

Abstract

Our previous studies into visible light-mediated aza-Henry reactions demonstrated that molecular oxygen played a vital role in catalyst turnover as well as the production of base to facilitate the nucleophilic addition of nitroalkanes. Herein, improved conditions for the generation of iminium ions from tetrahydroisoquinolines that allow for versatile nucleophilic trapping are reported. The new conditions provide access to a diverse range of functionality under mild, anaerobic reaction conditions as well as mechanistic insights into the photoredox cycle.
The catalytic oxidation of α-amino C−H bonds to generate reactive intermediates, specifically iminium ions, is a useful method in organic synthesis.1 Exploitation of these reactive intermediates via reaction with diverse nucleophiles can lead to biologically relevant structural motifs including β-amino acids and potential drug candidates such as noscapine.2 The pioneering work of Li3 and Murahashi4 has demonstrated the utility of metal catalysis to access such functionally distinct architectures via α-amino C−H activation.5,6 Herein we report the advancement of α-amino C−H functionalization via visible light-mediated photoredox catalysis.
Free-radical chemistry has played a crucial role in accessing complex molecular frameworks through chemoselective transformations including polyene cyclization cascades and reduction/oxidation of remote unsaturated carbons.7 Recent efforts emphasising benign, mild catalytic systems,8 including copper catalysis3 and visible light-mediated organic reactions,9 represent appealing alternatives.10,11 Visible light-mediated photoredox catalysis using substoichiometric quantities (typically 1 mol %) of metal complexes to facilitate redox cycles using mild stoichiometric oxidants has emerged at the forefront of this growing trend.12
Several synthetic organic groups13,14,15 have harnessed the inherent characteristics of light-active metal complexes such as Ru(bpy)3Cl2 to promote chemical transformations.16 These metal complexes hold advantages over alternative reagents for light/energy conversions since their photochemical properties may be fine-tuned through manipulation of ligand/metal combinations, thus enabling augmentations of redox potentials.13 As a consequence, a complete overhaul of reaction design can be avoided.
During our initial investigations into reductive dehalogenations using Ru(bpy)3Cl2 in the presence of HCO2H•iPr2NEt, we discovered, through deuterium labelling experiments, that iPr2NEt was a major hydrogen atom donor.17 As depicted in Figure 1, Ru2+* (Ered = +0.84 V vs. SCE) is able to oxidize tertiary amines to generate the corresponding amino radical cation. Accordingly, the bond dissociation energy (BDE) of the α-C–H bond is dramatically lowered (90.7 kcal/mol BDE drops to approximately 17 kcal/mol using triethylamine as an example).18 This, in turn, correlates to a calculated pKa of the α-C–H bond to be 26.7 pKa units.19 By exploiting this inherent physical characteristic of amino radical cations, we anticipated that we could generate iminium ions via direct H-atom abstraction or deprotonation and oxidation of the resultant α-amino radical. Due to several divergent pathways available to the α–amino radical, and slow catalyst turnover with oxygen, we have focused upon accelerating the α-C–H oxidation chemistry through modification of the stoichiometric oxidant driven by our mechanistic observations, thus biasing the pathway to the iminium ion.20,21,22
Figure 1.

Physical properties of amino radical cations and their potential for diversification under visible light-mediated photoredox catalysis
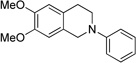
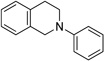
We began our investigation by choosing a suitable reaction for optimization, one capable of representing a broad set of nucleophiles. Based upon this requisite, the cyanation of tetrahydroisoquinolines was chosen. Our initial cyanation attempts involved using Ir(ppy)2(dtbbpy)PF6 (1 mol %) in N,N-dimethylformamide (DMF) under white light irradiation (Table 1). Ethyl α-bromoacetate (EtO2CCH2Br) was chosen as the stoichiometric oxidant with N-phenyl tetrahydroisoquinoline as the substrate and NaCN as the nucleophile.
Table 1.
