

Abstract
T cell receptor (TCR) engagement of peptide-major histocompatibility complex (MHC) is essential to adaptive immunity, but it is unknown if TCR signaling responses are influenced by the binding topology of the TCR-peptide-MHC complex. We developed yeast-displayed peptide-MHC libraries that enabled us to identify new peptide sequences reactive with a single TCR. Structural analysis showed that four peptides bound to the TCR with distinct 3-dimensional (3D) and 2D affinities, using entirely different binding chemistries. Three of the peptides that shared a common docking mode, where key TCR-MHC germline interactions are preserved, induced TCR signaling. The fourth peptide failed to induce signaling, and was recognized in a substantially different TCR-MHC binding mode that apparently exceeded geometric tolerances compatible with signaling. We suggest that the ‘stereotypical’ TCR-MHC docking paradigm evolved from productive signaling geometries, and that TCR signaling can be modulated by peptides that are recognized in alternative TCR-pMHC binding orientations.
Introduction
A fundamental question about transmembrane receptors is whether extracellular ligand binding architecture can influence the nature of receptor activation. This is especially pertinent to the ability of αβ T cell antigen receptors (TCR) to sense and differentially respond to the universe of peptides presented by major histocompatibility complex (MHC). T cell activation is initiated by TCR engagement of peptides displayed upon MHC (pMHC), but subsequent signaling is the product of a complex series of events involving the TCR-associated CD3, CD4 and CD8 coreceptors, and assembly into multimeric clusters that ultimately stimulate phosphorylation of intracellular immunoreceptor tyrosine-based activation motifs (ITAMs) (Beddoe et al., 2009; Gil et al., 2002; Kuhns et al., 2006; van der Merwe and Dushek, 2011; Wucherpfennig et al., 2010). Given the wide range of TCR binding geometries to peptide-MHC seen in TCR-pMHC complexes that fall within the limits of a loosely conserved docking orientation (Hahn et al., 2005; Rudolph et al., 2006), it has so far appeared that a pMHC binding event of sufficient affinity and duration can induce signaling regardless of the TCR-pMHC complex architecture. Furthermore, the lack of correlation between TCR-pMHC structural differences and the type of T cell signals induced implies that different TCR-pMHC binding modes do not generate distinct cellular signals (Ding et al., 1999).
It is clear that the chemistry of pMHC recognition by the TCR does influence signaling by virtue of its effect on the binding kinetics, half-life, affinity, and other biophysical parameters (Alam et al., 1996; Kersh et al., 1998; Qi et al., 2006). A single thermodynamic or kinetic parameter can sometimes qualitatively correlate with T cell responses (Aleksic et al., 2010; Govern et al., 2010), and recent methodologies accounting for receptor confinement and two-dimensional (2D) receptor kinetics in the membrane have shown correlations with activation (Huang et al., 2010; Huppa et al., 2010). It is also generally accepted that clustering of the TCR is critical for T cell activation within the immune synapse, as pMHC multimers are required for signaling in αβ T cells (Bromley et al., 2001; van der Merwe and Cordoba, 2011). Bivalent TCR and CD3 antibodies can substitute for pMHC to induce T cell responses through clustering. Importantly, however, not all TCR-CD3 specific antibodies stimulate T cells equally, suggesting that the receptor geometry required for full activation may not be completely permissive (Janeway, 1995; Yoon et al., 1994), as has been proposed (Cochran et al., 2001).
The TCR-pMHC docking geometry has been extensively studied in an effort to understand MHC restriction (Garcia et al., 2009; Godfrey et al., 2008; Marrack et al., 2008; Wilson and Stanfield, 2005; Wucherpfennig et al., 2009). The compendium of complex structures has revealed a loosely conserved docking paradigm, or binding ‘footprint.’ TCRs bind pMHC roughly on a diagonal with the TCR Vβ complementarity determining regions (CDRs) positioned over the MHC α1 helix and peptide C-terminus and the TCR Vα CDRs positioned over the MHC α2 (Class I) or β1 (Class II) helix or peptide N-terminus, thereby “polarizing” the TCR orientation on the MHC surface (Garcia and Adams, 2005). There is a wide range (+/− ~100°) of docking angles that conform to this ‘canonical’ docking polarity. That such a range of docking angles can support TCR signaling suggests that any constraints on signaling imposed by the geometry of TCR-pMHC engagement are either quite loose or non-existent.
The conservation of a TCR-MHC docking topology could be a product of coevolved TCR-MHC germline specificity (Mazza and Malissen, 2007), a consequence of extrinsic factors such as coreceptor steric influences (Buslepp et al., 2003; Collins and Riddle, 2008), or a product of CDR3-mediated peptide selection during thymic education (Huseby et al., 2005). Recent evidence supports the idea of a coevolved germline specificity as an important determinant of the TCR-MHC binding mode (Dai et al., 2008; Feng et al., 2007; Newell et al., 2011; Rubtsova et al., 2009; Scott-Browne et al., 2009). In particular, a series of structural and functional studies of the widely used murine Vβ8.2 germline segment showed that a similar set of TCR-MHC interfacial contacts are formed, which likely represent the evolutionary signature of TCR-MHC coevolution, and appear to play a role in orienting the TCR docking footprint on the MHC (Dai et al., 2008; Feng et al., 2007; Garcia et al., 2009). An alternative view is that coreceptors bias the TCR for pMHC recognition, and that TCRs can also recognize non-MHC antigens (Van Laethem et al., 2007).
A related issue is how TCRs cross-react with the universe of different peptide antigens they encounter during thymic selection and peripheral surveillance (Felix and Allen, 2006; Mason, 1998). Although we know that peptides have different potencies to activate TCR signaling, we do not know if peptide cross-reactivity is achieved through a single docking footprint or if a range of MHC docking modes exist that would have disparate impacts on signaling induced by each peptide (Felix et al., 2007; Yin and Mariuzza, 2009). Although cross-reactive TCR complexes so far have shown similar docking modes (Mazza et al., 2007), in many cases the peptides shared key TCR contact residues (Ding et al., 1999; Macdonald et al., 2009; Mazza et al., 2007), leaving open the question of how sequences of unrelated peptides are accommodated by the TCR.
Here we address the roles of TCR-pMHC binding geometry, interface chemistry, affinity and kinetics in TCR signaling. We describe the development of peptide-MHC libraries in yeast that allowed us to discover large collections of peptides reactive with a given TCR. We measured the 2D and 3D interaction parameters with four different peptides, assayed signaling properties, and determined the crystal structures of the complexes. Our results suggest that there are geometric constraints on TCR-MHC docking footprints compatible with signaling and, more generally, TCR signaling can be modulated by perturbations in the extracellular receptor-ligand architecture.
Results
42F3 versus 2C recognition of H2-Ld presenting the QL9 peptide
The 42F3 TCR is derived from an alloreactive cytotoxic T lymphocyte clone that recognizes the class I MHC molecule H2-Ld presenting the peptide p2Ca933–940 of endogenous mouse 2-oxoglutarate dehydrogenase (Accession NP_035086)(Hornell et al., 1999). 42F3 is related to another p2Ca-H2-Ld reactive TCR, 2C, whose structure and binding properties have been extensively investigated (Colf et al., 2007; Garcia et al., 1996; Holler et al., 2003; Sykulev et al., 1994). Both 42F3 and 2C also recognize the related nonamer epitope of 2-oxogluterate dehydrogenase, QL9. The variable domains of 42F3 are Vα3.3 and Vβ8.3, therefore encoding identical CDR1α and CDR2α, and nearly identical CDR1β and CDR2β to 2C (Figure 1A). However, 42F3 and 2C use different CDR3 sequences to recognize QL9-H2-Ld (Figure 1A). Thus, these two TCR represent an ideal pair to ask whether shared germline contacts would persist despite their distinct CDR3-peptide contacts.
