

Abstract
New drugs are urgently needed for the treatment of tropical and subtropical parasitic diseases, such as African sleeping sickness, Chagas' disease, leishmaniasis and malaria. Enzymes in polyamine biosynthesis and thiol metabolism, as well as polyamine transporters, are potential drug targets within these organisms. In the present review, the current knowledge of unique properties of polyamine metabolism in these parasites is outlined. These properties include prozyme regulation of AdoMetDC (S-adenosylmethionine decarboxylase) activity in trypanosomatids, co-expression of ODC (ornithine decarboxylase) and AdoMetDC activities in a single protein in plasmodia, and formation of trypanothione, a unique compound linking polyamine and thiol metabolism in trypanosomatids. Particularly interesting features within polyamine metabolism in these parasites are highlighted for their potential in selective therapeutic strategies.
Keywords: α-difluoromethylornithine, Leishmania, malaria, ornithine decarboxylase (ODC), polyamine, S-adenosylmethionine decarboxylase (AdoMetDC), spermidine synthase, Trypanosoma
Abbreviations: AdoDATO, S-adenosyl-1,8-diamino-3-thiooctane; AdoMao, S-(5′-deoxy-5′-adenosyl)-1-aminoxy-4-(methylsulfonio)-2-cyclopentene; AdoMet, S-adenosylmethionine; AdoMetDC, AdoMet decarboxylase; APA, 1-amino-oxy-3-aminopropane; CNS, central nervous system; CHA, cyclohexylamine; DAB, 1,4-diamino-2-butanone; dcAdoMet, decarboxylated AdoMet; DFMO, α-difluoromethylornithine; LDC, lysine decarboxylase; 4MCHA, 4-methylcyclohexylamine; MDL73811, 5′-{[(Z)-4-amino-2-butenyl]methylamino}-5′-deoxyadenosine; MGBG, methylglyoxal bis(guanylhydrazone); MTA, 5′-methylthioadenosine; OAT, ornithine aminotransferase; ODC, ornithine decarboxylase; PAT, putative amino acid transporter; PLP, pyridoxal-5′-phosphate; SpdS, spermidine synthase; SpmS, spermine synthase; TryR, trypanothione reductase; TryS, trypanothione synthetase; TS2, trypanothione disulfide; TSA, TryS-amidase; T(SH)2, dihydrotrypanothione; WHO, World Health Organization
INTRODUCTION
The polyamines (putrescine, spermidine and spermine) occur in increased concentrations in rapidly proliferating cells including cancerous cells [1–4] and parasitic organisms [5]. Moreover, their biosynthetic enzymes also present with increased activities in these cells. The polyamines and their derivatives mediate a variety of important functions and are therefore essential to cell growth and proliferation. Interference with polyamine biosynthesis has been successfully applied to the clinical treatment of West African sleeping sickness caused by Trypanosoma brucei gambiense and has prompted the notion that polyamine homoeostasis may also be an important drug target in the other major protozoan parasites of human importance. These include the trypanosomatids Trypanosoma cruzi (causing Chagas' disease) and Leishmania spp. (causing leishmaniasis), as well as the apicomplexan Plasmodium spp. causing malaria. These diseases afflict people globally, killing millions and disabling many more, and impose further threats due to the rapid appearance and spread of parasite strains (and associated insect vectors) that are becoming increasingly resistant to current drugs and insecticides. Even though distinct differences exist between them, the polyamine biosynthetic enzymes have been identified as possible drug targets in all of these parasites [5]. In the present review, recent discoveries in the metabolism of polyamines and their thiol derivatives are discussed, particularly highlighting the unique properties of T. brucei, T. cruzi, Leishmania spp. and Plasmodium spp. Moreover, the exploitation of these properties in the discovery of novel polyamine-related therapeutics is considered.
POLYAMINE HOMOEOSTASIS IN THE HUMAN HOST
ODC (ornithine decarboxylase) is a PLP (pyridoxal-5′-phosphate)-dependent enzyme that catalyses the conversion of L-ornithine into putrescine (1,4-diaminobutane), the first step in polyamine biosynthesis [6] (Figure 1). AdoMetDC (S-adenosylmethionine decarboxylase) catalyses the formation of dcAdoMet (decarboxylated AdoMet), which donates the aminopropyl group for the conversion of putrescine into spermidine, and subsequently of spermidine into spermine [7,8]. The latter reactions are catalysed by SpdS (spermidine synthase) and SpmS (spermine synthase) respectively [9]. In addition to the ability of mammalian cells to synthesize polyamines, they can also interconvert polyamines [10]; however, the latter possibility does not exist in any of the parasites included in the present review.
Figure 1. Polyamine biosynthetic pathways and polyamine transporters of trypanosomatid and apicomplexan parasites compared with those of their human host.

The parasites as well as their vectors are depicted. T. brucei is transmitted by tsetse flies and T. cruzi is transmitted by kissing bugs. These trypanosomatids cause sleeping sickness and Chagas' disease respectively. Leishmania spp. are transmitted by sandflies, whereas malaria-causing Plasmodium parasites are transmitted by Anopheles mosquitoes. Cell membrane transporters (cylinders) are sized approximately in proportion to their efficiencies. All images are in the public domain with photo images credited to the CDC Public Health Image Library (http://phil.cdc.gov/phil/) and the human body image to Wikimedia commons (http://commons.wikimedia.org/). Cad, cadaverine; homoT(SH)2, homotrypanothione; On, ornithine; Put, putrescine; ROS, reactive oxygen species; Spd, spermidine; Spm, spermine.
Human cells are not only capable of synthesizing and interconverting polyamines, but can also take up polyamines from their environment [11]. However, physiological fluids are not a rich source of polyamines and most cells (except for the mucosal cells of the digestive tract) are therefore likely to depend on endogenous synthesis. The mechanism of mammalian polyamine transport remains poorly understood, and no polyamine transporter has as yet been molecularly characterized.
The polyamine biosynthetic enzymes of the human host have been well characterized on a biochemical and structural level. Human ODC is an obligate homodimer as two shared active sites are formed at the dimer interface between the N-terminal domain of one monomeric subunit and the C-terminal domain of the other [12–14]. The PLP cofactor is bound in a Schiff-base linkage to the Lys69 residue in the N-terminal domain. In the C-terminal domain of the second subunit, the Cys360 residue plays an essential role in ensuring correct protonation of an intermediate in the decarboxylation reaction (Figure 2A).
Figure 2. Structural descriptions of the polyamine biosynthetic enzymes.

(A) Crystal structure of homodimeric human ODC (PDB code 1D7K, grey) superimposed with T. brucei ODC (PDB code 1QU4, monomers shown in orange and yellow). PLP molecules within the active sites are shown in green. An active site (magnified) of T. brucei ODC containing DFMO–PLP (PDB code 2TOD, pink) and putrescine (PDB code 1F3T, grey) is shown. The Cys360 and Lys69 residues from the DFMO-bound T. brucei enzyme (yellow) and the human apo crystal structure (PDB code 2OO0, orange) are shown. (B) Crystal structure of homodimeric human AdoMetDC (PDB code 1JEN, yellow and orange for the α- and β-subunits respectively) superimposed with the homology model of monomeric P. falciparum AdoMetDC (pink and grey for the α- and β-subunits respectively). Putrescine within the charged buried site is shown in green. An active site (magnified) of P. falciparum AdoMetDC containing putrescine from the human structure (PDB code 1I7M, grey) and the amino acid residues (numbered) that are predicted to mimic putrescine binding in the parasite (yellow) and the human residues that have been shown to interact with putrescine (orange). (C) Crystal structure of homodimeric human SpdS (PDB code 2O06, yellow) superimposed with the crystal structures of T. cruzi SpdS (PDB code 3BWC, grey) and P. falciparum SpdS (PDB code 2I7C, pink). MTA and putrescine within the active site are shown in green. The gate-keeping loops of human SpdS and P. falciparum SpdS are shown in blue in the closed positions. The active site (magnified) of SpdS from P. falciparum containing AdoDATO (grey), MTA (pink) and putrescine (orange) is shown.