Optimization of iminium ion formation
 | ||
|---|---|---|
| entry | conditions | yielda |
| 1 | Ir (1 mol %), EtO2CCH2Br (3 equiv), DMF, white Light, NaCN (5 equiv) | 36 |
| 2 | Ru (1 mol %), (EtO2C)2CHBr (3 equiv), DMF, blue LEDs, NaCN (5 equiv) | 95 |
| 3 | Ru (1 mol %), CCl4DMF (1:1), blue LEDs, NaCN (5 equiv) | 36 |
| 4 | Ru (1 mol %), CCl4 (3 equiv), CH3CN, blue LEDs, NaCN (5 equiv) | 53 |
| 5 | Ru (1 mol %), BrCCl3 (3 equiv), DMF, blue LEDs, NaCN (5 equiv) | 60 |
| 6 | Ru (1 mol %), BrCCl3 (3 equiv), DMF, blue LEDs then no light, NaCN (5 equiv) | 85 |
| 7 | Ru (1 mol %), BrCCl3 (3 equiv), DMF, blue LEDs then no light, Bu4NCN (5 equiv) | 17 |
| 8 | Ru (1 mol %), BrCCl3 (3 equiv), THF, blue LEDs then no light, NaCN (5 equiv) | NR |
| 9 | Ru (1 mol %), BrCCl3 (3 equiv), THF:H2O (2:1), blue LEDs then no light, NaCN (5 equiv) | 83 |
Isolated percent yields after chromatography on SiO2.
In the event, a low isolated yield of the product was obtained (36%, entry 1). As a result, we switched the photocatalyst to Ru(bpy)3Cl2 and the light source to blue LEDs. Diethyl bromomalonate [(Et2OC)2CHBr] was first selected as the oxidant in order to probe the reactivity for this process given the precedented ability of Ru1+ to reduce the C–Br bond. Subsequently, an encouraging 95% isolated yield of cyanation product 2 was obtained (entry 2). Unfortunately, general application of this oxidant is impractical due to its potential side reactivity arising from the resultant malonyl radical and diethylmalonate generated after hydrogen atom abstraction. Changing the stoichiometric oxidant to carbon tetrachloride (CCl4) significantly decreased the rate and overall yield, both in DMF and acetonitrile (CH3CN) (entries 3 and 4). In testing the conversion of starting material, BrCCl3 in DMF was found to be a suitable alternative for catalytic turnover and full conversion of tetrahydroisoquinoline to the iminium ion was observed in <3 h (vida infra).
However, reactions with one-pot additions of BrCCl3 and NaCN were found to give inconsistent results and it was soon discovered that these components reacted under the photoredox conditions to produce trichloroacetonitrile (Cl3CCN), thereby impeding the catalytic cycle. To prevent this undesired reactivity, excess NaCN was added after TLC analysis indicated full conversion to the iminium. Furthermore, the reaction flask was removed from blue LED irradiation. With the use of an inorganic nucleophile, a solvent screen was then needed to improve the nucleophilic trapping of more polar reactants. After evaluation of DMF, CH3CN, and tetrahydrofuran/water (THF/H2O) mixtures (Table 1, entries 6–9), the highest yield was obtained upon using a 2:1 THF/H2O mixture, providing 83% yield of cyanation product (entry 9). No reaction was observed in pure THF presumably due to the insolubility of the Ru(bpy)3Cl2 catalyst (entry 8).
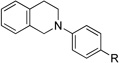
Having established our reaction conditions for the successful cyanation of N-phenyltetrahydroisoquinoline (1), we next applied the reaction parameters to a representative substrate and nucleophile scope. We first assessed the applicability of our newly developed reaction conditions to aza-Henry chemistry (Table 2, entries 1–3).
Table 2.
Functionalizaation of tetrahydroisoquinolines enabled by visible light-mediated photoredox catalysis
| entry | substrate | product | yielda |
|---|---|---|---|
 |
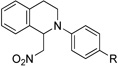 |
||
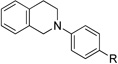
| 1b | 1 R = H | 4 R = H | 95 |
| 2b | 3 R = Br | 5 R = Br | 93 |
 |
 |
||
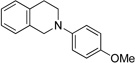
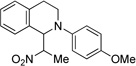
| 3b | 6 | 7 | 95 (d.r.= 2:1) |
 |
 |
||
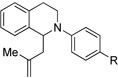
| 4c | 1 R = H | 8 R = H | 85 |
| 5c | 6 R = OMe | 9 R = OMe | 44 |
 |
 |
||
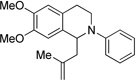
| 6c | 10 | 11 | 43 |
 |
 |
||
| 7d | 1 R = H | 12 R = H | 59 |
| 8d | 3 R = Br | 13 R = Br | 65 |
 |
 |
||
| 9e | 1 | 14 | 69 |
 |
 |
||
| 10e | 1 | 15 | 68 (d.r.= 3:2) |
Isolated percent yields after chromatography on SiO2.
Ru(bpy)3Cl2 (1 mol %), BrCCl3 (3 equiv), DMF, blue LEDs, 3 h, then no light, Et3N (5 equiv), nitroalkane (5 equiv).
