

Abstract
Rationale
In rodents, infarct size following ischemia/reperfusion (I/R) exhibits a circadian dependence on the time of coronary occlusion. It is not known if a similar circadian dependence of infarct size occurs in humans.
Objective
To determine if humans exhibit a circadian dependence of infarct size in the setting of ST-elevation myocardial infarction (STEMI).
Methods and Results
A retrospective analysis of 1031 patients with STEMI referred for primary PCI with known ischemic times between 1 and 6 hours identified 165 patients with occluded arteries on presentation without evidence of pre-infarction angina or collateral blood flow. Both ischemic duration and angiographic area-at-risk were not dependent on time of infarct onset. We observed that the extent of infarct size measured by CK release was significantly associated with time of day onset of infarction (p<0.0001). The greatest myocardial injury occurred at a 1 AM onset of ischemia and 5AM onset of reperfusion with the peak CK measured at the peak of the curve being 82% higher than that recorded at the trough. Similarly, left-ventricular ejection fraction (LVEF) measured within 2 days of infarction was also dependent on time of onset of STEMI with the absolute LVEF at peak more than 7% higher than at trough (43% vs. 51%), (p< 0.03). These findings were supported by a subgroup of patients (n=45) who underwent cardiac MRI measurements of infarct size and area-at-risk measurements.
Conclusions
The results of this study demonstrate for the first time in humans that myocardial infarct size and left-ventricular function following STEMI have a circadian dependence on the time of day onset of ischemia.
Keywords: circadian, infarct size, ischemia/reperfusion, STEMI
INTRODUCTION
Circadian rhythms exert a profound influence on cardiovascular physiology (1). Heart rate, blood pressure, and many circulating hormones such as aldosterone and cortisol (2) follow a circadian pattern. In a similar manner, adverse cardiovascular events also demonstrate a time of day dependence with episodes of myocardial infarction (3), sudden cardiac death (4) and stent thrombosis (5) all peaking in the early morning hours. This time of day dependence has mainly been attributed to non-cardiac factors such as changes in sympathetic tone (6) and circadian variation in coagulation factors (7).
Evidence has recently emerged that circadian clocks intrinsic to cardiac cells may contribute to time of day dependence of cardiovascular physiology. Multiple clock proteins appear to be regulated in a time-dependent manner in cardiomyocytes that may have profound effects on myocardial metabolism, function and response to injury (8). In animal models, the disruption of these circadian clocks has been implicated in the pathogenesis of various cardiovascular diseases (9). Recently, Durgan et al. (10) demonstrated that ischemia/reperfusion (I/R) tolerance is dependent on the time-of-day of coronary occlusion. Using a mouse I/R model, they demonstrated a 3.5-fold variation in infarct size that was dependent on the time of day onset of coronary occlusion. They observed that the largest injury occurred at the sleep to wake transition, which translated into reduced LV function when examined by echocardiography one month later. Furthermore, they demonstrated that the time of day variation in infarct size was abolished in a transgenic mouse with an ablated cardiomyocyte circadian clock.
We investigated if the phenomenon described by Durgan et al. (10) occurs in humans by analyzing if the time of day onset at which the heart is subjected to ischemia influences subsequent infarct size and left ventricular function in a cohort of patients with ST-segment elevation myocardial infarction (STEMI).
METHODS
Study Design
A retrospective analysis was performed on all patients who were admitted to The Minneapolis Heart Institute at Abbott Northwestern Hospital from January 2006 through September 2010 as part of the Level 1 Acute MI program. This is a regionalized transfer network for primary percutaneous coronary intervention (PCI) involving 31 community hospitals and emergency departments throughout Minnesota and Western Wisconsin (11). All STEMI patients who present to the referring hospitals are transferred to our institution for primary PCI regardless of time of presentation and no significant difference in pharmacologic therapy or transfer times occur in those patients transferred at night versus daytime (12) (See Supplemental Table I). All patients had well-defined ischemic times and the data was supported by a subgroup with cardiac MRI (cMRI) measurements of infarct size and area-at-risk (AAR). All medical records were reviewed by 2 of the authors blinded to time of presentation to ensure accuracy of the data.