Human AdoMetDC is synthesized as a pro-enzyme, which undergoes an autocatalytic internal serinolysis reaction [7,8]. This reaction generates the two subunits (α and β), as well as the active site pyruvoyl group, which is involved in the decarboxylation process. Human AdoMetDC is a homodimer with two individual active sites and two putrescine-binding sites (Figure 2B).
Both of the human aminopropyltransferases SpdS and SpmS exist as dimers of two identical subunits. Neither of the enzymes requires a cofactor for catalytic activity. SpdS and SpmS are highly specific for putrescine and spermidine respectively, as the amine acceptor for an aminopropyl group from dcAdoMet [15,16]. The active sites of the two enzymes are similar, but differ in the sizes of their amine substrate-binding pockets [15,16]. In SpdS (Figure 2C) [15], as well as in SpmS [16], a flexible gate-keeping loop keeps the substrate (putrescine or spermidine) in the correct position for subsequent catalysis and opens to release the product (Figure 2C).
AFRICAN SLEEPING SICKNESS
African sleeping sickness (African trypanosomiasis) is a fatal disease caused by infection with two of three subspecies of T. brucei. The disease is endemic in sub-Saharan Africa in areas populated by the tsetse fly (genus Glossina), which transmit the parasites with their bite. It is estimated that 70000–80000 people are infected and that approximately 30000 deaths from the disease occur annually [17]. The disease progresses from a haemolymphatic stage to a meningoencephalitic stage. More than 90% of the reported cases of African sleeping sickness are caused by T. b. gambiense [18]. This subspecies is responsible for the more chronic disease, occurring in the central and western regions, whereas T. b. rhodesiense causes the more acute disease and is encountered in the eastern and southern regions of Africa. A third subspecies, T. b. brucei, and several other species (Trypanosoma congolense, Trypanosoma vivax, Trypanosoma suis and Trypanosoma simiae) cause nagana, a wasting disease among domestic animals. T. b. brucei is non-pathogenic to humans, and serves as a model organism for drug discovery.
Despite the death toll and the heavy burden of illness, there is an acute lack of satisfactory drugs against African sleeping sickness since all of the drugs in use suffer from serious limitations. These include a lack of selectivity against the parasite, excessive host toxicity, emerging drug resistance, requirement for parenteral administration, limited availability and high cost of treatment.
Polyamine metabolism as a drug target in T. brucei
Putrescine and spermidine are synthesized in T. brucei parasites by ODC, AdoMetDC and SpdS (Figure 1). However, SpmS is missing in T. brucei as well as in the other parasites dealt with in the present review. T. brucei and other trypanosomatids are capable of synthesizing a unique conjugate between GSH and spermidine called trypanothione (N1,N8-bisglutathionylspermidine) (see below). Aspects of polyamine biosynthesis in T. brucei that are exploitable in possible therapeutic strategies are discussed below.
Inhibition of ODC in T. brucei
A large number of ODC inhibitors have been developed and characterized. DFMO (α-difluoromethylornithine or eflornithine), which functions as an enzyme-activated irreversible inhibitor [19], proved to be very effective against T. b. brucei [20] and T. b. gambiense [21]. Merrell Dow in fact filed its New Drug Application for DFMO (called Ornidyl™) in 1988, noting only one indication: T. b. gambiense. Eventually, DFMO has become the first-line drug against late-stage disease, i.e. when the parasites have invaded the CNS (central nervous system). It has been nicknamed ‘the resurrection drug’ because of its striking effects on comatose patients [22].
The effectiveness of DFMO against African sleeping sickness is probably attributable to the following facts: (i) the parasite ODC remains irreversibly inhibited due to its slow turnover rate [23], whereas the inactivated human host ODC is rapidly replaced by newly synthesized active enzyme; (ii) the extracellular parasite cannot obtain polyamines from the blood of the host because of its negligible polyamine uptake capability [24]; (iii) in the absence of spermidine, the parasites cannot make trypanothione, and are therefore seriously compromised in their antioxidant capability (see below); (iv) the polyamine-depleted parasites exhibit a marked decrease in protein synthesis, in particular the variant surface glycoprotein coat [25], which is essential for evasion of the host immune response; (v) AdoMet and dcAdoMet accumulate dramatically in the parasites and may cause aberrant methylation [26]; (vi) the polyamine-depleted parasites differentiate irreversibly into short stumpy forms, which are incapable of binary fission and have lost their infectivity for the vertebrate host [27]; and (vii) since these parasites spend their entire life-cycle extracellularly they are more directly exposed to DFMO than are parasites that replicate intracellularly (e.g. T. cruzi, Leishmania spp. and Plasmodium spp.).
Crystal structures have been solved for the native T. brucei ODC, as well as for the enzyme in complex with PLP, DFMO or putrescine [28,29] (Figure 2A). T. brucei ODC, like human ODC, is an obligate homodimer with two identical active sites formed at the dimer interface by interactions between the Cys360 residue in the C-terminal domain of one subunit and the Lys69 residue in the N-terminal domain of the other subunit [14,30]. The contacts at the dimer interface are formed primarily by the C-terminal domains, which interact through six aromatic rings that form a stacking interaction across the domain boundary [28]. In the absence of the substrate, the PLP cofactor forms a Schiff base with the Lys69 residue in the boundary between the two ODC subunits. Similarly to the corresponding Cys360 residue in the human enzyme [31], the conserved Cys360 residue in T. brucei ODC plays an essential role in the formation of a covalent adduct with DFMO. Within the dimer interface, DFMO forms a Schiff base with PLP on one of the monomers and a covalent bond to the Cys360 residue in the other monomer. The binding of DFMO induces a 145 ° rotation of the Cys360 residue toward the active site, compared with the native T. brucei ODC structure (Figure 2A) [28]. This mechanism of action explains the success of DFMO as a suicide inhibitor of ODC, causing polyamine depletion and subsequent inhibition of cell growth and proliferation.
Besides its remarkable success against T. brucei, DFMO unfortunately has poor pharmacokinetic characteristics. Recent clinical trials, using DFMO in combination with the trypanocidal agent nifurtimox, demonstrated that the course of DFMO treatment could be shortened and simplified (with infusion every 12 h for 7 days, instead of every 6 h for 14 days) without compromising the antiparasitic effect of DFMO [32]. This combination therapy is presently considered suitable for first-line use in the treatment of second-stage West African sleeping sickness.
The active sites of the human and T. brucei ODCs are very similar, which suggests that rational design of inhibitors selectively targeting the enzyme of the parasite may prove challenging. However, the prolonged half-life of T. brucei ODC compared with that of the human counterpart is obviously a beneficial characteristic for targeting this protein in the treatment of African sleeping sickness. Four novel inhibitor classes that are selective and specific for T. brucei ODC have recently been identified in a high-throughput screen [33]. The most active compounds, dithioamidines, were shown to be able to inhibit ODC activity by binding to allosteric sites and thereby controlling enzyme activity.