Ru(bpy)3Cl2 (1 mol %), BrCCl3 (3 equiv), DMF, blue LEDs, 3 h, then no light, methallyl trimethylsilane (5 equiv).
Ru(bpy)3Cl2 (1 mol %), BrCCl3 (3 equiv), DMF, blue LEDs, 3 h, then no light, Et3N (5 equiv), silyl enol ether (5 equiv).
Ru(bpy)3Cl2 (1 mol %), BrCCl3 (3 equiv), DMF, blue LEDs, 3 h, then no light, Et3N (5 equiv), 1,3-dicarbonyl (5 equiv).
Surprisingly, after generation of the iminium ion from the corresponding tetrahydroisoquinoline and removal of the blue LEDs, we injected a nitroalkane into the reaction as the nucleophile only to discover slow conversion to product. However, once we added an equal molar amount of Et3N23 in the absence of light, the reaction went to completion under 4 hours and the products were isolated in excellent yield.
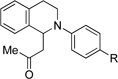
We further applied our newly developed reaction conditions to other nucleophiles and tetrahydroisoquinolines. We explored the potential for intermolecular allylations. Surprisingly, when attempting to trap the iminium with allyl trimethylsilane, we did not isolate any desired product. We believe that the putative β-silyl carbocation intermediate is not stable enough under the conditions to effectively deliver the desired product. As a result, we tried trapping with methallyl trimethylsilane, which would undergo addition via a tertiary carbocation. Fortunately, methallyl trimethylsilane addition was successful (Table 2, entries 4–6).
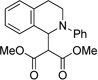
Next, we found that silylenol ethers (Table 2, 12 and 13) and 1,3-dicarbonyls including malonates and methylacetoacetates (Table 2, 14 and 15, respectively) successfully underwent addition. Acetoacetone also underwent addition, however, the resulting product readily decomposed upon purification presumably via a retro-Mannich pathway.
 |
(1) |
 |
(2) |
Interestingly, we can also use this method to synthesize polycyclic structures. For example, addition of a siloxyfuran following iminium ion formation produced butenolide 16 in good yield (eq 1). Furthermore, exposure of the iminium to indole results in Friedel-Crafts product 17 (eq 2). These diverse nucleophiles provide platforms for further expansion to more functionalized products.
The products listed in Table 2 compare favourably in both scope and yield to known methods for their synthesis including copper-mediated cross-dehydrogenative coupling1 and organic dye photocatalysis.14 However, our reaction conditions have the capacity to blend two complementary methods as a means to arrive at diverse functionality. Figure 2 illustrates this point with the visible light-mediated oxidation of N-phenyltetrahydroisoquinolines coupled with a copper-assisted alkynylation sequence to afford propargylic amines in good yield.
Figure 2.

Alkynylation coupled with photoredox catalysis
On the basis of our mechanistic analysis, we believe that nucleophilic trapping proceeds through the in situ generation of a reactive iminium ion of N-aryltetrahydroisoquinoline (see the Supporting Information). However, we speculate that formation of the iminium ion can arise via divergent pathways (see Figure 1). Our first hypothesis involves initial irradiation of Ru2+ to generate Ru2+* which subsequently oxidizes the N-aryltetrahydroisoquinoline to the corresponding radical cation. The resulting Ru1+, in turn, reduces bromotrichloromethane to the bromide ion and trichloromethyl radical. Hydrogen-atom abstraction by the trichloromethyl radical generates the reactive iminium ion susceptible to nucleophilic trapping under a variety of reaction conditions.24
Our second mechanistic hypothesis predicts a radical chain mechanism where initial irradiation of Ru2+ generates Ru2+*, which subsequently oxidizes N-aryltetrahydroisoquinoline to the corresponding radical cation.24 This radical cation can then undergo deprotonation to form the α-amino radical. Oxidation of the α-amino radical by BrCCl3 (electron transfer or atom transfer) to form the trichloromethyl radical.25 This radical can then abstract a H-atom from another N-aryltetrahydroisoquinoline to generate the α-amino radical and further propogate a radical chain process.
In conclusion, we have developed a new, more versatile approach to the functionalization of α-amino carbons using visible light-mediated photoredox catalysis. This method is highlighted by the compatibility of a broad range of nucleophiles resulting in functionally diverse products. The use of bromotrichloromethane as the stoichiometric oxidant has successfully promoted the photoredox cycle and, in the process, provided insight into the reaction mechanism. Further exploration of the mechanism and functionalization of α-amino carbons using visible light-mediated photoredox catalysis is currently underway.