A total of 1031 patients presented with STEMI and ischemic times between 1 and 6 hours. The ischemic time was defined as the time difference between the onset of chest pain to restoration of flow following the first balloon inflation during primary PCI. All patients were pre-medicated with aspirin (325 mg), clopidogrel (600 mg), heparin (60 Units/kg) or bivalrudin (0.75 mg/kg bolus followed by 1.75 mg/kg-hr) and underwent routine PCI of the culprit vessel with the majority of patients receiving drug-eluting stents.
A total of 866 patients were excluded from further analysis based on at least one of the following exclusion criteria (Figure 1): TIMI flow > 0 prior to coronary intervention or collateral filling of the infarct vessel (n=568), presence of pre-infarction angina (n=94), prior CABG or myocardial infarction in the same vascular distribution (n=72), cardiac arrest requiring more than 1 shock or CPR (n=70), peak of cardiac enzymes not recorded (n=44), postconditioning (13) performed during coronary intervention (n=10), vessel could not be opened (n=6), or acute stent thrombosis within 72hrs (n=2). A total of 165 patients (78% male, 59.5±12.7 yrs) with well-defined times of ischemia onset and duration (183 ± 66 mins) were included in the final analysis (Table 1).
Figure 1.
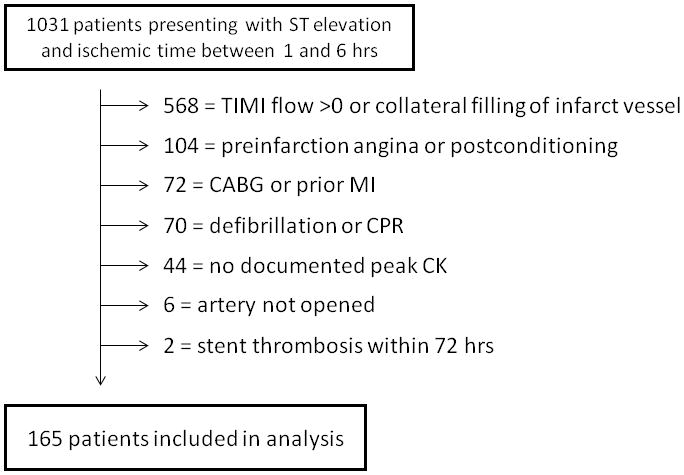
Study Design: Of 1031 patients presenting with ST elevation and an ischemic Time ≤ 6hrs, 866 patients were excluded for a variety of reasons. Patients could have more than 1 reason for exclusion but only 1 per patient is noted in the diagram.
TABLE 1.
Baseline Demographics
| Baseline Demographics (N=165) | |
| Age (years) | 59.5 ± 12.7 |
| % Male | 79 |
| Ischemic Time (mins) | 183 ± 66 |
| HTN (%) | 48 |
| Dyslipidemia (%) | 48 |
| Diabetes (%) | 12 |
| Infarct Artery (n) LAD |
65 |
| CX | 29 |
| RCA | 71 |
Measurement of Infarct Size and Area-at-Risk by Cardiac MRI
A subgroup of 45 patients underwent cardiac MRI (cMRI) measurements of infarct size and AAR within 3 days following STEMI. Infarct size was determined by delayed enhancement with gadolinium. Endocardial and epicardial borders were manually segmented from base to apex and infarct size was determined using the CIMRA software system (CSON Medical, www.csonmedical.com). AAR was estimated as the total amount of subendocardial surface area containing delayed enhancement as a percentage of the total subendocardial surface area (14). Both infarct size and AAR were expressed as percentage of total LV mass.
The peak creatine kinase (CK) level was used as a surrogate of infarct size as previously described (15–17). The validity of this assumption was also confirmed in the subgroup of patients in our cohort that underwent cardiac MRI, demonstrating a highly significant relationship between peak CK and delayed enhancement with gadolinium. Measurements of CK were performed on presentation and every 6 to 8 hrs for 24 to 48 hours following reperfusion with the highest value designated as the peak CK. Measurement of left-ventricular ejection fraction (LVEF) was obtained by echocardiography in 146 of the 165 patients within 48 hours following reperfusion.