Inhibition of AdoMetDC in T. brucei
Trypanosomatid genomes are unique in that they contain two types of AdoMetDC genes: (i) an orthologue to the gene that encodes a functional AdoMetDC in other species; and (ii) a paralogous gene, encoding a protein called prozyme, that is catalytically dead but allosterically active [34]. By forming a high-affinity heterodimer with AdoMetDC, prozyme can up-regulate the enzyme activity 1200-fold. It has been suggested that the prozyme has evolved through duplication of the ancestral AdoMetDC gene and subsequent mutational drift, losing catalytic activity in the process, but gaining allosteric regulatory function. Prozyme shares only approximately a 30% amino acid sequence identity with the catalytically active T. brucei AdoMetDC. In addition, it is missing several critical residues required for processing and catalytic activity. In this respect, AdoMetDC from T. brucei and other trypanosomatids represent a novel structural subclass of eukaryotic AdoMetDCs, that otherwise exist as monomers (e.g. plant AdoMetDC) or homodimers (e.g. mammalian AdoMetDC) [8].
Although not a substrate of AdoMetDC, putrescine was found to bind and strongly activate the homodimeric mammalian AdoMetDC, but not the monomeric plant enzyme [8]. There is positive co-operativity in putrescine and substrate (AdoMet) binding to the homodimeric form of human AdoMetDC [35]. Interestingly, putrescine was also found to stimulate the activity of homodimeric T. brucei AdoMetDC, but only had a negligible effect on the heterodimeric complex with prozyme [34]. It is conceivable that the allosteric activation of T. brucei AdoMetDC by prozyme and the co-operativity seen in the human enzyme may somehow be related [8].
The discovery of prozyme opens up new prospects for inhibiting AdoMetDC in trypanosomatids, either by blocking formation of the active AdoMetDC–prozyme complex or by stabilizing the inactive conformation of AdoMetDC. Inhibitors developed against the heterodimeric active site may block the AdoMetDC activity more effectively than inhibitors of the homodimeric active site [34]. It is anticipated that the crystal structures of the heterodimeric and the homodimeric forms of T. brucei AdoMetDC will provide valuable information for structure-based drug design, as well as for understanding of the evolution of the AdoMetDC gene.
AdoMetDC has been chemically validated as a drug target in T. brucei. MDL73811 (5′-{[(Z)-4-amino-2-butenyl]methylamino}-5′-deoxyadenosine), a structural analogue of dcAdoMet and an enzyme-activated irreversible inhibitor of AdoMetDC, was shown to be at least 100-fold more potent than DFMO against murine T. b. brucei infections [36]. MDL73811 treatment could even cure mice infected with a multidrug-resistant strain [36], as well as clinical isolates of T. b. rhodesiense [37]. In combination, DFMO and MDL73811 acted synergistically, curing T. b. rhodesiense-infected mice, thus permitting lower doses to be used [37].
The AdoMet analogue AdoMao [S-(5′-deoxy-5′-adenosyl)-1-aminoxy-4-(methylsulfonio)-2-cyclopentene] inhibits AdoMetDC irreversibly [38]. Of several diastereomeric forms synthesized, trans-1S,4S-AdoMao proved to be the most promising in all respects. It inhibited in vitro growth of T. b. brucei bloodforms with an IC50 of 0.9 μM, and was also effective against two clinical isolates of T. b. rhodesiense. Interestingly, growth inhibition was configuration-dependent, indicating that the unique trypanosomal AdoMet transporter [39] and/or the trypanosomal AdoMetDC prefer substrates in the S-configuration. Because of its minimal effects on human cells, trans-1S,4S-AdoMao may be considered parasite-specific. It is possible that the T. brucei parasites exhibit a facilitated uptake of AdoMet and its analogues, whereas only small quantities of these enter mammalian cells [39].
Lead optimization around MDL73811 resulted in an analogue (Genz-644131) that was 5-fold more potent than MDL73811 against the T. b. brucei AdoMetDC–prozyme complex [40]. This analogue exhibited a longer half-life than MDL73811 as well as improved potency and CNS penetration in T. b. brucei-infected mice. The mechanism by which AdoMetDC inhibitors kill the parasites is believed to be associated not only with depletion of spermidine and related thiol derivatives [41], but with the accumulation of AdoMet, which may cause aberrant methylation [42].
Inhibition of SpdS in T. brucei
Several SpdS inhibitors have been tested against T. brucei in vitro. These include a nucleoside bisubstrate (transition state) analogue, namely, AdoDATO (S-adenosyl-1,8-diamino-3-thiooctane) [43], a relatively large compound (423.5 Da) that fills both the dcAdoMet- and putrescine-binding sites [15]. Two other SpdS inhibitors, CHA (cyclohexylamine) and dicyclohexylamine, compete with putrescine binding [43], but off-target effects have also been suggested. Administration of dicyclohexylamine, the most potent of the SpdS inhibitors, to mice with trypanosomiasis did not increase their survival time. Nevertheless, RNA interference-mediated silencing of the SpdS gene in bloodstream form T. brucei led to depletion of polyamine pools and thiol derivatives, which resulted in cell growth cessation and ultimately cell death [44,45]. Thus SpdS may be considered a genetically validated drug target in African trypanosomes, but chemical validation will have to await the development of inhibitors that selectively and irreversibly inactivate the enzyme.
Polyamine-dependent redox metabolism as a drug target in T. brucei
Trypanosomatids have a unique thiol-based redox metabolism, depending on trypanothione as their principal thiol, in contrast with their human host, which utilizes GSH [46]. Trypanothione provides reducing equivalents to a variety of essential reactions in the parasites through oxidation of the dithiol form of trypanothione [T(SH)2 (dihydrotrypanothione/reduced trypanothionine)] into the disulfide form [TS2 (trypanothione disulfide/oxidized trypanothione)]. This is followed by regeneration of T(SH)2 by TryR (trypanothione reductase), an NADPH-dependent flavoenzyme (Figure 3). The transfer of the reducing equivalents from trypanothione is mediated by tryparedoxins, which are small highly abundant proteins with oxidoreductase activity. A major function of the trypanosomatid redox system is to overcome the oxidative stress induced by the host defence mechanisms. The hydroperoxides produced by the host are detoxified to their respective alcohols by a family of tryparedoxin-dependent peroxidases (Figure 3). For a comprehensive review of thiol redox homoeostasis see Krauth-Siegel and Comini [47].
Figure 3. Central role of trypanothione in detoxification of hydroperoxides.

Hydroperoxides (ROOH) produced by host defence mechanisms are detoxified in trypanosomatids to their respective alcohol (ROH) by tryparedoxin-dependent peroxidases (TXNPx). The reducing equivalents for the reaction are originally derived from NADPH through a thiol redox chain composed of TryR, T[SH]2/TS2 and tryparedoxin (TXN[SH]2/TXNS2), [SH]2 and S2 refer to the dithiol [reduced] and disulfide [oxidized] forms respectively. T[SH]2 is synthesized by a stepwise conjugation of two molecules of GSH to a spermidine (Spd) molecule, catalysed by TryS. Insert: chemical structure of T[SH]2 (the spermidine moiety is highlighted in red). An animated version of Figure 3 is available at http://www.BiochemJ.org/bj/438/0229/bj4380229add.htm.