Figure 1. The 42F3 and 2C TCRs and their interactions with H2-Ld–QL9.
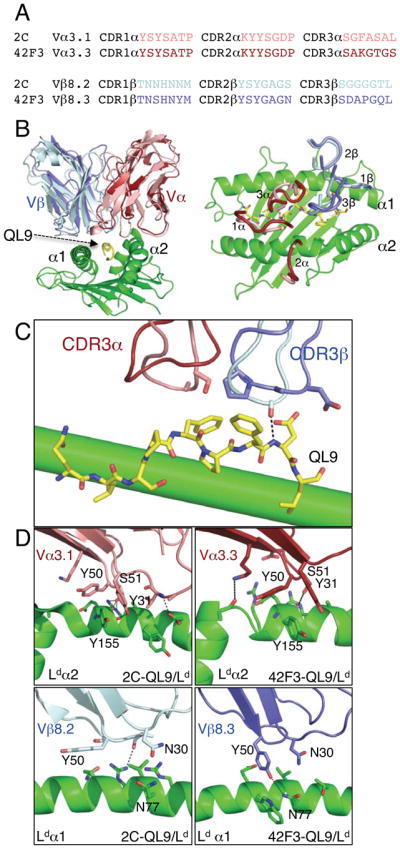
(A) Primary sequence alignment of the six CDR loops of 42F3 and 2C TCRs. (B) Ribbon representations of the 42F3 and 2C TCRs in complex with QL9-H2-Ld aligned on the MHC (left), Top-down perspective of the six CDR loops of 2C and 42F3 (loops) on the surface of H2-Ld (right). 2C is lighter shade, 42F3 is darker shade (C) Contacts made by the CDR3 loops to the QL9 peptide for both 2C (lighter shaded colors) and 42F3 (darker shaded colors) TCRs are depicted. (D) Contacts made by the Vα and Vβ CDR1 and CDR2 loops to the MHC helices. The MHC is colored green, QL9 is colored yellow, TCRα is colored firebrick and salmon and TCRβ is colored slate and palecyan for 42F3 and 2C, respectively. See also Figures S1 and S4.
We solved the crystal structure of a 42F3 single-chain Fv in complex with QL9-H2-Ld to 2.75Å resolution and found that it shared a highly similar overall binding footprint compared to the structure of 2C-QL9-H2-Ld (Figure 1B, Table S1)(Colf et al., 2007), with Vα binding contacts being much more similar than Vβ (Figure 1C). The Vα3s in both TCRs use a nearly identical set of contacts involving Tyr31α, Lys48α, Tyr50α, Ser51α and Gly52α to contact the H2-Ld α2 helix at Glu154 and Tyr155 (Figure 1D). In both interfaces, Ser51α hydrogen bonds to the MHC α2 helix backbone through its side chain hydroxyl while Tyr31α and Tyr50α bury Tyr155 on the MHC surface (Figure 1D). For Vβ8.2 and Vβ8.3 the 2C and 42F3 contacts with H2-Ld were highly divergent from one another (Figure 1D). In both TCRs, Asn30β and Tyr50β make contacts to the α1 helix, but the specific pair-wise residue contacts were different (Figure 1D).
Identification of new 42F3-reactive peptides
We developed a method to screen for 42F3 binding to yeast-displayed H2-Ld-peptide libraries (Figures 2A, S1). A variant of the ‘mini-MHC’ platform, previously used for biophysical studies (Colf et al., 2007; Jones et al., 2006), was fused through a Gly-Ser linker to the N-terminus of a nonamer peptide. In order to accomodate the Gly-Ser linker without disrupting TCR binding, we introduced a ‘knob-to-hole’ mutation (Trp167 to Ala) at the end of the MHC α2 helix that created a notch for the linker. The single-chain (sc) H2-Ld-peptide complexes were fused to the C-terminus of the yeast Aga2 protein (Gai and Wittrup, 2007) (Figure 2A). Key to this strategy is that selections are based on direct binding, in a cell free system with recombinant TCR, devoid of co-receptor or other potential endogenous influences on the mode of recognition by the TCR. To select and stain the pMHC yeast, we created TCR-tetramers by complexing C-terminally biotinylated TCR to streptavidin labeled with phycoerythrin (SA-PE). We first verified that an H2-Ld platform containing the cognate peptide QL9 would be specifically recognized by recombinant QL9-specific TCR using flow cytometry. Using an affinity-matured variant of 2C called m33 (Holler et al., 2003), we observed bright peptide dependent staining of yeast displaying Aga2-Ld-QL9 (Figure S1) (wild-type 2C TCR also showed positive staining). 42F3 tetramers poorly stained the same yeast population (Figure S1), indicative of a weak affinity for this TCR-pMHC complex, consistent with our SPR measurements (Figure S1).
Figure 2. Yeast surface display of peptide libraries presented by H2-Ld.

(A) Schematics of yeast displayed pMHC libraries. Inset is the stick representation of QL9 from the structure shown in Figure 1, showing the ‘up-facing’ recognition epitope of the 2C and 42F3 TCRs. (B) Two dimensional plot of the final enriched yeast populations of the random, TCR contact and MHC contact libraries enriched by FACS. Residues varied in each library are indicated in red where X represents 32 codons (NNK) encoding all 20 amino acids. Positions with fixed sequence are indicated in black. (C) Sequences of 42F3 peptides isolated from individual yeast colonies. Selected KD measurements are from SPR equilibrium values. See also Figure S1.
We created three classes of libraries for yeast-displayed sc H2-Ld-peptides. In the first, a random library consisted of peptides whose sequence was limited only to known anchor substitutions at the P2 and P9 anchor positions (Figure 2B)(Udaka et al., 2000). All other positions were allowed full amino acid diversity by using a degenerate NNK nucleotide codon set. A second library randomized only ‘up-facing’ TCR contact residues in the peptide based on the crystal structure (P4, P5, P7 and P8) (Figures 1C, 2B). A third library randomized ‘down-facing’ MHC contact positions (P1, P2, P3, P6) (Figures 1C, 2B).
After several rounds of enrichment by fluorescence activated cell sorting (FACS) with 42F3 tetramers (Figure S1), we recovered sequences of individual clones from the libraries that encoded diverse sets of peptides that were distinct from QL9 (Figure 2C). From the random library (Figures 2C, S1), we identified a single unique peptide sequence recognized by 42F3 (Figures 2B–C) that diverged from QL9 at every position. The TCR contact set encoded only conservative mutations at the P7 and P8 positions, while a new consensus arose at the P4 and P5 positions (Figure 2C). Encoded in the enriched MHC contact population were peptides that contained Trp at the P6 position and highly favored Asn at P3 (Figure 2C). We were surprised to find a Trp at P6, as it seemed incompatible with the pocket in the H2-Ld groove that the Pro occupied in QL9 (discussed below). BLAST sequence searches indicate that none of the peptides discovered showed substantial similarity to known proteins.
In vitro validation of library-derived peptides binding to 42F3
We expressed the respective recombinant pMHC complexes and evaluated affinity using surface plasmon resonance (SPR). Peptides selected from the combinatorial libraries bound to the recombinant 42F3 with affinities typical for TCR-pMHC interactions (KD ~ 5–50μM), and with very fast kinetics (Figure S1, Table S2). Only two of the tested pMHC complexes (3A1 and 5F1) (Figure S1, Table S2) exhibited fittable kinetics. All of the measured clones, including the random library clone 3A1, bound with higher affinity (KD ~ 4μM) to 42F3 than did the original agonist ligand presented by the single-chain H2-Ld-QL9 (Figures 2C, S1). These selected peptides provided a diverse collection of chemical features and affinities to assay for differences in signaling properties in a functional T cell assay.