Measurement of the Angiographic Area-at Risk
The angiographic AAR was calculated for each patient using the modified Alberta Provincial Project for Outcome Assessment in Coronary Heart Disease (APPROACH) score (18). This method is based on previous pathological studies estimating the percentage of myocardium perfused by each coronary artery. For each patient, an area-at-risk score, expressed as percentage of myocardium, was calculated based on the location of the culprit lesion in the infarct artery (proximal, mid, distal) in relation to major side branches and dominance of the vessel. This method has been previously validated by MRI measurements of AAR (14).
Statistical Analysis
The peak CK and left-ventricular ejection fraction (LVEF) for each patient was plotted against the time onset of ischemia expressed over a 24-hour interval. To determine if ischemic injury in the human heart is dependent on time of onset of infarction (circadian), a periodic sinusoidal regression analysis (19) was performed for both infarct size and LVEF using the online non-linear regression web page (http://statpages.org/nonlin.html) using the following equation:
| (1) |
with “a” indicating the MESOR (Midline Estimating Statistic Of Rhythm), a measure of the rhythm-adjusted mean value, “b” the amplitude and “c” the origin of the curve. A 24-hour period for circadian rhythm was assumed. Circadian rhythm was deemed to be present if the amplitude of the fitted curve was significantly different from 0 (Wald test).
RESULTS
Peak CK as a Correlation to Infarct Size as Measured by cMRI
Multiple studies have previously demonstrated that peak CK is highly correlated with myocardial infarct size assessed by cMRI (15,16) or SPECT (17). As demonstrated in Figure 2, there was excellent correlation between peak CK and infarct size by cMRI that supports our use of peak CK as a surrogate of infarct size for the entire cohort (r= 0.76, p < 0.0001).
Figure 2.

Correlation of peak CK with infarct size as measured by cardiac MRI: Infarct size was measured by cMRI (n=45), expressed as % of total myocardium and plotted against Peak CK. Correlation is highly significant (r=0.76, p<0.0001)
Determination of the Circadian Dependence of Myocardial Infarct Size Following STEMI
A significant association between time of onset of STEMI and myocardial infarct size as assessed by peak CK was demonstrated following nonlinear regression to a sinusoidal curve (Figure 3). Maximal injury was observed at the time of day onset of chest pain at 1AM. Amplitude of the sinusoidal curve was significantly different from 0 (p < 0.0001) with the peak CK of the fitted curve about 82% higher compared to trough suggesting a strong circadian influence. Similar results were obtained when peak troponin was used as a marker for myocardial damage (data not shown). A nearly identical relationship was observed when time of reperfusion was measured instead of onset of ischemia with the curve being shifted to the right by 4 hours (5 AM). Because infarct size is significantly affected by the AAR, we plotted the AAR over the 24-hour cycle and found no variation (Figure 4).
Figure 3.

Time of day dependence of myocardial infarct size with coronary occlusion: Peak CK was plotted against time onset of ischemia. Line represents the fitted sinusoidal curve: CK= 2543+739 × sin (2π x (onset hour-18.72)/24). Amplitude of the fitted curve is significantly different from 0 (p<0.0001) indicating Circadian influence on myocardial infarct size.
Figure 4.

Angiographic Area-at-Risk versus time of onset of ischemia over 24-hour cycle: No significant change in AAR is observed over time as expressed as four, 6-hour intervals
Although intra-hospital transfer times do not vary by time of day in our regionalized system (12), symptom-onset to emergency department presentation times may be delayed at night, potentially leading to longer ischemic times and myocardial injury (20). However, as depicted in Figure 5, ischemic times were relatively evenly distributed throughout the 24-hour day with no circadian relationship (p = 0.35) thereby excluding bias of myocardial injury by different ischemic times.
Figure 5.

Ischemic time (IT) versus time of day onset of myocardial infarction: IT was plotted against time of onset of ischemia. Line represents the fitted sinusoidal curve: IT= 183.36+7.61 × sin (2π x (onset hour-18.47)/24). Amplitude of the fitted curve is not significantly different from 0 (p=0.35) thereby demonstrating no Circadian influence on ischemic time.
Correlation of Angiographic Area-at-Risk to Measurements Obtained by cMRI
In the subgroup of patients that had paired cMRI measurements we confirmed that the angiographic method of the modified APPROACH score (18) was highly correlated with the AAR obtained by cMRI (Figure 6) (r=0.65, p <0.001). We then plotted the Peak CK normalized by the angiographic AAR (Figure 7a) or AAR measured by cMRI (Figure 7b) against the time of onset of ischemia to demonstrate maintenance of a circadian relationship.