Supplementary Material
Acknowledgment
Financial support for this research from the NIH-NIGMS (R01-GM096129), the Alfred P. Sloan Foundation, Amgen, and Boehringer Ingelheim is gratefully acknowledged. L. F. thanks the Novartis Institutes for BioMedical Research for a graduate fellowship. NMR (CHE-0619339) and MS (CHE-0443618) facilities at BU are supported by the NSF.
Footnotes
Supporting Information Available. Experimental procedures, 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews on cross dehydrogenative coupling, see: Yeung CS, Dong VM. Chem. Rev. 2011;111:1215. doi: 10.1021/cr100280d. Li C-J. Acc. Chem. Res. 2010;43:581. doi: 10.1021/ar9002587. Li C-J. Acc. Chem. Res. 2009;42:335. doi: 10.1021/ar800164n. Murahashi S-I, Zhang D. Chem. Soc. Rev. 2008;37:1490. doi: 10.1039/b706709g.
- 2.(a) Soriano MDPC, Shankaraiah N, Santos LS. Tetrahedron Lett. 2010;51:1770. [Google Scholar]; (b) Mahmoudian LS, Rahimi-Moghaddam P. Recent Patents on Anti-Cancer Drug Discovery. 2009;4:92. doi: 10.2174/157489209787002524. [DOI] [PubMed] [Google Scholar]
- 3.(a) Li Z, Li C-J. J. Am. Chem. Soc. 2005;127:3672. doi: 10.1021/ja050058j. [DOI] [PubMed] [Google Scholar]; (b) Li Z, Li C-J. J. Am. Chem. Soc. 2005;127:6968. doi: 10.1021/ja0516054. [DOI] [PubMed] [Google Scholar]; (c) Basle O, Li C-J. Green Chem. 2007;9:1047. [Google Scholar]; (b) Li Z-P, Li C-J. J. Am. Chem. Soc. 2004;126:11810. doi: 10.1021/ja0460763. [DOI] [PubMed] [Google Scholar]
- 4.(a) Murahashi S-I, Nakae T, Terai H, Komiya N. J. Am. Chem. Soc. 2008;130:11005. doi: 10.1021/ja8017362. [DOI] [PubMed] [Google Scholar]; (b) Murahashi S-I, Takaya H. Acc. Chem. Res. 2000;33:225. doi: 10.1021/ar980085x. [DOI] [PubMed] [Google Scholar]; (c) Murahashi S-I, Noata T. Bull. Chem. Soc. Jpn. 1996;69:1805. [Google Scholar]
- 5.(a) Sud A, Sureshkumar D, Klussmann M. Chem. Commun. 2009:3169. doi: 10.1039/b901282f. [DOI] [PubMed] [Google Scholar]; (b) Boess E, Sureshkumar D, Sud A, Wirtz C, Fares C, Klussmann M. J. Am. Chem. Soc. 2011;133:8106. doi: 10.1021/ja201610c. [DOI] [PubMed] [Google Scholar]
- 6.Shu X-Z, Xia X-F, Yang Y-F, Ji K-G, Liu X-Y, Liang YM. J. Org. Chem. . 2009;74:7464. doi: 10.1021/jo901583r. [DOI] [PubMed] [Google Scholar]
- 7.(a) Balzani V. Electron Transfer in Chemistry. vol. 1. Weinheim: Wiley-VCH; 2001. [Google Scholar]; (b) Curran DP, Rakiewicz DM. J. Am. Chem. Soc. 1985;107:1448. [Google Scholar]
- 8.For examples of the hazards and reactivity of other radical initiation systems, see: O’Mahony G. Synlett. 2004:572. Khattab MA, Elgamal MA, El-Batouti M. Fire Mater. 1996;20:253. Molander GA. Org. React. 1994;46:211.
- 9.For recent reviews on photoredox catalysis in organic synthesis see: Narayanam JMR, Stephenson CRJ. J. Chem. Soc. Rev. 2011;40:102. doi: 10.1039/b913880n. Teplý F. Collect. Czech. Chem. Commun. 2011;76:859. Yoon TP, Ischay MA, Du J. Nature Chem. 2010;2:527. doi: 10.1038/nchem.687.
- 10.For a representative example of photochemical reactions used as key steps in natural product syntheses see: Bach T, Hehn JP. Angew. Chem. Int. Ed. 2011;50:1000. doi: 10.1002/anie.201002845.