T cell activation
We generated stable CD8+ and CD8− 42F3 T cell hybridoma transfectants using retroviral-mediated gene transduction. Antigen presenting cells (APCs) pulsed with five TCR contact peptides produced IL-2 in the presence or absence of CD8, consistent with their affinities and the affinity threshold (KD ~ 1 to 5 μM) for CD8 independence (Chervin et al., 2009) (Figures 3A–B). In contrast, the four MHC contact peptides tested and the native QL9 required CD8 for stimulating 42F3 T cells (Figures 3A-B). While this result is expected for the peptides QL9 and p5E8, which have lower affinities for the 42F3 TCR (KD ~300 and 48 μM in the single-chain peptide-H2-Ld format, respectively), the CD8-dependence was not expected for peptides p5F1 and p5F2, which had affinities for 42F3 equal to or better than the CD8-independent TCR contact peptides (Figure 2C). It is possible that the binding to cell surface H2-Ld is reduced for the MHC contact peptides compared to the TCR contact peptides, and this property influences the requirement for CD8. Consistent with this notion, the TCR contact peptide p4B3 was active at low peptide concentrations in the presence of CD8 (Figure S2A).
Figure 3. T cell activation by peptide antigens.

IL-2 release for (A) CD8-negative and (B) CD8-positive 42F3 T cells stimulated by peptide (10μM) loaded APCs with an effector:target ratio of 1:1. Full dose-response curves are shown in Figure S4 (C) IL-2 release for CD8+ 42F3 T cells stimulated with peptide loaded H2-Ld-Ig dimers. (D) p3A1 antagonist assay where 42F3 T cells stimulated with 100nM p4B3 were assayed for IL-2 production over a range of p3A1 concentrations. The stimulatory anti-CD3 (2C11) antibody and/or the endogenous peptide QL9 were used to assay IL-2 sensitivity in each ELISA. The addition of no peptide and the irrelevant H2-Ld restricted MCMV epitope (YPHFMPTNL) were used to assay peptide independent IL-2 production in each experiment. Peptides showing CD8 dependence were colored cyan, peptides showing CD8 independence were colored green,. See also Figure S2.
A peptide from the random library, SPLDSLWWI (p3A1), failed to induce a strong IL-2 response for either 42F3 T cell line up to a concentration of 100μM peptide (Figures 3A–B, S2B-C), despite having one of the highest measured SPR affinities for the 42F3 TCR (Figures 2C, S1). To determine if the lack of IL-2 stimulation was due to inefficient loading of p3A1 onto H2-Ld on the APC, we assayed p3A1’s ability to compete with a biotinylated agonist (Figure S2A) for MHC binding on the APC surface. We labeled the biotinylated agonist with SA-PE and measured APC binding using flow cytometry (Figure S2D). The unlabelled p3A1 peptide effectively competed for MHC binding on the APCs at experimental conditions (10μM) comparably to several agonist peptides (Figure S2E), indicating loading in H2-Ld. We then asked whether peptide-loaded recombinant H2-Ld molecules, as opposed to peptide-loaded H2-Ld on APC, could activate 42F3 transfectants. Recombinant H2-Ld-Ig dimers presenting p3A1 failed to stimulate the 42F3 T cells, whereas agonist peptides loaded in the H2-Ld-Ig dimers effectively stimulated (Figure 3C). Finally, we asked whether the lack of stimulation was due to an APL-type of downstream antagonism that could inhibit the function of co-presented agonist peptide (Hogquist et al., 1994; Stone et al., 2011). However, agonist (p4B3) induced activation was not influenced by the p3A1 peptide presented by the same APC (Figure 3D). The p3A1 peptide, then, binds to 42F3 with comparable affinity to agonists, but does not appear to deliver either an activating or inhibitory signal.
Two-dimensional (2D) affinity and tetramer binding to cells
Recently, kinetic measurements using intact T cells have been shown to correlate with proliferative T cell responses for the 5c.c7 and OT1 TCR systems (Huang et al., 2010; Huppa et al., 2010). The advantage of this approach over 3D kinetic measurements such as SPR is that the membrane confinement properties of the TCR and pMHC are preserved. Since 3D kinetics of the 42F3 peptides poorly correlated with activation, we carried out experiments to measure the 2D kinetic parameters of p3A1-H2-Ld binding to CD8αβ− 42F3 T cells and compared it to several agonists (Figures 4A, S3). In this format, 42F3 transfectants are used as the source of TCR, and SA-coated red blood cells are decorated with the same N-terminally biotinylated sc H2-Ld-peptide complexes used for SPR measurements. In situ, the agonist peptides bound to the T cells with fast Ackon and fast 2D-koff creating greater than 2-fold difference in 2D affinity compared to p3A1 (Figure 4A), consistent with the IL-2 activation (Figures 3A–B). While all measured peptides had fast off-rates (t1/2=0.2–0.09s), the H2-Ld-p3A1 interaction had the slowest 2D-koff (Figure S3). The rank orders of peptides in the 2D format correlated better with peptide biological activity (Figure 3) than 3D (Figures 4B, S3). Both the lack of 42F3 T cell activation and the weak 2D-Ackon (Figure S3) suggest that p3A1 association with the 42F3 TCR is less favorable on the T cell surface compared to isolated TCR in solution.
Figure 4. 2D and 3D measurements of peptide-MHC interactions with 42F3.

(A) 2D affinities (AcKA) derived from T cell adhesion frequency and thermal fluctuation assays using pMHC coated RBCs. (B) SPR derived 3D affinities (expressed as KA for comparison to 2D) using sc H2-Ld constructs (p3A1 has the highest affinity, QL9 the lowest). Direct comparison of 2D and 3D kinetics also shown in Figure S5. (C) Fluorescent 42F3-tetramer staining of peptide loaded APCs. (D) Fluorescent sc H2-Ld-tetramer staining of 42F3 T cells. Peptides presented by H2-Ld were CD8 dependent (cyan) agonists, CD8 independent agonists (green), or the non-stimulatory peptide p3A1 (red). (E) Correlation between TCR tetramer staining and the 3D affinity of the pMHC ligand is compared to the correlation between pMHC tetramer staining and the 2D affinity (F). Open bars represent tetramer staining, and closed bars represent 2D and 3D affinities shown in panel A. See also Figure S3.
We further investigated the reduced 2D affinity of the p3A1 interface by probing the epitope availability on the peptide-loaded APCs and the 42F3 T cell line, respectively. 42F3-tetramers stain p3A1-loaded APCs (Figure 4C) while H2-Ld-p3A1-tetramers did not stain 42F3 T cells (Figure 4D). TCR tetramer staining is in good correlation with the 3D affinities measured by SPR (Figures 4E) while the pMHC tetramer binding correlated better with the 2D affinities (Figure 4F). Collectively, the 2D and tetramer staining experiments show that the engagement of 42F3 TCR on cells appears to be spatially constrained during engagement of p3A1 compared to agonist peptides. When the TCR was not presented in the context of a T cell membrane during ‘3D’ SPR measurements, either as a tetramer or monomer in solution, the binding affinities and kinetics of several agonist peptides and p3A1 were more similar (Figure 4E).