Figure 6.

Correlation of area-at-risk assessment (AAR) by MRI with AAR assessment by angiography: AAR measured by MRI and angiography are plotted against each other. Correlation is highly significant (r=0.65, p<0.0001).
Figure 7.


Circadian variation of myocardial infarct size when corrected for area at risk: Peak CK was divided by area at risk measured by angiography (a) and MRI (b). Lines represent the fitted sinusoidal curves. CK/AAR(angio)=99.95+22.16 × sin (2π x (onset hour-18.22)/24) (a) and CK/AAR(MRI)=73.2+16.64 × sin (2π x (onset hour-17.28)/24) (b). Amplitude of the fitted curves are significantly different from 0 (p<0.001(a) and p<0.02(b) confirming Circadian influence on myocardial infarct size when corrected for area at risk.
Assessment of Circadian Dependence of Left-Ventricular Function Following STEMI
LVEF was plotted against time of onset of infarction as a nonlinear (periodic) regression to a sinusoidal curve (Figure 9) in 146 of the 165 patients who had left-ventricular function measurements performed within 48 hours following STEMI. Amplitude (LVEF) of the resulting curve was significantly different from 0 (p < 0.03), demonstrating circadian influence with phase of the peak LVEF correlated with the trough of the peak CK curve (Figure 3). The absolute LVEF at peak was more than 7% higher than at trough (43% vs. 51%).
DISCUSSION
This study details one of the first descriptions in humans of a clear circadian distribution of myocardial infarct size in the setting of STEMI that is dependent on the time of coronary occlusion. The results are bolstered by a similar, but time-shifted circadian pattern of the degree of LV dysfunction that results following myocardial infarction. These findings build upon previous descriptions of circadian cardiovascular influences in humans such as onset of myocardial infarction (3) or sudden cardiac death (4). However, whereas many previous descriptions are likely related to non-cardiac circadian processes such as sympathetic tone (6) and coagulation factors (7), recent animal studies (9,10) demonstrate circadian influences on transcriptional events in cardiomyocytes that may lead to varying degrees of myocardial protection throughout a 24-hour cycle. These pre-clinical observations and our findings may be relevant to clinical trials examining interventions to reduce infarct size since they demonstrate that the time of coronary occlusion will influence the subsequent degree of infacrtion. In agreement with the findings of Muller et al. (1), we did observe that the majority of STEMIs (31%) occurred between 6AM and noon in our Level 1 patient population of more than 3000 subjects.
Many factors may ultimately affect final infarct size following prolonged ischemia. These include the occurrence of pre-infarction angina (21) or presence of collateral blood flow to the dependent myocardial region (22) among others, that may reduce infarct size for a given duration of ischemia and obscure potential circadian effects of myocardial protection based on the time of onset of ischemia. We carefully reviewed each chart and angiogram so that patients with these infarct-sparing mechanisms were excluded. Additionally, all patients had occluded arteries on presentation during their primary PCI so that the ischemic duration could be reasonably assessed.
We observed that the peak myocardial injury occurred at 1 AM when measured by time of symptom onset, and at 5 AM when measured by time of reperfusion. These findings are similar to those of De Luca et al. (23) who measured infarct size by enzyme analysis in 1548 consecutive patients with STEMI referred for primary PCI. By dividing the time of reperfusion into six, 4-hour increments, they observed that the largest infarctions occurred between 4 AM and 8 AM, a time period that was associated with the highest platelet aggregation. Our data also correlates well with the data of Durgan et al. (10) who observed that peak myocardial injury occurred near the sleep to wake transition in the nocturnal rodent following a 45-min coronary occlusion. Our analysis of LV function also reveals that the greatest decrease in LV function occurred in those patients whose infarction began around 1 AM, consistent with previous human data showing a peak incidence of CHF in those patients whose infarct began at nighttime (24).