Trypanothione is synthesized from GSH and spermidine by an ATP-dependent monomeric C–N ligase [TryS (trypanothione synthetase)], in a two-step reaction with glutathionylspermidine as an intermediate (Figures 3 and 4). In addition to a TryS domain, the enzyme contains an amidase domain, which catalyses the hydrolysis of T(SH)2 into GSH and spermidine [48]. This bifunctional TSA (TryS-amidase) is a key enzyme in trypanothione homoeostasis, catalysing both synthesis and degradation of T(SH)2 (Figure 4). Experiments with a tetracycline-dependent conditional double-knockout of TSA in T. brucei, revealed a marked reduction of T(SH)2 together with an accumulation of GSH, when the parasites were grown in vitro without tetracycline [48]. These effects were followed by inhibition of growth and eventually cell death. In the absence of tetracycline, the parasites were unable to infect mice, further validating TSA as a drug target in T. brucei. If the amidase activity of TSA was knocked out alone, the inhibitory effects on parasite growth in vitro and virulence in vivo were less pronounced, indicating that the TryS function is a better drug target than the amidase function. TryS has also been chemically validated as a drug target [49].
Figure 4. Simplified model of TSA structure and function.

The L. major enzyme has two catalytic domains, a C-terminal TryS domain (orange) and an N-terminal amidase domain (grey) and catalyses four reactions [96]. The TryS active site is a triangular-shaped cavity that accommodates each of the three substrates ATP (brown), GSH (pink) and spermidine (Spd) (blue). Reaction I: the γ-phosphate of ATP, the glycine carboxylate of GSH and the N1-amine group of spermidine are positioned in the ortho-centre of the TryS active site. The GSH carboxy group is activated by phosphorylation and the resulting anionic intermediate is subjected to a nucleophilic attack by the N1-amine. Consequently, N1-glutathionylspermidine (Spd–GSH) is formed, and ADP and Pi are released. Reaction II: new ATP and GSH molecules occupy the vacated pockets, and Spd–GSH is positioned with its N8-amine group in the catalytic centre and with the peptide chain directed away from the active site. The subsequent reactions are the same as in Reaction I, producing ADP and Pi, but T[SH]2 instead of Spd–GSH. Reaction III: in the amidase active site, a thioester is formed with T[SH]2, followed by hydrolytic cleavage and release of Spd–GSH and GSH. Reaction IV: a thioester is formed with Spd–GSH, followed by hydrolytic cleavage and release of GSH and Spd.
In addition, TryR has been genetically validated as a drug target in T. b. brucei [50]. Trypanosomes lacking TryR are unable to infect mice, and show increased sensitivity to oxidative stress [50]. In cell cultures, TryR activity has to be reduced by >90% to considerably reduce the growth rate. Therefore any drug designed to target this enzyme would have to be exceptionally effective to be pharmaceutically relevant. Studies of small-molecule inhibitors of T. brucei TryR identified several competitive inhibitors, the most promising being clomipramine (Ki=3 μM), a CNS-active drug [51]. However, this compound failed to extend survival time in an acute mouse model of African trypanosomiasis. Since reversible TryR inhibitors may be displaced by accumulation of TS2, occurring as a consequence of further cellular metabolism of T(SH)2 (Figure 3), irreversible active-site-directed TryR inhibitors may be required in a clinical setting. Another major challenge with druggability of the TryR target is the large size of its active site, which must accommodate the substrates, TS2 (721 Da) or glutathionylspermidine disulfide (867 Da) [51]. It has been estimated that, for an orally bioavailable drug, the molecular mass should ideally be under 500 Da [52]. Nevertheless, the trypanothione pathway and the linked redox pathways (Figure 3) appear to be essential for T. brucei function, which makes the enzymes in these pathways suitable drug targets [47,53].
Polyamine transport as a drug target in T. brucei
T. brucei exhibits very low rates of putrescine uptake and no evidence of any polyamine transporter has been found in the genome [24]. The parasite is therefore unlikely to circumvent the effect of DFMO by obtaining putrescine from the host. On the other hand, T. brucei transfectants expressing LmPOT1, a high-affinity transporter for both putrescine and spermidine derived from L. major (see below), were markedly stimulated in their putrescine uptake [24], indicating that there is a major difference between these two organisms in their capacity to use extracellular putrescine.
CHAGAS' DISEASE
Chagas' disease (American trypanosomiasis) is a devastating and potentially life-threatening illness caused by infection with T. cruzi. It is endemic and widespread in Latin America (19 countries), with 8–11 million chronically infected individuals mainly residing in rural areas with abject poverty. It is estimated that 100 million people are at risk and that 14000 deaths occur annually [17]. Recently, the disease has spread to other continents because of population mobility and failure to cure the chronic disease. The parasite is most often transmitted to humans by the infected faeces of ‘kissing bugs’ (mainly by species belonging to the Triatoma, Rhodnius and Panstrongylus genera). The bugs hide in cracked walls of adobe houses and come out at night to suck blood. Reservoirs of parasites are present both in wild and domestic animals. An individual can also become infected with T. cruzi through contaminated food, blood transfusion, organ transplantation, vertical transmission (passed from an infected mother to her child during pregnancy or birth) or accidental injection.
Benznidazole and nifurtimox are the only drugs available against Chagas' disease. Although almost 100% effective if given soon after infection, these drugs cause adverse reactions in up to 40% of treated patients, and are ineffective against the chronic form of the disease. Therefore there is a pressing need for new and better drugs. One problem to consider when designing new drugs against T. cruzi, is that these parasites circulate in the blood in high numbers only during the initial acute phase. During the chronic phase, which is thus far incurable, the parasites reside intracellularly, mainly in the heart muscle and in the smooth muscle of the digestive tract.
Polyamine metabolism as a drug target in T. cruzi
The polyamine requirement of T. cruzi is met in a strikingly different way than in T. brucei. The former trypanosomatid lacks ODC, the first enzyme in polyamine biosynthesis, and is therefore utterly dependent on uptake of putrescine from the host (Figure 1). Although T. cruzi does not possess a complete polyamine biosynthetic pathway, the parasite does express both AdoMetDC [54,55] and SpdS, implying that once putrescine is transported into the parasite, it can be converted into spermidine (and spermine) [56]. Although other trypanosomatids do not contain spermine, it appears as if T. cruzi SpdS may be somewhat promiscuous like the Thermotoga maritima SpdS [15] and able to convert some spermidine into spermine [56].
Inhibition of AdoMetDC in T. cruzi
The auxotrophic nature of T. cruzi for putrescine makes the polyamine pathway an especially attractive drug target. Obviously the lack of ODC in T. cruzi excludes the possibility of using any of the ODC inhibitors, but the AdoMetDC inhibitor MDL73811 has been shown to decrease the capability of T. cruzi to infect and multiply in rat heart myoblasts [57]. Since the inhibition of infectivity was not bypassed by the addition of spermidine or spermine, it is more likely that the adverse effect of MDL73811 on infectivity is due to the intracellular accumulation of AdoMet than to polyamine depletion.
As in T. brucei, AdoMetDC is allosterically activated by prozyme (a catalytically dead homologue), but the T. cruzi AdoMetDC–prozyme heterodimer also requires putrescine for activation to reach a similar efficiency as that of the fully activated T. brucei heterodimeric complex [58]. It is interesting to note that heterodimers formed between prozyme from T. brucei and AdoMetDC from T. cruzi, and prozyme from T. cruzi and AdoMetDC from T. brucei, are functional. It has been suggested that the stimulation of the T. cruzi AdoMetDC–prozyme heterodimer by putrescine, may represent a regulatory mechanism related to the lack of ODC in T. cruzi and its need for putrescine import from the host cell [58]. Human blood, in which T. brucei parasites reside, is a poor source of putrescine, whereas the cells in which T. cruzi parasites reside are rich in putrescine. The fact that the AdoMetDC–prozyme heterodimer interaction is unique to the trypanosomatids offers a species-selective mechanism that can be targeted in development of new types of drugs against these parasites.