Structural features of peptide-MHC recognition
We determined crystal structures of the 42F3 TCR bound to each of the four H2-Ld-peptide complexes: p3A1-H2-Ld (2.1 Å), QL9-H2-Ld (2.75 Å), p4B10-H2-Ld (2.9Å) and p5E8-H2-Ld (3.1Å) (Figures 5A, S4, and Table S1). Each peptide was well defined in the electron density and presented a unique structural epitope and chemical surface to the TCR (Figures 5A, S4). In fact, the interaction chemistries of each of the four peptides with the 42F3 CDR3s were nearly entirely distinct, ranging from largely hydrophobic (p3A1, QL9), to polar-hydrophobic (p5E8), to charged (p4B10), highlighting a remarkable ability of a single binding site to accommodate a range of structural chemistries (Figure 5A). While we do not describe each interface here in detail, of particular note are unexpected conformations of the p5E8 and p3A1 peptides, which contain either a Trp or Leu substitution at P6 respectively (P6 is Pro in QL9) (Figure 2C). In p5E8, the peptide backbone has flipped, such that P6-Trp is now an up-facing TCR contact instead of a down-facing MHC anchor, apparently due to a lack of space for the Trp side chain in the H2-Ld peptide-binding (Figure 5A). The p5E8 P5-Phe, a TCR contact in QL9, is flipped down into the MHC anchor pocket previously filled with the QL9 P6-Pro (Figure 5A). Thus, the TCR epitope of the MHC contact peptide is completely altered even though none of the TCR contact residues were randomized in this library. In the case of p3A1, the P6-Leu also becomes a TCR contact. However, the p3A1 peptide fails to fill the P6-pocket, instead forming an arched conformation (Figures 5A, S4). These observations highlight the unpredictable manifestations of peptide substitutions on T cell recognition, and that MHC anchors can greatly impact the TCR epitope in unexpected fashions. Superposition of the four TCR structures from the complexes shows that CDR3 conformational variability in 42F3 primarily lies in CDR3α, not CDR3β (Figure 5B). This illustrates TCR cross-reactivity through a relatively rigid TCR binding site in the absence of significant CDR3β flexibility.
Figure 5. Cross-reactivity through distinct peptide recognition chemistries by the 42F3 TCR.

(A) Left column, shows ribbon diagrams of the VαVβ domains of 42F3 bound to QL9, p5E8, p4B10 and p3A1 presented by the α1α2 of H2-Ld. Center column shows accompanying interactions of the respective TCR CDR3 loops with the presented peptides (MHC is colored green, peptides are colored yellow). Right column shows two-dimensional contact maps of 42F3 CDR3α and CDR3β loop contacts. The TCR structures in the p3A1, p4B10, and p5E8 complexes included Constant domains, but these are not depicted in the figures. (B) Aligned structures of the 42F3 TCR Variable domains shows superpositions of CDR loop structures from the four different complexes. 42F3α is colored firebrick (QL9), lightpink (p5E8), raspberry (p4B10) and magenta (p3A1). The 42F3β is colored slate (QL9), lightblue (p5E8), cyan (p4B10) and blue (p3A1). See also Figure S4.
TCR-pMHC docking geometry
All three of the agonist peptides share a similar overall docking footprint where the Vα and Vβ are positioned diagonally across the surface of the pMHC (Figure 6A). In contrast, the non-stimulatory p3A1 docking footprint diverged markedly (Figures 6A, 7A). In the agonist complexes, comparison of the Vα3.3 contact points with the α2 helix revealed the same conserved contact set previously observed for four 2C complexes with agonist peptides (Figures 1D, 6B) involving Vα germline residues Lys48α, Tyr31α, Tyr50α and Ser51α (Figures 6C, S5). Through small-scale rolling and tilting adjustments of the TCR in each complex, Vα3.3 formed a variety of auxiliary polar and van der Waals contacts unique to each pMHC’s interface, whilst retaining a shared core of contacts (Figures 6C, S5). For example, a C-terminal roll in the QL9 complex allowed Lys48α to hydrogen bond to the MHC α2 backbone, whereas in the p4B10 complex, the Vα makes additional hydrogen bonds from Tyr50α and Ser51α to the Glu154 carboxyl group (Figure S5). These adjustments show that the Vα germline contacts adapt to peptide-specific sequence differences while maintaining a strikingly conserved contact set with the MHC α2 helix (Figures 6B–C). This peripheral plasticity in the midst of a conserved core interaction network was also noted for the Vβ8/I-A complexes (Dai et al., 2008; Feng et al., 2007).
Figure 6. Stimulatory TCR/pMHC docking geometries mediated by a Vα3 germline ‘codon.’.

(A) The TCR docking footprints of 42F3 recognizing three stimulatory and one non-stimulatory peptide. Dashed lines connecting the Cα of Y50α and Y50β carbons are used to illustrate the relative orientations of the respective complexes 42F3-QL9-H2-Ld (black) and 42F3-p3A1-H2-Ld (red). Black and red dots indicate the center of masses of the 42F3 TCR in the QL9 versus p3A1 complexes, respectively. (B) The convergent docking footprints of seven different Vα3Vβ8 TCR complexes with H2-Ld-peptide agonists including four previous 2C-complexes and three 42F3 complexes recognizing stimulatory peptides. Inset (top) is a color-coded legend for each TCR and the corresponding peptide recognized in each H2-Ld complex. Inset (bottom) is a contact map depicting conserved contacts between the Vα3.1 or Vα3.3 and the α2-helix in seven agonist complexes. (C) Detailed contacts of CDR1 and CDR2 of the Vα3.1 and Vα3.3 to the α2 helix (green) in seven agonist complexes. van der Waals contacts are depicted as sticks while hydrogen bonds and salt bridges are depicted with dashed lines. See also Figure S5.
Figure 7. Altered TCR-pMHC docking geometry in a non-stimulatory complex.

(A) The docking footprint of 42F3 recognizing the p3A1 non-stimulatory peptide. As in Figure 6, a dashed line connects the Cα of Y50α and Y50β carbons (red, black for stimulatory complexes) to illustrate the binding orientation. (B) The germline contacts made by either the Vα (left panel) or the Vβ (right panel) to the MHC helices of H2-Ld presenting p3A1. Residues making van der Waals contacts are represented as sticks and dashed lines represent hydrogen bonds. (C) Range of docking footprints in 40 Class I TCR-pMHC agonist complexes (firebrick and slate) aligned on the MHC together with the non-stimulatory p3A1 peptide footprint (magneta and blue). The CDR loops of 42F3 in the p3A1 complex are highlighted as thick tubes, and the docking angle is shown as a dashed yellow line, with the center of mass between the CDR3α and CDR3β of 42F3 denoted by the yellow dot. (D) Theoretical models for how TCR-pMHC docking geometry in the p3A1 (red peptide) complex could influence signaling compared to the productive agonist (green peptide).
The structures show that 42F3 Vβ8, which appeared to be key germline contacts in prior Vβ8 complexes, does not form many shared or conserved contacts with the H2-Ld α1 helix between the different peptide complexes (Figures 6A–B, S5). While Tyr50β and Asn31β continue to play prominent roles in each of these interfaces, and the overall location of the Vβ-MHC contact patch is very similar between the structures (Figure 6B), the pairwise contacts are chemically distinct (Figure S5). The residue Tyr50β, central to the previously described Vβ8.2-I-A ‘codon’ (Feng et al., 2007), uses a variety of side chain rotamers to recognize the different pMHC surfaces (Figure S5). In the p5E8 complex, the Tyr50β rotamer lies planar to the α1 helix as seen in the 2C-QL9-H2-Ld complex (Figure S5). In the QL9 and p4B10 complexes, Tyr50β points deep into the peptide-binding groove to make a hydrogen bond to peptide at the P8-Asp position (Figure S5). These structures highlight the important role, and remarkable ability of Tyrosine residues in TCR V-regions to form multifarious germline MHC contacts (Garcia et al., 2009; Marrack et al., 2008).