Studies from human cardiac tissue taken at the time of transplant surgery reveal significant circadian transcriptional variation in the principal cardiomyocyte clock genes, with PER1 and PER2 peaking in the morning and BMAL1 peaking in the evening (8). These variations in clock gene expression are 12 hours out of phase with the nocturnal rodent and are consistent with the time difference in their respective sleep to wake cycles. The results of Durgan et al (10) demonstrated significant diurnal variations in the phosphorylation status of proteins well known to modulate I/R injury including Akt and glycogen synthase kinase-3b (GSK-3b), which influences the opening of the mitochondrial permeability transition pore, the end-effecter of reperfusion injury (25). In their study, the nadir of the phosphorylation status of Akt and GSK-3b occurred at the time-point of greatest myocardial injury and overall phosphorylation status of both proteins were linearly-related to infarct size. In a secondary study, these findings were supported by observations in a circadian clock mutant mouse that is resistant to infarction due to chronic elevations in the phosphorylation status of these two proteins.
In a recent publication, Suarez-Barrientos et al. (26) measured myocardial injury by peak CK or troponin-I in patients admitted to their institution with their first STEMI treated with primary PCI or fibrinolytic therapy. When they divided the presentation of symptom-onset into four, 6-hour blocks, they observed that the largest infarctions occurred when their onset of symptoms began between 6AM and noon with a secondary peak between noon and 6PM. This is several hours later than our observations of peak myocardial injury. However, it is likely that significant differences in methodologies are relevant since no apparent effort was made to exclude patients with modifiers of infarct size such as collateral blood flow or pre-infarction angina, and no measurements of AAR were performed to allow determination if a circadian tolerance to ischemia exists in the human heart.
Study Limitations
Our use of peak CK as a surrogate of infarct size to demonstrate a circadian injury response in the human heart in the setting STEMI warrants comment. The use of peak CK has been validated by multiple groups as an excellent estimate of infarct size (27) using different imaging modalities such as cMRI (15,16) and SPECT (17). Haase et al. (15) prospectively measured infarct size with delayed contrast-enhanced cMRI in 45 patients who underwent primary PCI for STEMI and observed a strong correlation between the absolute size of infarct (grams) and peak CK (r= 0.72, p < 0.001). Similar findings were also reported by Pride et al. (16) in 78 patients with STEMI utilizing cMRI measurements of infarct size (r= 0.93, p < 0.001). In the EVOLVE trial, Chia et al. (17) found that peak CK was highly correlated (r=0.73) with infarct size as determined by SPECT 5 days following primary PCI in 378 patients. Importantly, the subgroup of patients in our cohort with cMRI measurements of infarct size demonstrated excellent correlation with peak CK and had the same circadian relationship as obtained in the overall study cohort.
We have stipulated the onset of ischemia to be the onset of chest pain but recognize that in some patients this may not be the same timepoint, particularly if the patient was asleep when the infarction began. We cannot rule out that this may have produced a small effect on our data since the duration of ischemia would be longer and time-of-onset of ischemia would be shifted to an earlier timepoint. However, we did have access to all emergency room records where the patients initially presented and all subjects were interviewed the following day as part of our Level 1 follow-up data collection program. Additionally, we have assumed that the occluded artery on presentation to the catheterization laboratory was closed throughout the entire duration of ischemia. Because the anticoagulation strategy was not controlled for in this cohort of patients, we cannot rule out that it may have had some small effect on the results of this investigation.
To rule out a possible selection bias of patients as a cause for our findings, we compared the baseline demographic data of patients in the Circadian cohort versus all patients in the Level 1 STEMI program admitted during the same time period (Supplemental Table 1). This analysis showed no important differences between groups except for age (59.5 vs 62.8 yrs), slightly greater Killips class 2–4 (10.4 vs 13.8%) and higher mortality in the overall STEMI population due to our prospective exclusion of cardiac arrest, previous bypass surgery and myocardial infarction in the circadian group. Additionally, no important differences were observed between those patients admitted through our emergency department versus those transferred from outside hospitals (Supplemental Table II).
Conclusions
Our findings demonstrate that humans display a circadian response to myocardial injury and LV function in the setting of STEMI. Maximal injury occurs when infarction begins in early morning hours and reperfusion occurs approximately 4 hours later approaching the sleep to wake transition. These findings imply that there is a time-dependent mechanism of myocardial protection in the human heart that may have implications in clinical trials evaluating therapies to reduce infarct size in the setting of I/R injury. Because this report is from a single center, these results should be confirmed in a larger, prospectively designed, multi-center trial with exclusive use of cMRI to measure infarct size and AAR.