Another group of compounds, MGBG [methylglyoxal bis(guanylhydrazone)] and a series of its aromatic derivatives, are much less potent inhibitors of T. cruzi AdoMetDC than of the human enzyme [54]. Although one such derivative (CGP40215A) had an IC50 of 4.5 nM for T. b. rhodesiense, it was inactive against T. cruzi amastigotes in murine macrophages in vitro [59]. Surprisingly, the treatment of T. cruzi AdoMetDC with another MGBG derivative and substrate analogue (CGP48664A) at low concentrations and in the absence of putrescine resulted in an unexpected increase in enzyme activity [60]. These results suggested that the analogue can bind to the putrescine-binding site and that, in the presence of putrescine or at high analogue concentrations, binding is shifted to that of the active site. This feature is a specific characteristic of T. cruzi AdoMetDC and provides important avenues for drug design where activity can be inhibited by targeting both the putrescine- and substrate-binding sites.
Inhibition of SpdS in T. cruzi
In addition to AdoMetDC, SpdS may provide an essential targeting point in T. cruzi polyamine metabolism. However, very limited information is available regarding the specific inhibition of the T. cruzi SpdS. Therefore structure-based inhibitor discovery may provide novel inhibitors to target SpdS and this may have particular application to the inhibition of T. cruzi. The previously published crystal structures of T. cruzi SpdS could provide important insights into inhibitor identification, as the co-inhibition of AdoMetDC and SpdS would have severe consequences on the availability of polyamines for these parasites. To date structures of the apo form of SpdS as well as the protein with bound AdoMet have been solved (Structural Genomics of Pathogenic Protozoa Consortium, http://www.sgpp.org/description.shtml) [60a]. The structures are highly similar to that of the mammalian enzyme but, for both of these structures, the gate-keeping loops were not defined, which could be due to the absence of bound dcAdoMet. The relatively large size of the SpdS active site [10–15 Å (1 Å=0.1 nm), Figure 2C] could again prove problematic for the design of inhibitors that conform to Lipinski's rule [52]. Innovative strategies therefore need to be considered in order to effectively inhibit the enzyme, and at the same time achieve high selectivity.
Inhibition of polyamine function in T. cruzi with polyamine analogues
In addition to biosynthesis inhibition through the targeting of specific enzyme active sites, polyamine analogues have been investigated in various organisms to perturb polyamine homoeostasis. Ample evidence is available to substantiate that counteraction of polyamine function by these analogues is a rational approach for anticancer drug development [2]. Although the mechanisms of action of polyamine analogues differ between human cells and parasites, some analogues clearly exhibit antiparasitic effects. Ideally the analogue should (i) use a polyamine transporter to gain entry into the parasite, thus competing with the natural polyamines for uptake; (ii) down-regulate the polyamine biosynthetic enzymes; (iii) reduce the pools of the natural polyamines; (iv) counteract the natural polyamines in their infection- and growth-related functions; and (v) exhibit parasite-selective activity (reviewed in [2,61,62]).
Among the first compounds that were synthesized to meet several of these criteria were the N,N′-bis(benzyl)polyamine analogues [63]. Two of these (MDL27695 and MDL27699) [64] and two N,N′-bis(ethylthiophene)polyamine analogues (MDL28302 and MDL29431) [65] were found to inhibit host cell invasion and intracellular amastigote multiplication of T. cruzi. These compounds are all analogues of spermine, suggesting that they may interfere with the intracellular binding and thus the normal functions of spermine and the other polyamines. In fact MDL27695 binds to DNA and may compete for spermine- or spermidine-binding sites on DNA [63].
Polyamine-dependent redox metabolism as a drug target in T. cruzi
T. cruzi, like other trypanosomatids, has a bifunctional TSA with separate active sites for the synthesis of T(SH)2 and for the amidase reaction, hydrolysing T(SH)2 into GSH and spermidine (Figure 4) [66,67]. However, T. cruzi is uniquely capable of converting the alternative polyamine cadaverine (1,5-diaminopentane) into aminopropylcadaverine, bis(aminopropyl)cadaverine and N1,N9-bis(glutathionyl)aminopropylcadaverine (homotrypanothione) [56]. Thus SpdS and TryS in T. cruzi accept cadaverine and aminopropylcadaverine as substrates, in addition to putrescine and spermidine respectively. A source of cadaverine may be the cadaverine-containing actinomycetes [68], which are known to reside in the gut of the Rhodnius vector [69]. The fact that homotrypanothione disulfide is readily reduced by T. cruzi TryR, with similar kinetic parameters to those of TS2, suggests that it is a physiological substrate. Another novel thiol, N1,N12-bis(glutathionyl)spermine has also been identified in T. cruzi [66], indicating that the T. cruzi TryS site in TSA can use spermine as a substrate. Interestingly, N1,N12-bis(glutathionyl)spermine disulfide is also a physiological substrate of TryR [66]. The crystal structure of T. cruzi TryR has been solved and is nearly identical with the structure of the T. b. brucei enzyme [51].
Polyamine transport as a drug target in T. cruzi
As mentioned above, T. cruzi is a diamine (putrescine/cadaverine) auxotroph. The rate of putrescine uptake is approximately 40 times higher in T. cruzi than in other trypanosomatids [70], and exceeds that of spermidine by about as much. Biochemical characterization of polyamine transport in T. cruzi epimastigotes showed that the transporter exhibits a high specificity for putrescine and cadaverine, but low specificity for spermidine and spermine [71]. The parasite can obtain putrescine, but not cadaverine, from the human host, which lacks this polyamine. However, as mentioned above, a source of cadaverine may be the actinomycetes [68] residing in the gut of the vector [69].
The first cell-surface polyamine transporter to be molecularly identified in eukaryotic cells was a high-affinity putrescine/spermidine transporter (LmPOT1) from L. major [24] (see below). Results from the T. cruzi genome-sequencing project was screened for amino acid/auxin permeases and 60 PAT (putative amino acid transporter) genes clustered in 12 groups (TcPAT1–12) were identified [72]. Further investigation of TcPAT12, the most divergent member in terms of amino acid sequence, revealed 55% amino acid identity with LmPOT1 [73]. Functional expression of TcPAT12 in Xenopus laevis oocytes indicated that it is a high-affinity spermidine transporter (Km=14–26 μM). It also transports putrescine (and L-arginine), but at a 7-fold lower rate than spermidine [73].
Two orthologues (TcPOT1.1 and TcPOT1.2) of LmPOT1 were recently identified within the T. cruzi genome [74]. Both genes are heterozygous alleles located on chromosome 11, and their open reading frames predict polypeptides of 613 and 627 amino acids with a 95% identity. Hydropathy profiles indicate that both proteins have 12 transmembrane domains with intracellular N- and C-termini [74]. One of these orthologues, TcPOT1.2, is the TcPAT12 gene previously characterized by Carrillo et al. [73]. In contrast with the earlier findings, TcPOT1.2 and its paralogue TcPOT1.1 recognize putrescine and cadaverine (but not spermidine or spermine) as ligands [74]. Interestingly, transporter function and localization is regulated by the polyamine content of the extracellular milieu [74]. TcPOT1.1, and to some extent TcPOT1.2, are able to cover the entire cell surface and thus increase the cell's putrescine uptake when putrescine becomes scarce. This suggests that T. cruzi uses a signalling system that can sense putrescine availability. Collectively, the signalling and transport systems seem to enable the parasite to overcome its inability to synthesize polyamines de novo. Given the auxotrophy for putrescine, these systems are very promising targets [74].