A germline-encoded motif for TCR Vα3 recognition of H2-Ld
Analysis of seven total TCR-H2-Ld-peptide complexes: three 42F3 agonist complexes, a 2C complex with H2-Ld (Colf et al., 2007) and three CDR3α mutants of 2C (m6, m67, m13) in complex with H2-Ld (Jones et al., 2008) revealed a conserved contact set between these Vα3 CDRs 1α and 2α with the α2 helix of H2-Ld (Vα3.3 in the case of 42F3, Vα3.1 in the cases of 2C, m67, m6 and m13)(Figure 6B). Central to the Vα3.1 and Vα3.3 motif are Tyr31α and Tyr50α van der Waals contacts to H2-Ld Tyr155 and the TCR Ser51α hydrogen bond to the helical backbone at H2-Ld Glu154 (Figure 6C). Y31α and Y50α are among the most energetically important in the 2C TCR interaction with H2-Ld-QL9 (Manning et al., 1998). The Vα3.3 interaction motif accommodates slight differences in TCR roll and tilt, a variety of CDR3 sequences (2C, m6, m67, m13 and 42F3) and considerable peptide variation (QL9, pB10, pE8) (Figure 6C). We conclude that 2C and 42F3 TCR germline recognition of H2-Ld is centered on a Vα-centric ‘codon’, as compared to the apparently Vβ-centric recognition of the class II I-A complexes (Figure 6C). It appears that different TCR complexes can be more or less Vα or Vβ-centric in the utilization of conserved germline-contacts.
Unusual docking geometry of the p3A1 non-stimulatory complex
p3A1 peptide recognition required a global reorientation of the entire 42F3-H2-Ld interface compared to the agonist complexes, rotating ~38° clockwise and translating ~7Å from the center of the groove so that 42F3 lies over the α1 helix, resulting in a TCR orientation nearly parallel to the peptide (Figures 6A, 7A). 42F3 recognition of H2-Ld did not involve the Vα codon seen in the stimulatory complexes (Figure 6A, C). Instead, the TCR Vα3.3 makes a single contact to the α2 helix at H2-Ld Tyr155 through 42F3 Tyr50α, and straddles both helices of the peptide-binding groove, forming a hydrogen bond between 42F3 Thr29α to H2-Ld Gln65 of the α1 helix (Figures 7B, S5). The parallel docking topology also enables Vβ8.3 to straddle the groove, with contacts on both the α1 and α2 helices, including Tyr50β interacting with the α2-helix. (Figures 7B, S5). Also, a slightly ‘squeezed’ conformation of p3A1 peptide-binding groove results in a narrower distance between α1 and α2 helices, facilitating Asn31β bridging the MHC groove to form a bifurcated hydrogen bond with both Thr80 of the Ld α1 and Lys146 of α2 helices (Figure 7B). The docking footprint appears to be at the extreme clockwise end of the range compared to other class I agonist TCR-pMHC complexes (Figure 7C). Importantly, despite the unusual MHC footprint, 42F3 CDR3α and CDR3β are focused on the peptide, and the global polarity of the complex does not violate the previous paradigm of Vα lying over the N-terminal end of the groove, and Vβ lying over the C-terminal end (Figures 5A, 7A).
Discussion
In a classical study, Janeway and colleagues showed that different TCR and CD3 antibodies had varied agonistic properties, prompting the suggestion that TCR signaling was dependent upon the overall architecture of the TCR-CD3 complex (Janeway, 1995; Yoon et al., 1994). In contrast, other studies, such as those using chemically defined oligomerization agents concluded that intermolecular proximity alone was the key determinant for TCR activation (Cochran et al., 2001). Here we developed a peptide-based approach to investigate the interrelationship between TCR-pMHC binding chemistry, docking geometry, 2D and 3D binding parameters, and signaling. Our principal findings are that: 1) not every binding orientation is compatible with signaling, and 2) a TCR utilizing entirely distinct chemistries to recognize different peptides exhibits highly persistent germline-mediated contacts. That alternative TCR-MHC binding modes are accessed by certain peptides is an important extension to prior studies on TCR cross-reactivity where the germline MHC contacts have largely remained intact, albeit often undergoing adjustments (Borbulevych et al., 2009; Ding et al., 1999; Garcia et al., 1998; Macdonald et al., 2009; Mazza et al., 2007; Reiser et al., 2003; Wucherpfennig et al., 2009; Yin and Mariuzza, 2009). Here we find that peptide cross-reactivity can influence signaling through gross alterations of TCR binding geometry.
What is the mechanistic basis for the p3A1 lack of activity? We do not know if the manifestation of an altered TCR docking topology as a lack of signaling is reflective of conformational change(s), altered CD3 associations, clustering or other events known to be critical for TCR signaling (van der Merwe and Dushek, 2011; Gil et al., 2002; Minguet et al., 2007). Although not providing detailed answers, the 2D versus 3D experiments are informative. The 2D method clearly demonstrated that H2-Ld-p3A1 is binding to the 42F3 TCR on the cell surface, albeit with less frequency than the agonist peptides. The H2-Ld-p3A1 bound with high affinity to soluble monomeric 42F3 using SPR (3D), but neither H2- Ld-p3A1 tetramers or p3A1 loaded dimers (H2-Ld-Ig) stained or stimulated 42F3 T cells respectively. Thus, when cellular spatial constraints are absent, there is a high affinity interaction between H2-Ld-p3A1 and 42F3, yet each of the three ‘constrained methods’ yielded reduced signaling and binding by p3A1 versus a panel of agonist peptides. Collectively, these 2D and 3D data suggest that the spatial orientation of the pre-bound states of the TCR and pMHC on the cell membrane are critical factors determining TCR-MHC associations and TCR signaling.
We can propose several speculative, but equally plausible, models for p3A1’s lack of activity. In one, in order for the TCR to engage in the unusual H2-Ld docking angle on the APC, it is required to twist out of a signaling-productive oligomeric or ultrastructural arrangement (Figure 7D). The docking angle in the p3A1 complex may have exceeded the allowable range of signaling-competent binding modes. The minimal signaling TCR ligand is a pMHC dimer (Boniface et al., 1998; Cochran et al., 2001), and a study proposed a specific topology of the corresponding αβ-TCR-CD3 dimer (Kuhns et al., 2010). Engagement of p3A1 may either produce unproductive TCR-pMHC dimers or inhibit, or ‘poison’, the formation of productive p3A1 dimers by blocking the recruitment of a second TCR or MHC, the latter scenario being an ‘oligomer exclusion’ model (Figure 7D). In short, the p3A1 docking topology may be incompatible with the TCR-CD3 signaling dimer architecture (Figure 7D). However, we currently know very little about the ultra-structure of clustered TCR and how these geometric considerations influence the engagement of distinct pMHC orientations. Since the majority of class I MHC on the APC surface present self-peptides, it is rare for a T cell receptor dimer to encounter two foreign agonist peptides in MHC simultaneously. Rather, T cells can apparently overcome this limitation by signaling via an MHC “pseudodimer” containing only one agonist peptide, but whether there are geometric constraints placed upon agonist signaling by the endogenous coagonist is not known (Juang et al., 2010). Unclustering the TCR by disrupting lipid rafts or actin polymerization has been shown to change the 2D-affinity for TCR-pMHC (Huang et al., 2010; Huppa et al., 2010), suggesting an important role for TCR membrane organization in antigen recognition. It is also possible that simultaneous, multi-point attachment of CD3, CD8, and MHC to the TCR within a unitary signaling complex in these signaling clusters can occur only within a range of TCR-pMHC docking angles that are accommodated by canonical orientations. The p3A1 docking angle may have exceeded these tolerances so that in order for 42F3 to bind to p3A1, either the signaling competent TCR dimer or oligomer or its associations with co-receptors (CD3, CD4 or CD8) are disrupted. It is puzzling that while the association of p3A1 was weaker in situ than agonists, the resulting 2D AcKa, Ackon and koff were equivalent to that of stimulatory pMHC associations on OT1 CD8+ T cells (Huang et al., 2010). Thus, the lack of activation is not fully explained by decreased 2D kinetics alone.