Supplementary Material
Figure 8.

Left-ventricular ejection fraction versus time of day onset of STEMI. Ejection Fraction (EF) was plotted against time onset of ischemia. Line represents the fitted sinusoidal curve: EF=46.83+3.41 × sin (2π x (onset hour-6.20)/24). Amplitude of the fitted curve is significantly different from 0 (p<0.03) indicating circadian influence on ejection fraction following STEMI.
Novelty and Significance.
What is Known?
Circadian rhythms exert a profound influence on cardiovascular physiology including heart rate, blood pressure, and circulating hormones.
Cardiovascular events such as myocardial infarction, sudden cardiac death and stent thrombosis also demonstrate a circadian dependence.
In rodents, infarct size following ischemia/reperfusion is dependent on the time of coronary occlusion, with the largest injury occurring at the sleep to wake transition; however, it is not known if this occurs in humans.
What New Information Does This Article Contribute?
Humans display a circadian rhythm with respect to myocardial infarct size and LV function in the setting of ST-elevation myocardial infarction (STEMI).
Maximal injury occurs when infarction begins in early morning hours (1 AM) and reperfusion occurs approximately 4 hours later, approaching the sleep to wake transition.
Recent studies demonstrate that myocardial infarct size following ischemia/reperfusion in rodents follows a circadian distribution, with the largest infarct size occurring at the sleep to wake transition. To determine if this occurs in humans, we performed a retrospective of analysis of all patients presenting to our institution with their first STEMI for primary percutaneous coronary intervention (PCI) who had well defined ischemic times between 1 and 6 hours. All patients had an occluded artery on presentation, with no evidence of pre-infarction angina or collateral blood flow. Infarct size was measured using peak CK, which was highly correlated with delayed enhancement in a subgroup of patients with cardiac MRI. Angiographic area-at-risk did not vary over the 24-hour period. We observed that infarct size was significantly associated with time of day onset of infarction. The greatest myocardial injury occurred at a 1 AM onset of ischemia and 5AM onset of reperfusion, with the peak CK measured at the peak of the circadian curve being 82% higher than that recorded at the trough. Similarly, the absolute LVEF at peak (measured 12 hours later) was more than 7% higher than at trough (43% vs. 51%). The results of this study demonstrate for the first time in humans that myocardial infarct size and LV function following STEMI have a circadian dependence on the time of day onset of ischemia. These findings may have implications for clinical trials evaluating therapies to reduce infarct size following ischemia/reperfusion.
Acknowledgments
The authors acknowledge Dr. John C. Pezzulo of Georgetown University and Ross Garberich, BS of the Minneapolis Heart Institute Foundation for their assistance with the statistical analysis.
Sources of Funding: Supported by funding from the National Institutes of Health 1-RO1 HL103927 (JHT) and the Jon Holden DeHaan Foundation, Naples, FL
Non-standard Abbreviations and Acronyms
- AAR
Area-at-Risk
- Akt
Protein kinase B
- CHF
Congestive heart failure
- CK
Creatine kinase
- cMRI
cardiac magnetic resonance imaging
- GSK-3b
glycogen synthase kinase-3b
- I/R
ischemia/reperfusion
- LVEF
Left-ventricular ejection fraction
- MESOR
Midline Estimating Statistic Of Rhythm
- PCI
Percutaneous coronary intervention
- SPECT
Single photon emission computer tomography
- STEMI
ST-elevation myocardial infarction
Footnotes
Disclosures: none
References
- 1.Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation. 1989;79:733–743. doi: 10.1161/01.cir.79.4.733. [DOI] [PubMed] [Google Scholar]