- 11.For a review on current synthetic utility of photochemical processes, see: Albini A, Fagnoni M, editors. Handbook of Synthetic Photochemistry. Weinheim: Wiley-VCH; 2010.
- 12.For reviews on the photophysical properties of photoredox catalysts see: Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, Von Zelewsky V. Coord. Chem. Rev. 1988;84:85. Kalyanasundaram K. Coord. Chem. Rev. 1982;46:159.
- 13.(a) Pham PV, Nagib DA, MacMillan DWC. Angew. Chem. Int. Ed. 2011;50:6119. doi: 10.1002/anie.201101861. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicewicz D, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Hurtley AE, Cimesia MA, Ischay MA, Yoon TP. Tetrahedron. 2011;67:4442. doi: 10.1016/j.tet.2011.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ischay MA, Anzovino ME, Du J, Yoon TP. J. Am. Chem. Soc. 2008;130:12886. doi: 10.1021/ja805387f. and references therein. [DOI] [PubMed] [Google Scholar]
- 15.(a) Dai C, Narayanam JMR, Stephenson CRJ. Nature Chem. 2011;3:140. doi: 10.1038/nchem.949. [DOI] [PubMed] [Google Scholar]; (b) Furst L, Narayanam JMR, Stephenson CRJ. Angew. Chem. Int. Ed. 2011;50:9655. doi: 10.1002/anie.201103145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For other selected examples of photoredox catalysis in organic applications see: Andrews RS, Becker JJ, Gagné MR. Org. Lett. 2011;13:2406. doi: 10.1021/ol200644w. Maji T, Karmakar A, Reiser O. J. Org. Chem. 2011;76:736. doi: 10.1021/jo102239x. Andrews SR, Becker JJ, Gagné MR. Angew. Chem. Int. Ed. 2010;49:7274. doi: 10.1002/anie.201004311. Koike T, Akita M. Chem. Lett. 2009;38:166.
- 17.Narayanam JMR, Tucker JW, Stephenson CRJ. J. Am. Chem. Soc. 2009;131:8756. doi: 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]
- 18.Wayner DDM, Dannenberg JJ, Griller D. Chem. Phys. Lett. 1986;131:189. [Google Scholar]
- 19.See the Supporting Information for pKa approximation derived from the following: Zhang X, Bordwell FG. J. Org. Chem. 1992;57:4163. Xu W, Mariano PS. J. Am. Chem. Soc. 1991;113:1431. Dombrowski GW, Dinnocenzo JP, Farid S, Goodman JL, Gould IR. J. Org. Chem. 1991;64:427. Nelsen SP, Ippoliti JT. J. Am. Chem. Soc. 1986;108:4879. Nicholas M. de P., Arnold DR. Can. J. Chem. 1982;60:2165.
- 20.Condie AG, González-Gómez JC, Stephenson CRJ. J. Am. Chem. Soc. 2010;132:1464. doi: 10.1021/ja909145y. [DOI] [PubMed] [Google Scholar]
- 21.For advances in α-C–H oxidation of tertiary amines using photoredox catalysis, see: Zou Y-Q, Lu L-Q, Fu L, Chang N-J, Rong J, Chen J-R, Xiao W-J. Angew. Chem. Int. Ed. 2011;50:7171. doi: 10.1002/anie.201102306. Xuan J, Cheng Y, An J, Lu L-Q, Zhang X-X, Xiao W-J. Chem. Commun. 2011;47:8337. doi: 10.1039/c1cc12203g. Rueping M, Zhu S, Koenigs RM. Chem. Commun. 2011;47:8679. doi: 10.1039/c1cc12907d. Rueping M, Vila C, Koenigs RM, Poscharny K, Fabry DC. Chem. Commun. 2011;47:2360. doi: 10.1039/c0cc04539j.
- 22.For a recent example of α-C–H oxidation of tertiary amines using organic dye-mediated photoredox catalysis, see: Hari DP, König B. Org. Lett. 2011;13:3852. doi: 10.1021/ol201376v.
- 23.For a discussion on the effects of triethylamine in the electrochemical oxidation of N-arylisoquinolines, see: Baslé O, Borduas N, Dubois P, Chapuzet JM, Chan T-H, Lessard J, Li C-J. Chem. Eur. J. 2010;16:8162. doi: 10.1002/chem.201000240.
- 24.See the Supporting Information for a detailed mechanism.
- 25.For a selected electron transfer/atom transfer case study, see: Curran DP, Guthrie DB, Geib SJ. J. Am. Chem. Soc. 2008;130:8437. doi: 10.1021/ja8012962.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.