of Bcl-2 may be a target of pathogenic proteins, such as mutant SOD1, which result in devastating neurodegenerative disorders like ALS.
HIGHLIGHTS.
Inhibition of Bcl-2 by the BH3 mimetic HA14-1 selectively depletes mitochondrial glutathione.
Bcl-2 interacts with glutathione in intact cells and this interaction is antagonized by the BH3 mimetic HA14-1.
The BH3 mimetic HA14-1 and recombinant Bim inhibit 3H-glutathione uptake into isolated mitochondria.
Bcl-2 interacts with the 2-oxoglutarate carrier (OGC) and this interaction is enhanced by glutathione.
Co-expression of Bcl-2 and OGC increase mitochondrial glutathione.
The protective effect of Bcl-2 against hydrogen peroxide depends on OGC function.
Acknowledgments
This study was supported by a Merit Review grant from the Department of Veterans Affairs (to D.A.L.) and an R01 grant NS062766 from the National Institutes of Health (to D.A.L.). The authors acknowledge Juliane Schwarz and Natalie A. Kelsey for experimental support.
Footnotes
ABBREVIATIONS: [3H]GSH, tritiated glutathione; BH3, Bcl-2 homology 3-domain; CGNs, cerebellar granule neurons; CHO, Chinese hamster ovarian cells; CNS, central nervous system; DIC, dicarboxylate carrier; DMSO, dimethylsulfoxide; DSS, disuccinimidyl suberate; DTT, dithiothreitol; EA, ethacrynic acid; GSH, reduced glutathione; GSH-MEE, Glutathione monoethylester; GST, glutathione S-transferase; HA14-1, 2-Amino-6-bromo-α-cyano-3-(ethoxycarbonyl)-4H-1-benzopyran-4-acetic acid ethyl ester; IMM, inner mitochondrial membrane; MOS, mitochondrial oxidative stress; OGC, 2-oxoglutarate carrier; PBS, phosphate buffered saline; PhS, phenylsuccinate; SOD1, Cu/Zn superoxide dismutase; γ-GCS, gamma-glutamyl cysteine synthase.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lin MT, Beal MF. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature. 2006;443:787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 2.Franco R, Cidlowski JA. Apoptosis and Glutathione: Beyond an Antioxidant. Cell Death Differ. 2009;16:1303–14. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 3.Muyderman H, Nilsson M, Sims NR. Highly Selective and Prolonged Depletion of Mitochondrial Glutathione in Astrocytes Markedly Increases Sensitivity to Peroxynitrite. J Neurosci. 2004;24:8019–28. doi: 10.1523/JNEUROSCI.1103-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muyderman H, Wadey AL, Nilsson M, Sims NR. Mitochondrial glutathione protects against cell death induced by oxidative and nitrative stress in astrocytes. J Neurochem. 2007;102:1369–82. doi: 10.1111/j.1471-4159.2007.04641.x. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong JS, Jones DP. Glutathione Depletion Enforces the Mitochondrial Permeability Transition and Causes Cell Death in Bcl-2 Overexpressing Hl60 Cells. Faseb J. 2002;16:1263–5. doi: 10.1096/fj.02-0097fje. [DOI] [PubMed] [Google Scholar]
- 6.Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically Altered Mice to Evaluate Glutathione Homeostasis in Health and Disease. Free Radic Biol Med. 2004;37:1511–26. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 7.Sims NR, Nilsson M, Muyderman H. Mitochondrial Glutathione: A Modulator of Brain Cell Death. J Bioenerg Biomembr. 2004;36:329–33. doi: 10.1023/B:JOBB.0000041763.63958.e7. [DOI] [PubMed] [Google Scholar]
- 8.Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid Redox Signal. 2009;11:2685–700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffith OW, Meister A. Origin and Turnover of Mitochondrial Glutathione. Proc Natl Acad Sci U S A. 1985;82:4668–72. doi: 10.1073/pnas.82.14.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lash LH. Mitochondrial Glutathione Transport: Physiological, Pathological and Toxicological Implications. Chem Biol Interact. 2006;163:54–67. doi: 10.1016/j.cbi.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Z, Lash LH. Evidence for Mitochondrial Uptake of Glutathione by Dicarboxylate and 2-Oxoglutarate Carriers. J Pharmacol Exp Ther. 1998;285:608–18. [PubMed] [Google Scholar]