That the non-stimulatory peptide is not a naturally occurring sequence, and binds in an unconventional orientation, has potential implications for the notion of germline TCR-MHC co-evolution. Germline-specificity presumably evolved in the context of coreceptors and natural peptide antigen sequences, in part, resulting in productive TCR-MHC-CD3-CD8(or CD4) signaling geometries. p3A1, being an ‘unnatural’ peptide could have accessed geometric limits of TCR-pMHC orientation incompatible with signaling on T cells. In this respect, the synthetic approach may sidestep evolutionary pressures experienced in the context of the endogenous peptide milieu to achieve a ‘unnatural’ docking footprint. However, it is unclear if the docking mode we see for p3A1 is ‘germline-encoded,’ or not, since the overall the canonical binding polarity is not violated compared to other TCR-pMHC complexes, and important germline residues (e.g. Tyr31α, Tyr51α, Tyr50β) still mediate the MHC contacts. It is intriguing to consider that non-productive peptides and binding modes analogous to p3A1 could exist in nature but have evaded experimental detection using TCR signaling-based identification methods.
With reference to prior studies showing conserved germline Vβ8.2-I-A contacts in over eight TCR-pMHC complexes, the Vα3.3 and Vα3.1 recognition of H2-Ld shown here for seven different agonist peptide complexes (42F3 and 2C) illustrates that both Vβ and Vα centric germline motifs, which we have euphemistically referred to as ‘codons’ exist. In the case of H2-Ld, the Vα codon centers on MHC position 155, which has been previously identified as a component of the MHC restriction triad, and is contacted in nearly all Class I TCR-pMHC complex structures (Tynan et al., 2007), highlighting the astonishing ability of each V-segment to form structurally distinct sets of contacts with different classes of MHC. Such diversity appears conserved within a given MHC, but very different across different MHC.
On one hand, elucidation of an alternative docking mode suggests that peptide can influence the TCR to form dissimilar germline contacts with MHC. On the other hand, three of the four peptides are recognized in a convergent and apparently germline-encoded Vα docking position. That this convergence was achieved with distinct peptides selected in the absence of co-receptors strongly supports the influence of an intrinsic MHC specificity. We suggest these seemingly incongruent results are consistent and reconcile the relative roles of intrinsic (germline engrafted specificity) and extrinsic (steric influence of co-receptors) in positioning the TCR-MHC docking orientation. Germline TCR-MHC specificity probably arose within the context of a higher order TCR-CD3-MHC-CD4 (or CD8) complex where the TCR was positioned topologically, through multi-point attachment, for productive signaling. In this sense, the interfacial TCR/MHC contacts mediating signaling competent binding geometries, have been selected during evolution. These higher order geometric constraints would certainly involve co-receptor influences that could enforce the invariant TCR-MHC docking polarity. However, this interfacial specificity was engrafted in the germline, which obviated the need for co-receptors to actively ‘position’ the TCR.
Taken together, our data demonstrate a relationship between TCR-MHC docking geometry, peptide cross-reactivity, germline bias, and signaling. The innate cross-reactivity of T cell recognition is apparently more than a means of simply enabling a ‘structurally agnostic’ bimolecular interaction between a TCR-MHC. Rather, the chemistry of the peptide can modulate docking footprints as well as kinetics and affinity, which in turn can modulate signaling. Our future studies will attempt to more deeply understand the generality and ultrastructural mechanism of this unexpected finding.
Materials and Methods
Cloning and expression of 42F3 TCR
RNA was isolated from primary 42F3 T cells in order to prepare cDNA of the TCR α and β chains. For recombinant expression, the V regions of the α and β chain were spliced by overlapping extension to form a Vα-(Gly4Ser)4-Vβ scFv expressed in pET22b to allow for periplasmic secretion. Two stabilizing mutations in the Vα3.3 and five mutations in the Vβ8.3 were introduced as previously described for 2C (Colf et al., 2007). For full-length ectodomains, the wt Vα3.3 and Vβ8.3 regions of the 43F3 TCR were fused in frame with human constant domains (Boulter et al., 2003) and expressed in baculovirus with a C-terminal acidic GCN4 zipper-BAP-6xHis tag (α) or a C-terminal basic GCN4-zipper-6xHis tag (β).
Yeast displayed H2-Ld-peptide libraries
H2-Ld was displayed on yeast by converting a minimal α1α2 variant of H2-Ld called m31, that was previously used for structural studies (Jones et al., 2006) into a single chain pMHC with a C-terminal tethered peptide (Aga2-LdW167A-pep) by incorporating the mutation W167A. A degenerate primer containing the peptide was used to PCR amplify combinatorial libraries. The yeast libraries were constructed by standard methods (Chao et al., 2006). The number of transformed yeast for each library were 1.8×108, 1.0×108 and 5.7×107 for the Random, TCR contact and MHC contact libraries respectively.
42F3-Tetramer selection of yeast clones
After induction of the yeast libraries, the yeast cells were incubated with 470nM preformed 42F3-tetramers assembled on SA-PE (Invitrogen) and 1:100 dilution of anti-HA-Alexa488 (Invitrogen) sorted on a FACS Aria (BD). During the first round of selections the brightest 2.5% of the TCR-Tet+/HA+ population was sorted into a single culture. The sorted cells were grown overnight and the process repeated. After the first round of selection, the brightest 1% of TCR-Tet+/HA+ was sorted until greater than 10% of the yeast population could be resolved from the unstained population by flow cytometry.
2D kinetics measurements
Procedures for coupling pMHC to red blood cells (RBC) or glass beads has been described (Huang et al., 2007). Briefly, RBCs were first biotinylated with biotin-X-NHS (EMD chemicals) and then reacted to streptavidin; borosilicate beads were first cleaned, silanized, and then reacted to streptavidin-maleimide (Sigma-Aldrich, St. Louis, MO). Streptavidinized RBC or beads were finally coupled to the single-chain pMHC through their biotin-tags. Site densities of TCR on hybridoma cells and pMHC on RBC or bead surfaces were measured with flow cytometry. Micropipette adhesion frequency assay and Thermal fluctuation assay were carried out in a similar manner as previously described (Chesla et al., 1998) and described in detail in Supplementary Methods.
Supplementary Material
Acknowledgments
We thank Natasha Goriatcheva, Engin Özkan, Dane Wittrup, Evan Newell, and Mark Davis for helpful discussions. We also gratefully acknowledge the staff and resources of the Stanford Synchrotron Radiation Laboratory (SSRL) and the UC-Berkeley Advanced Light Source (ALS). JJA was supported by a Canadian Institutes of Health Research (CIHR) post-doctoral fellowship, MB is supported by a National Science Foundation and Stanford Graduate pre-doctoral fellowship and is a member of the Stanford program in Immunology. This work was supported by NIH grants AI48540 (KCG) and GM55767 (DMK). KCG is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Accession Numbers
Atomic coordinates have been deposited in the PDB under the accession numbers 3TPU, 3TJH, 3TFK, and 3TF7 for the 42F3-p5E8, 42F3-p3A1, 42F3-p4B10, and 42F3-QL9 TCR-pMHC complexes, respectively.