- 2.Nomura S, Fujitaka M, Sakura N, Ueda K. Circadian rhythms in plasma cortisone and cortisol and the cortisone/cortisol ratio. Clinica Chimica Acta. 1997;266:83–91. doi: 10.1016/s0009-8981(97)00142-3. [DOI] [PubMed] [Google Scholar]
- 3.Muller JE, Stone PH, Turi ZG, et al. Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med. 1985;313:1315–22. doi: 10.1056/NEJM198511213132103. [DOI] [PubMed] [Google Scholar]
- 4.Muller JE, Ludmer PL, Willich SN, Tofler GH, Aylmer G, Kangos I. Circadian variation in the frequency of sudden cardiac death. Circulation. 1987;75:131–38. doi: 10.1161/01.cir.75.1.131. [DOI] [PubMed] [Google Scholar]
- 5.Mahmoud KD, Lennon RJ, Ting HH, Rihal CS, Holmes DR. Circadian variation in coronary stent thrombosis. J Am Coll Cardiol Intv. 2011;4:183–90. doi: 10.1016/j.jcin.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 6.Panza JA, Epstein SE, Quyyumi AA. Circadian variation in vascular tone and its relation to alpha-sympathetic vasoconstrictor activity. N Engl J Med. 1991;325:986–90. doi: 10.1056/NEJM199110033251402. [DOI] [PubMed] [Google Scholar]
- 7.Tofler GH, Brezinski D, Schafer AI, Czeisler CA, Rutherford JD, Willich SN, Gleason RE, Williams GH, Muller JE. Concurrent morning increase in platelet aggregability and the risk of myocardial infarction and sudden cardiac death. N Engl J Med. 1987;316:1514–8. doi: 10.1056/NEJM198706113162405. [DOI] [PubMed] [Google Scholar]
- 8.Leibetseder V, Humpeler S, Svoboda M, Schmid D, Thalhammer T, Zuckermann A, Marktl W, Ekmekcioglu C. Clock genes display rhythmic expression in human hearts. Chronobiol Int. 2009;26:621–36. doi: 10.1080/07420520902924939. [DOI] [PubMed] [Google Scholar]
- 9.Bray MS, Shaw C, Moore M, Garcia R, Zanquetta M, Durgan D, Jeong W, Tsai J, Bugger H, Zhang D, Rohrwasser A, Rennison J, Dyck J, Litwin S, Hardin P, Chow C, Chandler M, Abel E, Young ME. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol. 2008;294:H1036–H1047. doi: 10.1152/ajpheart.01291.2007. [DOI] [PubMed] [Google Scholar]
- 10.Durgan DJ, Pulinikunnil T, Villegas-Montoya C, Garvey ME, Frangogiannis NG, Michael LH, Chow C-W, Dyck JRB, Young ME. Ischemia/Reperfusion tolerance is time-of-day-dependent. Circ Res. 2010;106:546–550. doi: 10.1161/CIRCRESAHA.109.209346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henry TD, Sharkey SW, Burke N, Chavez IJ, Graham KJ, Henry CR, Lips DL, Madison JD, Menssen KM, Mooney MR, Newell MC, Pedersen WR, Poulose AK, Traverse JH, Unger BT, Wang YL, Larson DM. A regional system to provide timely access to percutaneous coronary intervention for ST-elevation myocardial infarction. Circulation. 2007;116:721–728. doi: 10.1161/CIRCULATIONAHA.107.694141. [DOI] [PubMed] [Google Scholar]
- 12.Miedema MD, Newell MC, Duval S, Garberich RF, Handran CB, Larson DM, Mulder S, Wang YL, Lips DL, Henry TD. Causes of delay and associated mortality in patients transferred with ST-segment-elevation myocardial infarction. Circulation. 2011 doi: 10.1161/CIRCULATIONAHA.111.033118. (in press) [DOI] [PubMed] [Google Scholar]
- 13.Garcia S, Henry TD, Wang YL, Chavez IJ, Pedersen WR, Lesser JR, Shroff GR, Moore L, Traverse JH. Long-term follow-up of patients undergoing postconditioning during ST-elevation myocardial infarction. J Cardiovasc Transl Res. 2011;4:92–98. doi: 10.1007/s12265-010-9252-0. [DOI] [PubMed] [Google Scholar]
- 14.Ortiz-Perez JT, Meyers SN, Lee DC, Kansai P, Klocke FJ, Holly TA, Davidson CJ, Bonow RO, Wu E. Angiographic estimates of myocardium at risk during acute myocardial infarction: validation study using cardiac magnetic resonance imaging. Eur Heart J. 2007;28:1750–1758. doi: 10.1093/eurheartj/ehm212. [DOI] [PubMed] [Google Scholar]