- 12.Chen Z, Putt DA, Lash LH. Enrichment and Functional Reconstitution of Glutathione Transport Activity from Rabbit Kidney Mitochondria: Further Evidence for the Role of the Dicarboxylate and 2-Oxoglutarate Carriers in Mitochondrial Glutathione Transport. Arch Biochem Biophys. 2000;373:193–202. doi: 10.1006/abbi.1999.1527. [DOI] [PubMed] [Google Scholar]
- 13.Coll O, Colell A, Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC. Sensitivity of the 2-Oxoglutarate Carrier to Alcohol Intake Contributes to Mitochondrial Glutathione Depletion. Hepatology. 2003;38:692–702. doi: 10.1053/jhep.2003.50351. [DOI] [PubMed] [Google Scholar]
- 14.Zhong Q, Putt DA, Xu F, Lash LH. Hepatic Mitochondrial Transport of Glutathione: Studies in Isolated Rat Liver Mitochondria and H4iie Rat Hepatoma Cells. Arch Biochem Biophys. 2008;474:119–27. doi: 10.1016/j.abb.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lash LH, Putt DA, Matherly LH. Protection of Nrk-52e Cells, a Rat Renal Proximal Tubular Cell Line, from Chemical-Induced Apoptosis by Overexpression of a Mitochondrial Glutathione Transporter. J Pharmacol Exp Ther. 2002;303:476–86. doi: 10.1124/jpet.102.040220. [DOI] [PubMed] [Google Scholar]
- 16.Xu F, Putt DA, Matherly LH, Lash LH. Modulation of Expression of Rat Mitochondrial 2-Oxoglutarate Carrier in Nrk-52e Cells Alters Mitochondrial Transport and Accumulation of Glutathione and Susceptibility to Chemically Induced Apoptosis. J Pharmacol Exp Ther. 2006;316:1175–86. doi: 10.1124/jpet.105.094599. [DOI] [PubMed] [Google Scholar]
- 17.Wadey AL, Muyderman H, Kwek PT, Sims NR. Mitochondrial glutathione uptake: characterization in isolated brain mitochondria and astrocytes in culture. J Neurochem. 2009;109(Suppl 1):101–08. doi: 10.1111/j.1471-4159.2009.05936.x. [DOI] [PubMed] [Google Scholar]
- 18.Kamga CK, Zhang SX, Wang Y. Dicarboxylate Carrier-Mediated Glutathione Transport Is Essential for Reactive Oxygen Species Homeostasis and Normal Respiration in Rat Brain Mitochondria. Am J Physiol Cell Physiol. 299:C497–505. doi: 10.1152/ajpcell.00058.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 Functions in an Antioxidant Pathway to Prevent Apoptosis. Cell. 1993;75:241–51. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 20.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 Inhibition of Neural Death: Decreased Generation of Reactive Oxygen Species. Science. 1993;262:1274–7. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 21.Ellerby LM, Ellerby HM, Park SM, Holleran AL, Murphy AN, Fiskum G, Kane DJ, Testa MP, Kayalar C, Bredesen DE. Shift of the Cellular Oxidation-Reduction Potential in Neural Cells Expressing Bcl-2. J Neurochem. 1996;67:1259–67. doi: 10.1046/j.1471-4159.1996.67031259.x. [DOI] [PubMed] [Google Scholar]
- 22.Voehringer DW, Meyn RE. Redox Aspects of Bcl-2 Function. Antioxid Redox Signal. 2000;2:537–50. doi: 10.1089/15230860050192314. [DOI] [PubMed] [Google Scholar]
- 23.Hochman A, Sternin H, Gorodin S, Korsmeyer S, Ziv I, Melamed E, Offen D. Enhanced Oxidative Stress and Altered Antioxidants in Brains of Bcl-2-Deficient Mice. J Neurochem. 1998;71:741–8. doi: 10.1046/j.1471-4159.1998.71020741.x. [DOI] [PubMed] [Google Scholar]
- 24.Zimmerman AK, Loucks FA, Schroeder EK, Bouchard RJ, Tyler KL, Linseman DA. Glutathione binding to the Bcl-2 homology-3 domain groove: a molecular basis for Bcl-2 antioxidant function at mitochondria. J Biol Chem. 2007;282:29296–304. doi: 10.1074/jbc.M702853200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding WX, Ni HM, DiFrancesca D, Stolz DB, Yin XM. Bid-Dependent Generation of Oxygen Radicals Promotes Death Receptor Activation-Induced Apoptosis in Murine Hepatocytes. Hepatology. 2004;40:403–13. doi: 10.1002/hep.20310. [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Lu H, Shi H, Du Y, Yu J, Gu S, Chen X, Liu KJ, Hu CA. Puma Overexpression Induces Reactive Oxygen Species Generation and Proteasome-Mediated Stathmin Degradation in Colorectal Cancer Cells. Cancer Res. 2005;65:1647–54. doi: 10.1158/0008-5472.CAN-04-1754. [DOI] [PubMed] [Google Scholar]