Additional Methods
More detailed descriptions of methods for protein expression and purification, SPR measurements, protein crystallization and structure refinement, T cell activation assays, and MHC-TCR tetramer staining can be found in the supplemental information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NR. T-cell-receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- Aleksic M, Dushek O, Zhang H, Shenderov E, Chen JL, Cerundolo V, Coombs D, van der Merwe PA. Dependence of T cell antigen recognition on T cell receptor-peptide MHC confinement time. Immunity. 2010;32:163–174. doi: 10.1016/j.immuni.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beddoe T, Chen Z, Clements CS, Ely LK, Bushell SR, Vivian JP, Kjer-Nielsen L, Pang SS, Dunstone MA, Liu YC, et al. Antigen ligation triggers a conformational change within the constant domain of the alphabeta T cell receptor. Immunity. 2009;30:777–788. doi: 10.1016/j.immuni.2009.03.018. [DOI] [PubMed] [Google Scholar]
- Boniface JJ, Rabinowitz JD, Wulfing C, Hampl J, Reich Z, Altman JD, Kantor RM, Beeson C, McConnell HM, Davis MM. Initiation of signal transduction through the T cell receptor requires the multivalent engagement of peptide/MHC ligands. Immunity. 1998;9:459–466. doi: 10.1016/s1074-7613(00)80629-9. [DOI] [PubMed] [Google Scholar]
- Borbulevych OY, Piepenbrink KH, Gloor BE, Scott DR, Sommese RF, Cole DK, Sewell AK, Baker BM. T cell receptor cross-reactivity directed by antigen-dependent tuning of peptide-MHC molecular flexibility. Immunity. 2009;31:885–896. doi: 10.1016/j.immuni.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, Jakobsen BK. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein engineering. 2003;16:707–711. doi: 10.1093/protein/gzg087. [DOI] [PubMed] [Google Scholar]
- Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- Buslepp J, Wang H, Biddison WE, Appella E, Collins EJ. A correlation between TCR Valpha docking on MHC and CD8 dependence: implications for T cell selection. Immunity. 2003;19:595–606. doi: 10.1016/s1074-7613(03)00269-3. [DOI] [PubMed] [Google Scholar]
- Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- Chervin AS, Stone JD, Holler PD, Bai A, Chen J, Eisen HN, Kranz DM. The impact of TCR-binding properties and antigen presentation format on T cell responsiveness. J Immunol. 2009;183:1166–1178. doi: 10.4049/jimmunol.0900054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesla SE, Selvaraj P, Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophysical journal. 1998;75:1553–1572. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran JR, Cameron TO, Stone JD, Lubetsky JB, Stern LJ. Receptor proximity, not intermolecular orientation, is critical for triggering T-cell activation. The Journal of biological chemistry. 2001;276:28068–28074. doi: 10.1074/jbc.M103280200. [DOI] [PubMed] [Google Scholar]
- Colf LA, Bankovich AJ, Hanick NA, Bowerman NA, Jones LL, Kranz DM, Garcia KC. How a single T cell receptor recognizes both self and foreign MHC. Cell. 2007;129:135–146. doi: 10.1016/j.cell.2007.01.048. [DOI] [PubMed] [Google Scholar]
- Collins EJ, Riddle DS. TCR-MHC docking orientation: natural selection, or thymic selection? Immunol Res. 2008;41:267–294. doi: 10.1007/s12026-008-8040-2. [DOI] [PubMed] [Google Scholar]
- Dai S, Huseby ES, Rubtsova K, Scott-Browne J, Crawford F, Macdonald WA, Marrack P, Kappler JW. Crossreactive T Cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity. 2008;28:324–334. doi: 10.1016/j.immuni.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding YH, Baker BM, Garboczi DN, Biddison WE, Wiley DC. Four A6-TCR/peptide/HLA-A2 structures that generate very different T cell signals are nearly identical. Immunity. 1999;11:45–56. doi: 10.1016/s1074-7613(00)80080-1. [DOI] [PubMed] [Google Scholar]
- Felix NJ, Allen PM. Learning from the lost: new insights into TCR specificity. Nat Immunol. 2006;7:1127–1128. doi: 10.1038/ni1106-1127. [DOI] [PubMed] [Google Scholar]
- Felix NJ, Donermeyer DL, Horvath S, Walters JJ, Gross ML, Suri A, Allen PM. Alloreactive T cells respond specifically to multiple distinct peptide-MHC complexes. Nat Immunol. 2007;8:388–397. doi: 10.1038/ni1446. [DOI] [PubMed] [Google Scholar]
- Feng D, Bond CJ, Ely LK, Maynard J, Garcia KC. Structural evidence for a germline-encoded T cell receptor-major histocompatibility complex interaction ‘codon’. Nat Immunol. 2007;8:975–983. doi: 10.1038/ni1502. [DOI] [PubMed] [Google Scholar]
- Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol. 2007;17:467–473. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia KC, Adams EJ. How the T cell receptor sees antigen--a structural view. Cell. 2005;122:333–336. doi: 10.1016/j.cell.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Garcia KC, Adams JJ, Feng D, Ely LK. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol. 2009;10:143–147. doi: 10.1038/ni.f.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia KC, Degano M, Pease LR, Huang M, Peterson PA, Teyton L, Wilson IA. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA. An αβ T cell receptor structure at 2.5Å and its orientation in the TCR-MHC complex. Science. 1996;274:209–219. doi: 10.1126/science.274.5285.209. [DOI] [PubMed] [Google Scholar]
- Gil D, Schamel WW, Montoya M, Sanchez-Madrid F, Alarcon B. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 2002;109:901–912. doi: 10.1016/s0092-8674(02)00799-7. [DOI] [PubMed] [Google Scholar]
- Godfrey DI, Rossjohn J, McCluskey J. The fidelity, occasional promiscuity, and versatility of T cell receptor recognition. Immunity. 2008;28:304–314. doi: 10.1016/j.immuni.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Govern CC, Paczosa MK, Chakraborty AK, Huseby ES. Fast on-rates allow short dwell time ligands to activate T cells. Proc Natl Acad Sci U S A. 2010;107:8724–8729. doi: 10.1073/pnas.1000966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. 2005;6:490–496. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Holler PD, Chlewicki LK, Kranz DM. TCRs with high affinity for foreign pMHC show self-reactivity. Nat Immunol. 2003;4:55–62. doi: 10.1038/ni863. [DOI] [PubMed] [Google Scholar]
- Hornell TM, Solheim JC, Myers NB, Gillanders WE, Balendiran GK, Hansen TH, Connolly JM. Alloreactive and syngeneic CTL are comparably dependent on interaction with MHC class I alpha-helical residues. J Immunol. 1999;163:3217–3225. [PubMed] [Google Scholar]
- Huang J, Edwards LJ, Evavold BD, Zhu C. Kinetics of MHC-CD8 interaction at the T cell membrane. Journal of immunology. 2007;179:7653–7662. doi: 10.4049/jimmunol.179.11.7653. [DOI] [PubMed] [Google Scholar]
- Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, Zhu C. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 2010;464:932–936. doi: 10.1038/nature08944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huseby ES, White J, Crawford F, Vass T, Becker D, Pinilla C, Marrack P, Kappler JW. How the T cell repertoire becomes peptide and MHC specific. Cell. 2005;122:247–260. doi: 10.1016/j.cell.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Janeway C. Ligands for the T-cell receptor: Hard times for the avidity models. Immunol Today. 1995;16:223–225. doi: 10.1016/0167-5699(95)80163-4. [DOI] [PubMed] [Google Scholar]
- Jones LL, Brophy SE, Bankovich AJ, Colf LA, Hanick NA, Garcia KC, Kranz DM. Engineering and characterization of a stabilized alpha1/alpha2 module of the class I major histocompatibility complex product Ld. J Biol Chem. 2006;281:25734–25744. doi: 10.1074/jbc.M604343200. [DOI] [PubMed] [Google Scholar]