DAB (1,4-diamino-2-butanone) is a putrescine analogue that inhibits axenic growth of T. cruzi epimastigotes in a dose-dependent manner, leading to oxidative stress and subsequent mitochondrial damage (Table 1) [75]. Co-incubation with putrescine reversed the inhibitory effects of DAB. Since T. cruzi is dependent on polyamine uptake for survival, it is postulated that DAB competes for or inhibits the uptake of putrescine by the cell-surface putrescine transporter, which consequently leads to depletion of putrescine, spermidine and trypanothione. It is therefore conceivable that the oxidative stress observed is due to lack of trypanothione.
Table 1. Selected inhibitors of ODC, AdoMetDC, SpdS and polyamine functions that have shown efficacy against parasites.
Inhibitory concentrations (IC50,μM) of these drugs against the parasites in vitro are indicated. Values are from references as indicated. The promastigote [(p)] and amastigote [(a)] forms of Leishmania are indicated.
| T. brucei | T. cruzi | L. donovani | P. falciparum | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Inhibitor | IC50 | Reference(s) | IC50 | Reference | IC50 | Reference(s) | IC50 | Reference(s) | |
| ODC inhibitors | |||||||||
| DFMO | 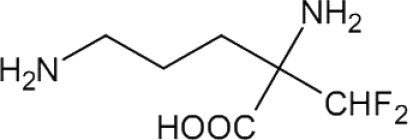 |
81 | [21] | – | – | ~30 (p) | [84] | 1250 | [108,114,115] |
| APA | 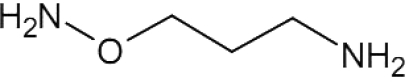 |
– | – | – | – | 42 (p), 5 (a) | [86] | 1 | [108] |
| AdoMetDC inhibitors | |||||||||
| MDL73811 | 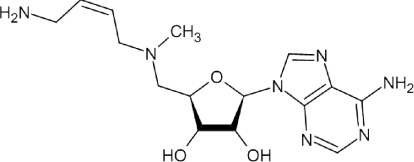 |
– | – | – | – | 40 (p) | [82] | 3 | [108] |
| MGBG |  |
32 | [139,140] | 500 | [54] | 67 (p) | [141] | – | [120] |
| CGP48664A | 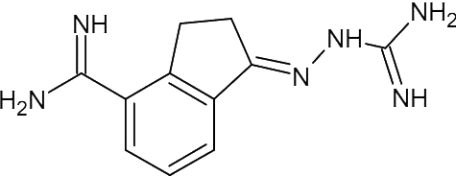 |
0.49 | [59] | ~30 | [59] | >90 (a) | [59] | 8.8 | [108] |
| CGP40215A | 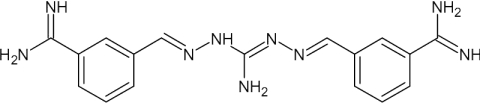 |
0.0045 | [59] | >90 | [59] | 18 (p), >90 (a) | [93] [59] | 1.8 | [108] |
| SpdS inhibitors | |||||||||
| CHA | 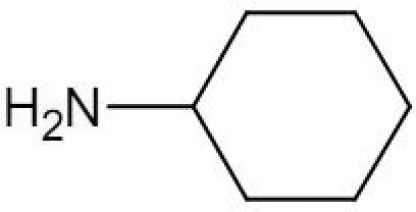 |
– | – | – | – | – | – | 19.7 | [109] |
| 4-MCHA | 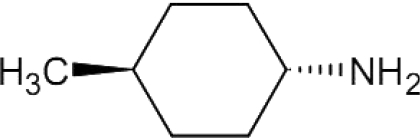 |
– | – | – | – | – | – | 1.4 | [109] |
| Dicyclohexylamine | 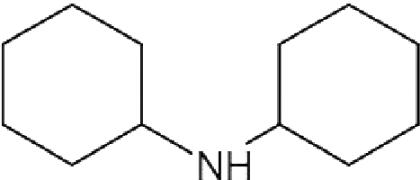 |
– | – | – | – | – | – | >1 000 | [109] |
| Polyamine analogues/transport inhibitors | |||||||||
| MDL27695 | 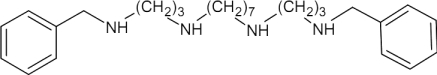 |
– | – | <1 | [64] | 18 (p) | [142] | 3 | [127] |
| DAB | 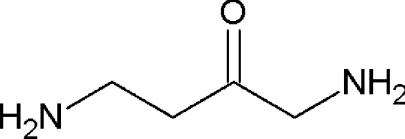 |
– | – | 1180 | [75] | 144 (p) | [103] | – | – |
LEISHMANIASIS
Leishmaniasis is a complex of diseases caused by protozoan parasites of the genus Leishmania [76]. The disease is transmitted through the bite of infected sand flies of the genera Phlebotomus and Lutzomyia, which are the most common vectors in the Old and New World respectively. Leishmaniasis is mainly a zoonotic disease (with reservoirs in rodents and canines). However, in Africa and the Indian subcontinent, visceral leishmaniasis caused by Leishmania donovani is an anthroponotic disease transmitted from human-to-human by sandflies [77].
The main forms of the disease are cutaneous, mucocutaneous and visceral leishmaniasis, with clinical spectra ranging from skin ulcers to liver damage and anaemia. The most severe form is visceral leishmaniasis (also known as kala azar or black fever), in which the parasites cause hepatosplenomegaly, fever, weight loss and anaemia [76]. Visceral leishmaniasis is lethal unless promptly diagnosed and treated. Leishmaniasis is considered endemic in 88 countries with 350 million people at risk [17]. The number of people infected is estimated to be more than 12 million with 1–2 million new cases of the cutaneous form and 0.5 million new cases of the visceral form occurring each year. It is estimated that visceral leishmaniasis causes more than 50000 deaths worldwide annually.
There are approximately 20 species of Leishmania that infect humans. Visceral leishmaniasis is mainly caused by three species of the L. donovani complex (L. donovani, Leishmania infantum and Leishmania chagasi) and is endemic in approximately 70 countries, although the majority of new cases are confined to only relatively few countries (India, Bangladesh, Nepal, Brazil, Kenya, Sudan and Ethiopia) [76]. Leishmania parasites occur in two different forms, extracellular promastigotes (infectious form) in the sandfly vector, and intracellular amastigotes (replicative form) within the phagolysosomes of the human host macrophages.
Current drug treatments of visceral leishmaniasis suffer from various drawbacks such as host toxicity, high cost, difficulty to administer and drug-resistance development. Pentavalent antimonials have been the first-line drugs in the treatment of visceral leishmaniasis for more than 70 years, except in India due to the extensive development of resistance [78–80]. The antifungal drug amphotericin B has become the second-line treatment of visceral leishmaniasis, in spite of its severe and potentially lethal side effects [79,80]. Lipid formulations of amphotericin B with reduced toxicity have been developed and are now the choice of treatment for visceral leishmaniasis in developed countries [79,80]. However, due to high cost, this treatment is not an option in most of the endemic countries. There are drugs such as miltefosine and paromomycin in the pipeline with promising therapeutic effects [80]. Unfortunately, they also suffer from limitations such as high cost, toxicity, difficult administration route, long treatment schedule as well as the generation of drug resistance.