- 27.Zimmermann AK, Loucks FA, Le SS, Butts BD, Florez-McClure ML, Bouchard RJ, Heidenreich KA, Linseman DA. Distinct Mechanisms of Neuronal Apoptosis Are Triggered by Antagonism of Bcl-2/Bcl-X(L) Versus Induction of the BH3-Only Protein Bim. J Neurochem. 2005;94:22–36. doi: 10.1111/j.1471-4159.2005.03156.x. [DOI] [PubMed] [Google Scholar]
- 28.Ravindranath V, Shivakumar BR, Anandatheerthavarada HK. Low glutathione levels in brain regions of aged rats. Neuroscience Letters. 1989;101:187–190. doi: 10.1016/0304-3940(89)90528-4. [DOI] [PubMed] [Google Scholar]
- 29.Sagara JI, Miura K, Bannai S. Maintenance of neuronal glutathione by glial cells. J Neurochem. 1993;61:1672–6. doi: 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- 30.Huang J, Philbert MA. Distribution of glutathione and glutathione-related enzyme systems in mitochondria and cytosol of cultured cerebellar astrocytes and granule cells. Brain Res. 1995;680:16–22. doi: 10.1016/0006-8993(95)00209-9. [DOI] [PubMed] [Google Scholar]
- 31.Milanesi E, Costantini P, Gambalunga A, Colonna R, Petronilli V, Cabrelle A, Semenzato G, Cesura AM, Pinard E, Bernardi P. The Mitochondrial Effects of Small Organic Ligands of Bcl-2: Sensitization of Bcl-2-Overexpressing Cells to Apoptosis by a Pyrimidine-2,4,6-Trione Derivative’. J Biol Chem. 2006;281:10066–72. doi: 10.1074/jbc.M513708200. [DOI] [PubMed] [Google Scholar]
- 32.Janssen A, Trijbels F, Sengers R, Smeitink J, Van Den Heuvel L, Wintjes L, Stoltenborg-Hogenkamp B, Rodenburg R. Spectrophotometric Assay for Complex I of the Respiratory Chain in Tissue Samples and Cultured Fibroblasts. Clinical Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- 33.Chen MJ, Yap YW, Choy MS, Koh C, Seet SJ, Duan W, Whiteman M, Cheung NS. Early induction of calpains in rotenone-mediated neuronal apoptosis. Neuroscience Letters. 2006;397:69–73. doi: 10.1016/j.neulet.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Papadopoulos MC, Koumenis IL, Xu L, Giffard RG. Potentiation of Murine Astrocyte Antioxidant Defence by Bcl-2: Protection in Part Reflects Elevated Glutathione Levels. Eur J Neurosci. 1998;10:1252–60. doi: 10.1046/j.1460-9568.1998.00134.x. [DOI] [PubMed] [Google Scholar]
- 35.Satoh T, Sakai N, Enokido Y, Uchiyama Y, Hatanaka H. Free radical-independent protection by nerve growth factor and Bcl-2 of PC12 cells from hydrogen peroxide-triggered apoptosis. J Biochem. 1996;120:540–6. doi: 10.1093/oxfordjournals.jbchem.a021447. [DOI] [PubMed] [Google Scholar]
- 36.Chipuk JE, Green DR. How Do Bcl-2 Proteins Induce Mitochondrial Outer Membrane Permeabilization? Trends Cell Biol. 2008;18:157–64. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voehringer DW. Bcl-2 and Glutathione: Alterations in Cellular Redox State That Regulate Apoptosis Sensitivity. Free Radic Biol Med. 1999;27:945–50. doi: 10.1016/s0891-5849(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 38.Meredith MJ, Cusick CL, Soltaninassab S, Sekhar KS, Lu S, Freeman ML. Expression of Bcl-2 Increases Intracellular Glutathione by Inhibiting Methionine-Dependent Gsh Efflux. Biochem Biophys Res Commun. 1998;248:458–63. doi: 10.1006/bbrc.1998.8998. [DOI] [PubMed] [Google Scholar]
- 39.Jang JH, Surh YJ. Bcl-2 Attenuation of Oxidative Cell Death Is Associated with up-Regulation of Gamma-Glutamylcysteine Ligase Via Constitutive Nf-Kappab Activation. J Biol Chem. 2004;279:38779–86. doi: 10.1074/jbc.M406371200. [DOI] [PubMed] [Google Scholar]