- Jones LL, Colf LA, Stone JD, Garcia KC, Kranz DM. Distinct CDR3 conformations in TCRs determine the level of cross-reactivity for diverse antigens, but not the docking orientation. J Immunol. 2008;181:6255–6264. doi: 10.4049/jimmunol.181.9.6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang J, Ebert PJ, Feng D, Garcia KC, Krogsgaard M, Davis MM. Peptide-MHC heterodimers show that thymic positive selection requires a more restricted set of self-peptides than negative selection. J Exp Med. 2010;207:1223–1234. doi: 10.1084/jem.20092170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersh GJ, Kersh EN, Fremont DH, Allen PM. High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. Immunity. 1998;9:817–826. doi: 10.1016/s1074-7613(00)80647-0. [DOI] [PubMed] [Google Scholar]
- Kuhns MS, Davis MM, Garcia KC. Deconstructing the form and function of the TCR/CD3 complex. Immunity. 2006;24:133–139. doi: 10.1016/j.immuni.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Kuhns MS, Girvin AT, Klein LO, Chen R, Jensen KD, Newell EW, Huppa JB, Lillemeier BF, Huse M, Chien YH, et al. Evidence for a functional sidedness to the alphabetaTCR. Proc Natl Acad Sci U S A. 2010;107:5094–5099. doi: 10.1073/pnas.1000925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald WA, Chen Z, Gras S, Archbold JK, Tynan FE, Clements CS, Bharadwaj M, Kjer-Nielsen L, Saunders PM, Wilce MC, et al. T cell allorecognition via molecular mimicry. Immunity. 2009;31:897–908. doi: 10.1016/j.immuni.2009.09.025. [DOI] [PubMed] [Google Scholar]
- Manning TC, Schlueter CJ, Brodnicki TC, Parke EA, Speir JA, Garcia KC, Teyton L, Wilson IA, Kranz DM. Alanine scanning mutagenesis of an αβ T cell receptor: mapping the energy of antigen recognition. Immunity. 1998;8:413–425. doi: 10.1016/s1074-7613(00)80547-6. [DOI] [PubMed] [Google Scholar]
- Marrack P, Scott-Browne JP, Dai S, Gapin L, Kappler JW. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu Rev Immunol. 2008;26:171–203. doi: 10.1146/annurev.immunol.26.021607.090421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today. 1998;19:395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- Mazza C, Auphan-Anezin N, Gregoire C, Guimezanes A, Kellenberger C, Roussel A, Kearney A, van der Merwe PA, Schmitt-Verhulst AM, Malissen B. How much can a T-cell antigen receptor adapt to structurally distinct antigenic peptides? EMBO Journal. 2007;26:1972–1983. doi: 10.1038/sj.emboj.7601605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazza C, Malissen B. What guides MHC-restricted TCR recognition? Semin Immunol. 2007;19:225–235. doi: 10.1016/j.smim.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Minguet S, Swamy M, Alarcon B, Luescher IF, Schamel WW. Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity. 2007;26:43–54. doi: 10.1016/j.immuni.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Newell EW, Ely LK, Kruse AC, Reay PA, Rodriguez SN, Lin AE, Kuhns MS, Garcia KC, Davis MM. Structural Basis of Specificity and Cross-Reactivity in T Cell Receptors Specific for Cytochrome c-I-E. Journal of immunology. 2011;186:5823–5832. doi: 10.4049/jimmunol.1100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi S, Krogsgaard M, Davis MM, Chakraborty AK. Molecular flexibility can influence the stimulatory ability of receptor-ligand interactions at cell-cell junctions. Proc Natl Acad Sci U S A. 2006;103:4416–4421. doi: 10.1073/pnas.0510991103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser JB, Darnault C, Gregoire C, Mosser T, Mazza G, Kearney A, van der Merwe PA, Fontecilla-Camps JC, Housset D, Malissen B. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nature Immunology. 2003;4:241–247. doi: 10.1038/ni891. [DOI] [PubMed] [Google Scholar]
- Rubtsova K, Scott-Browne JP, Crawford F, Dai S, Marrack P, Kappler JW. Many different Vbeta CDR3s can reveal the inherent MHC reactivity of germline-encoded TCR V regions. Proc Natl Acad Sci U S A. 2009;106:7951–7956. doi: 10.1073/pnas.0902728106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annual Review of Immunology. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- Scott-Browne JP, White J, Kappler JW, Gapin L, Marrack P. Germline-encoded amino acids in the alphabeta T-cell receptor control thymic selection. Nature. 2009;458:1043–1046. doi: 10.1038/nature07812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JD, Aggen DH, Chervin AS, Narayanan S, Schmitt TM, Greenberg PD, Kranz DM. Opposite Effects of Endogenous Peptide-MHC Class I on T Cell Activity in the Presence and Absence of CD8. Journal of immunology. 2011;186:5193–5200. doi: 10.4049/jimmunol.1003755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykulev Y, Brunmark A, Tsomides TJ, Kageyama S, Jackson M, Peterson PA, Eisen HN. High-affinity reactions between antigen-specific T-cell receptors and peptides associated with allogeneic and syngeneic major histocompatibility complex class I proteins. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tynan FE, Reid HH, Kjer-Nielsen L, Miles JJ, Wilce MC, Kostenko L, Borg NA, Williamson NA, Beddoe T, Purcell AW, et al. A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat Immunol. 2007;8:268–276. doi: 10.1038/ni1432. [DOI] [PubMed] [Google Scholar]
- Udaka K, Wiesmuller KH, Kienle S, Jung G, Tamamura H, Yamagishi H, Okumura K, Walden P, Suto T, Kawasaki T. An automated prediction of MHC class I-binding peptides based on positional scanning with peptide libraries. Immunogenetics. 2000;51:816–828. doi: 10.1007/s002510000217. [DOI] [PubMed] [Google Scholar]
- van der Merwe PA, Cordoba SP. Late arrival: recruiting coreceptors to the T cell receptor complex. Immunity. 2011;34:1–3. doi: 10.1016/j.immuni.2011.01.001. [DOI] [PubMed] [Google Scholar]
- van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. 2011;11:47–55. doi: 10.1038/nri2887. [DOI] [PubMed] [Google Scholar]
- Van Laethem F, Sarafova SD, Park JH, Tai X, Pobezinsky L, Guinter TI, Adoro S, Adams A, Sharrow SO, Feigenbaum L, Singer A. Deletion of CD4 and CD8 coreceptors permits generation of alphabetaT cells that recognize antigens independently of the MHC. Immunity. 2007;27:735–750. doi: 10.1016/j.immuni.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Stanfield RL. MHC restriction: slip-sliding away. Nat Immunol. 2005;6:434–435. doi: 10.1038/ni0505-434. [DOI] [PubMed] [Google Scholar]
- Wucherpfennig KW, Call MJ, Deng L, Mariuzza R. Structural alterations in peptide-MHC recognition by self-reactive T cell receptors. Curr Opin Immunol. 2009;21:590–595. doi: 10.1016/j.coi.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig KW, Gagnon E, Call MJ, Huseby ES, Call ME. Structural biology of the T-cell receptor: insights into receptor assembly, ligand recognition, and initiation of signaling. Cold Spring Harb Perspect Biol. 2010;2:a005140. doi: 10.1101/cshperspect.a005140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Mariuzza RA. The multiple mechanisms of T cell receptor cross-reactivity. Immunity. 2009;31:849–851. doi: 10.1016/j.immuni.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Yoon ST, Dianzani U, Bottomly K, Janeway CA., Jr Both high and low avidity antibodies to the T cell receptor can have agonist or antagonist activity. Immunity. 1994;1:563–569. doi: 10.1016/1074-7613(94)90046-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.