Polyamine metabolism as a drug target in Leishmania spp.
Similarly to T. brucei, Leishmania parasites are able to synthesize putrescine and spermidine since ODC, AdoMetDC and SpdS activities are present in the parasite (Figure 1) and trypanothione is synthesized for redox control.
Systematic experiments by Ullman and colleagues [81–83] genetically validated the polyamine biosynthetic enzymes in L. donovani as drug targets. Knockout strains of L. donovani promastigotes, deficient in ODC, AdoMetDC or SpdS, were all demonstrated to be auxotrophic for polyamines. The ODC-null mutants were dependent on either putrescine or spermidine for their growth in vitro [83], whereas the AdoMetDC- and SpdS-null mutants were shown to be auxotrophic for spermidine only [81,82]. Taken together, these results indicate that spermidine, not putrescine (unless converted into spermidine), is the essential polyamine for growth of L. donovani promastigotes.
Inhibition of ODC in Leishmania
The potential of polyamine biosynthesis as a drug target in Leishmania was indicated by the findings that DFMO effectively inhibited the growth of L. donovani and Leishmania infantum promastigotes in culture [84,85]. In addition, APA (1-amino-oxy-3-aminopropane), a putrescine analogue that inhibits ODC in the nanomolar range, also reduced growth of L. donovani promastigotes (and amastigotes) (Table 1) [86,87]. The growth-inhibitory effect of APA was completely reversed by the addition of putrescine or spermidine. Interestingly, parasites overexpressing ODC showed an increased resistance to the antimonial drug Pentostam, concomitantly with resistance to APA, indicating a role for ODC in the development of Leishmania drug resistance (see below) [86].
Although in vivo treatment with DFMO suppressed the number of parasites in the liver and spleen of mice and hamsters infected with L. infantum and L. donovani, the effect was limited. These results suggested at the time that ODC, and possibly the other enzymes in the polyamine biosynthetic pathway, was not a useful drug target for treatment of visceral leishmaniasis [88,89]. Since the parasites live and replicate within the macrophages of the human host, it seemed conceivable that they might escape polyamine depletion by obtaining polyamines from the host cell [90].
To determine whether ODC deficiency also affects the growth of the intracellular amastigote form, Ullman and colleagues [91] investigated the capacity of ODC-knockout strains of virulent L. donovani to infect mice. The number of parasites in the liver and spleen of mice infected with the ODC-null mutants were found to be 6–3 orders of magnitude lower respectively than the number found in mice infected with the wild-type L. donovani. The infectivity was restored by episomal complementation of the ODC knockouts with an ODC expression plasmid, demonstrating the importance of ODC for growth and survival of L. donovani amastigotes in the mammalian host [91]. Recently, it was also shown that provision of oral putrescine could restore the virulence of the ODC-deficient parasites [92]. Thus despite earlier contrary indications, targeting ODC and the other polyamine biosynthetic enzymes of L. donovani could be a viable approach for treatment of visceral leishmaniasis. Unfortunately, structural information of any of the leishmanial polyamine biosynthetic enzymes is not currently available.
Inhibition of AdoMetDC in Leishmania
Potent inhibitors of AdoMetDC, such as CGP40215A and MDL73811, have been shown to inhibit growth of L. donovani promastigotes (Table 1) [82,93]. The effects of the enzyme inhibitors could be reversed by supplementation of putrescine or spermidine to the growth medium. In addition, it was demonstrated that overexpression of AdoMetDC in L. donovani promastigotes increased their resistance to the growth-inhibitory effect of MDL73811 [82]. However, no effect on the growth of L. donovani amastigotes in mouse macrophages was observed using CGP40215A at concentrations up to 90 μM (Table 1) [59].
Genomic analysis of the trypanosomatid AdoMetDC family indicates that Leishmania spp. also express prozyme [34], although a functional role for prozyme as an activator of AdoMetDC has yet to be demonstrated for this parasite [34,58].
Inhibition of SpdS in Leishmania
Very limited information is available regarding effects of SpdS inhibitors on Leishmania growth and survival. The only SpdS inhibitor tested is n-butylamine, which is as potent as the methylated CHA derivative 4MCHA (trans-4-methylcyclohexylamine), against the mammalian enzyme [94]. When the effect of n-butylamine was tested against L. donovani strains overproducing ODC, AdoMetDC or SpdS, it was shown that not only the SpdS overproducer exhibited resistance to n-butylamine, but also the ODC overproducer [95]. This may suggest that n-butylamine is not a specific inhibitor of SpdS, but may also act on ODC. A more probable explanation is that the increased levels of putrescine found in the ODC overproducers make these cells resistant to the inhibitor, which competes with putrescine for the same binding site of SpdS. This suggestion is also supported by the finding that putrescine supplementation protected wild-type parasites from n-butylamine toxicity [95].
Inhibition of polyamine function in Leishmania with polyamine analogues
The N,N′-bis(benzyl)polyamine analogue MDL27695, was shown to have potent antileishmanial activity, eliminating 77–100% of L. donovani amastigotes from mouse macrophages at a 1 μM concentration [63]. In mice and hamsters, the parasite burden was effectively suppressed by MDL27695, and in hamsters this analogue was equally effective against antimony-resistant L. donovani. The mechanism of action of MDL27695 was thought to involve interference with DNA and RNA synthesis [63].
Several novel types of polyamine analogues, synthesized by Cellgate, have been shown to inhibit L. donovani growth in vitro [95]. The oligoamines CGC-11211 and CGC-11226, and the macrocyclic polyamine analogue CGC-11235, all exhibited IC50 values of less than 1 μM. By screening various antileishmanial compounds against L. donovani strains overproducing ODC, AdoMetDC or SpdS, it was possible to identify those compounds that exert their effect predominantly by interfering with the polyamine biosynthetic enzymes [95].
Polyamine-dependent redox metabolism as a drug target in Leishmania
Structural analysis of L. major TSA revealed two catalytic domains: a C-terminal synthetase domain that generates T(SH)2 and an N-terminal amidase domain that can hydrolyse T(SH)2 [96]. The C-terminal active site of TryS is a triangular-shaped cavity that accommodates each of the three substrates (ATP, GSH and spermidine). The N-terminal amidase active site catalyses the cleavage of T(SH)2 and glutathionylspermidine, releasing GSH and spermidine. The four reactions catalysed by TSA are shown in Figure 4. The TryS and amidase activities of this bifunctional TSA obviously must be balanced to achieve the optimal concentrations of GSH, spermidine, glutathionylspermidine and T(SH)2. By restricting access to the amidase active site, the C-terminus may provide a mechanism by which the levels of these metabolites can be controlled [96]. This indicates an additional target for interference with polyamine and related metabolic processes in this parasite.
The use of pentavalent antimonials as first-line drugs in the treatment of visceral leishmaniasis is threatened by rapid development of drug resistance. The mechanism by which Leishmania spp. develop resistance to antimonials has not yet been fully clarified [97]. It is generally believed that pentavalent antimonials have to be reduced to the trivalent form Sb(III) before being biologically active. The mechanism(s) by which this occurs is still unknown. However, treatment of L. donovani with Sb(III) causes a rapid depletion of intracellular T(SH)2 and GSH as well as inhibition of TryR, resulting in a concomitant accumulation of the disulfide forms. This imbalance in thiol homoeostasis is believed to be lethal to the parasite. Experimentally selected antimonial-resistant strains of Leishmania spp. often show increased levels of T(SH)2 and GSH. These are caused by overexpression of γ-glutamylcysteine synthase and ODC, the rate-limiting enzymes in the biosynthesis of the two precursors to trypanothione, GSH and putrescine [98,99]. It is conceivable that increased synthesis of these thiols renders the cells more resistant to the perturbing effects of Sb(III) on thiol homoeostasis. Overexpression of ODC has been found in antimony-resistant field isolates of L. donovani [86]. Antimony-resistant field isolates have also been shown to exhibit elevated levels of tryparedoxin and tryparedoxin peroxidase, which are involved in hydroperoxide detoxification (Figure 3), strongly indicating that amplified antioxidative defences play a major role in clinical resistance to antimonials [100]. Interestingly, tryparedoxin peroxidase has been genetically validated as a drug target in T. b. brucei [53].