- 15.Haase J, Bayer R, Hackenbroch M, Stoger H, Hofmann M, Schwarz C-E, Reinmer H, Schwarz F, Ruef J, Sommer T. Relationship between size of myocardial infarctions assessed by contrast-enhanced MRI after primary PCI, biochemical markers, and time to intervention. J Interven Cardiol. 2004;17:367–373. doi: 10.1111/j.1540-8183.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- 16.Pride YB, Giuseffi JL, Mohanavelu S, Harrigan CJ, Manning WJ, Gibson CM, Appelbaum E. Relation between infarct size in ST-segment elevation myocardial infarction treated successfully by percutaneous coronary intervention and left ventricular ejection fraction three months after the infarct. Am J Cardiol. 2010;106:635–640. doi: 10.1016/j.amjcard.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 17.Chia S, Senatore F, Raffel OC, Lee H, Wackers FJT, Jang I-K. Utility of cardiac biomarkers in predicting infarct size, left ventricular function, and clinical outcome after primary percutaneous coonary intervention for ST-segment elevation myocardial infarction. J Am Coll Cardiol Intv. 2008;1:415–423. doi: 10.1016/j.jcin.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Graham MM, Faris PD, Ghali WA, Galbraith PD, Norris CM, Badry JT, Mitchell LB, Curtis MJ, Knudtson ML. Validation of three myocardial jeopardy scores in a population-based cardiac catheterization cohort. Am Heart J. 2001;142:254–261. doi: 10.1067/mhj.2001.116481. [DOI] [PubMed] [Google Scholar]
- 19.Bell KNI. Analyzing cycles in biology and medicine-a practical introduction to circular variables and periodic regression. Razorbill Press; St John’s, Newfoundland: 2008. [Google Scholar]
- 20.Holmes DR, Aguirre FV, Aplin R, Lennon RJ, Nestler DM, Bell MR, Rihal CS, Ting HH. Circadian rhythms in patients with ST-elevation myocardial infarction. Circ Cardiovasc Qual Outcomes. 2010;3:382–389. doi: 10.1161/CIRCOUTCOMES.109.913343. [DOI] [PubMed] [Google Scholar]
- 21.Kloner RA, Shook T, Antman EM, Cannon CP, Przyklenk K, Yoo K, McCabe CH, Braunwald E. Prospective temporal analysis of the onset of preinfarction angina versus outcome. Ciculation. 1998;97:1042–1045. doi: 10.1161/01.cir.97.11.1042. [DOI] [PubMed] [Google Scholar]
- 22.Elsman P, van’t Hof AWJ, deBoer MJ, Hoorntje JCA, Suryapranata H, Dambrink JHE, Zijlstra F. Role of collateral circulation in the acute phase of ST-segment myocardial infarction treated with primary coronary intervention. Eur Heart J. 2004;25:854–858. doi: 10.1016/j.ehj.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 23.De Luca G, Suryapranata H, Ottervanger JP, van’t Hof AWJ, Hoorntje JCA, Gosselink ATM, Dambrink J-HE, Zijlstra F, de Boer M-J. Circadian variation in myocardial perfusion and mortality in patients with ST-segment elevation myocardial infarction treated by primary angioplasty. Am Heart J. 2005;150:1185–9. doi: 10.1016/j.ahj.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 24.Mukamal KJ, Muller JE, Maclure M, Sherwood JB, Mittleman MA. Increased risk of congestive heart failure among infarctions with nighttime onset. Am Heart J. 2000;140:438–442. doi: 10.1067/mhj.2000.108830. [DOI] [PubMed] [Google Scholar]
- 25.Lim SY, Davidson SM, Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res. 2007;75:530–5. doi: 10.1016/j.cardiores.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suarez-Barrientos A, Lopez-Romero P, Vivas D, Castro-Ferreira F, Nunez-Gil I, Franco E, Ruiz-Mateos B, Garcia-Rubira JC, Fernandez-Ortiz A, Macaya C, Ibanez B. Circadian variations of infarct size in acute myocardial infarction. Heart. 2011;97:970–6. doi: 10.1136/hrt.2010.212621. [DOI] [PubMed] [Google Scholar]
- 27.Turer AT, Mahaffey KW, Gallup D, Weaver WD, Christenson RH, Every NR, Ohman EM. Enzyme estimates of infarct size correlate with functional and clinical outcomes in the setting of ST-segment elevation myocardial infarction. Curr Control Trials Cardiovasc Med. 2005;6:12. doi: 10.1186/1468-6708-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.