- 40.Merad-Saidoune M, Boitier E, Nicole A, Marsac C, Martinou JC, Sola B, Sinet PM, Ceballos-Picot I. Overproduction of Cu/Zn-Superoxide Dismutase or Bcl-2 Prevents the Brain Mitochondrial Respiratory Dysfunction Induced by Glutathione Depletion. Exp Neurol. 1999;158:428–36. doi: 10.1006/exnr.1999.7108. [DOI] [PubMed] [Google Scholar]
- 41.Rimpler MM, Rauen U, Schmidt T, Moroy T, de Groot H. Protection against Hydrogen Peroxide Cytotoxicity in Rat-1 Fibroblasts Provided by the Oncoprotein Bcl-2: Maintenance of Calcium Homoeostasis Is Secondary to the Effect of Bcl-2 on Cellular Glutathione. Biochem J. 1999;340(Pt 1):291–7. [PMC free article] [PubMed] [Google Scholar]
- 42.Mirkovic N, Voehringer DW, Story MD, McConkey DJ, McDonnell TJ, Meyn RE. Resistance to Radiation-Induced Apoptosis in Bcl-2-Expressing Cells Is Reversed by Depleting Cellular Thiols. Oncogene. 1997;15:1461–70. doi: 10.1038/sj.onc.1201310. [DOI] [PubMed] [Google Scholar]
- 43.Howard AN, Bridges KA, Meyn RE, Chandra J. Abt-737, a Bh3 Mimetic, Induces Glutathione Depletion and Oxidative Stress. Cancer Chemother Pharmacol. 2009;65:41–54. doi: 10.1007/s00280-009-1001-1. [DOI] [PubMed] [Google Scholar]
- 44.Belzacq AS, Vieira HL, Verrier F, Vandecasteele G, Cohen I, Prevost MC, Larquet E, Pariselli F, Petit PX, Kahn A, Rizzuto R, Brenner C, Kroemer G. Bcl-2 and Bax Modulate Adenine Nucleotide Translocase Activity. Cancer Res. 2003;63:541–6. [PubMed] [Google Scholar]
- 45.Voehringer DW, McConkey DJ, McDonnell TJ, Brisbay S, Meyn RE. Bcl-2 Expression Causes Redistribution of Glutathione to the Nucleus. Proc Natl Acad Sci U S A. 1998;95:2956–60. doi: 10.1073/pnas.95.6.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kirkland RA, Windelborn JA, Kasprzak JM, Franklin JL. A Bax-induced pro-oxidant state is critical for cytochrome c release during programmed neuronal death. J Neurosci. 2002;22:6480–90. doi: 10.1523/JNEUROSCI.22-15-06480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starkov AA, Polster BM, Fiskum G. Regulation of Hydrogen Peroxide Production by Brain Mitochondria by Calcium and Bax. J Neurochem. 2002;83:220–8. doi: 10.1046/j.1471-4159.2002.01153.x. [DOI] [PubMed] [Google Scholar]
- 48.Gallo M, Park D, Luciani DS, Kida K, Palmieri F, Blacque OE, Johnson JD, Riddle DL. MISC-1/OGC Links Mitochondrial Metabolism, Apoptosis, and Insulin Secretion. PloS One. 2011;6:e17827. doi: 10.1371/journal.pone.0017827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chi L, Ke Y, Luo C, Gozal D, Liu R. Depletion of Reduced Glutathione Enhances Motor Neuron Degeneration in Vitro and in Vivo. Neuroscience. 2007;144:991–1003. doi: 10.1016/j.neuroscience.2006.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of Familial Als-Linked Sod1 Mutants from Selective Recruitment to Spinal Mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 51.Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic Lateral Sclerosis-Associated Sod1 Mutant Proteins Bind and Aggregate with Bcl-2 in Spinal Cord Mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 52.Pedrini S, Sau D, Guareschi S, Bogush M, Brown RH, Jr, Naniche N, Kia A, Trotti D, Pasinelli P. Als-Linked Mutant Sod1 Damages Mitochondria by Promoting Conformational Changes in Bcl-2. Hum Mol Genet. 19:2974–86. doi: 10.1093/hmg/ddq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muyderman H, Hutson PG, Matusica D, Rogers ML, Rush RA. The Human G93a-Superoxide Dismutase-1 Mutation, Mitochondrial Glutathione and Apoptotic Cell Death. Neurochem Res. 2009;34:1847–56. doi: 10.1007/s11064-009-9974-z. [DOI] [PubMed] [Google Scholar]
- 54.Rizzardini M, Mangolini A, Lupi M, Ubezio P, Bendotti C, Cantoni L. Low Levels of Als-Linked Cu/Zn Superoxide Dismutase Increase the Production of Reactive Oxygen Species and Cause Mitochondrial Damage and Death in Motor Neuron-Like Cells. J Neurol Sci. 2005;232:95–103. doi: 10.1016/j.jns.2005.02.004. [DOI] [PubMed] [Google Scholar]