Polyamine transport as a drug target in Leishmania
Leishmania promastigotes as well as amastigotes are able to take up polyamines from their environment [101,102]. In Leishmania spp., the polyamine transport systems are often stage-specific, with different polyamine transport systems for the promastigote and amastigote stages [102]. In promastigotes of L. donovani and Leishmania mexicana and in amastigotes of L. mexicana, polyamine transport was found to be temperature- and pH dependent [102]. It was also saturable and independent of the Na+ concentration, but dependent on the protonmotive force. Results from kinetic analyses of polyamine transport indicate the existence of separate transport systems for putrescine and spermidine in Leishmania. Putrescine transport in L. infantum promastigotes has been shown to occur via a specific energy dependent and saturable carrier [85].
The cloning of the gene LmPOT1 encoding a high-affinity putrescine/spermidine transporter from L. major by Hasne and Ullman [24] was the first molecular identification of a eukaryotic cell-surface polyamine transporter. Sequence analysis predicted a protein of 803 amino acids with 9–12 transmembrane domains and also indicated that LmPOT1 belongs to the APC (amino acid/polyamine/organocation) superfamily of membrane transporters [24]. Expression of LmPOT1 in X. laevis oocytes induced high-affinity saturable transport of both putrescine and spermidine. However, when expressed in T. brucei (which normally transport polyamines poorly) LmPOT1 appeared to mainly transport putrescine with a Km value similar to that obtained in the Xenopus oocytes. Immunocytochemical analysis revealed a high expression of LmPOT1, which was localized to the cell surface and flagellum of L. major promastigotes [24].
DAB has been shown to be active against L. major promastigotes by targeting ODC as well as putrescine uptake into these parasites (Table 1). Similarly to the effect of DFMO treatment in other organisms, pretreatment of the parasites with DAB results in an increased uptake of putrescine [103].
MALARIA
Malaria is recognised by the WHO (World Health Organization) as one of the three most important infectious killers in the world, with around 350–500 million clinical malaria episodes occurring annually [104,105]. More than 80% of malaria-associated deaths occur in Africa south of the Sahara. In endemic African countries, malaria accounts for 25–35% of all outpatient visits, half of all hospitalizations and 35% of all hospital deaths. In some African countries, this disease constitutes 40% of the public health expenditures. Malaria is also prevalent in many other subtropical and tropical countries, e.g. in Southeast Asia and Latin America. This disease therefore rates as one of the major health, socioeconomic and developmental challenges facing many of the world's poorest countries.
Malaria is caused in humans by four major species of plasmodia that are transferred by the bite of a blood-feeding female mosquito of the Anopheles genus. Plasmodium vivax, Plasmodium ovale and Plasmodium malariae are less virulent than Plasmodium falciparum, which causes the most lethal form of this disease. The recent discovery of the zoonotic transfer of Plasmodium knowlesi (simian malaria) to humans indicates the evolving nature of the plasmodia and highlights our need for tight control of the spread of these parasites to humans [106]. Malaria parasites mature intracellularly in hepatocytes before infecting erythrocytes to commence the asexual life cycle. Treatment of severe malaria is negated by the alarming increase in resistance of the malaria parasite to all known antimalarials. Traditionally, malaria prophylaxis relied on antifolates (e.g. pyrimethamine and sulfadoxine) and chloroquine or its derivatives (e.g. mefloquine). Current treatment regimes advocated by the WHO include new antimalarials such as artemisinin in combination with conventional drugs to prolong the useful lifetime of the limited antimalarial dispensary. Ideally an antimalarial should be selective and essentially non-toxic to the human host. It should also have good oral bioavailability and be curative in a few days to limit the development of resistance.
Polyamine metabolism as a drug target in P. falciparum
The polyamines are present as major metabolites within the parasite and, with a total concentration of 10 mM, the polyamines make up 14% of the P. falciparum metabolome [107]. P. falciparum-infected erythrocytes contain high levels of putrescine and spermidine, but only low levels of spermine (Figure 1) [108]. Like in T. cruzi, spermine is synthesized by a somewhat promiscuous P. falciparum SpdS, which cannot only use putrescine, but also spermidine as a substrate (Figure 1) [109]. There is no evidence of trypanothione synthesis in these apicomplexan parasites.
Inhibition of the bifunctional AdoMetDC/ODC of P. falciparum
Possibly the most distinctive feature of polyamine biosynthesis in plasmodia is that a single open reading frame encodes both ODC and AdoMetDC in a unique bifunctional protein of ~330 kDa molecular mass (AdoMetDC/ODC) (Figure 5) [110]. The advantage of this bifunctional AdoMetDC/ODC protein probably is not a metabolic channelling of substrates to optimize downstream synthesis, because AdoMetDC and ODC do not catalyse consecutive reactions in the polyamine biosynthetic pathway. Instead, the bifunctionality may be important for the regulation of polyamine pools in the parasite [111]. In contrast with their single-enzyme orthologues in other organisms, ODC activity is strongly feedback-inhibited by putrescine, and AdoMetDC activity is not stimulated by putrescine in the bifunctional enzyme [112,113]. Protein–protein interactions, as well as long-range communication, are proposed to modulate the activities of ODC and AdoMetDC within the bifunctional complex [111], although each decarboxylase active site in the respective domains is able to function independently of the other [112]. The ODC activity seems to be allosterically stimulated by the presence of the AdoMetDC domain [114] and conversely AdoMetDC activity is inhibited by the presence of the ODC domain [112]. In this manner, it seems that the production of equimolar levels of putrescine and dcAdoMet is achieved.
Figure 5. The unique bifunctional AdoMetDC/ODC of malaria parasites.

(A) AdoMetDC (N-terminus) is associated via a hinge region to the C-terminal ODC domain. Two of these ~170 kDa AdoMetDC/ODC polypeptides associate to form the functional ~330 kDa AdoMetDC/ODC complex. aa, amino acid. (B) Predicted structural description of the bifunctional arrangement of AdoMetDC/ODC (middle panel) [143]. An ODC monomer (grey ribbon diagram in the right-hand panel) associates with another ODC monomer in a head-to-tail fashion to form the two shared active sites of the obligate ODC dimer (right-hand panel). To this, the areas that would be occupied by two AdoMetDC domains are presented as spheres indicating preferred centre of mass of AdoMetDC docked to ODC. Each individual AdoMetDC domain is formed by α- and β-subunits depicted in pink and yellow respectively in the ribbon diagram (left-hand panel).
The potential of AdoMetDC/ODC as an antimalarial target is largely based on the efficacy of DFMO to prevent malaria parasite proliferation in vitro [115]. However, DFMO does not appear to be as therapeutically useful as against African trypanosomiasis, because it is not curative of Plasmodium berghei infections in vivo [116]. Despite the therapeutic failure of DFMO against malaria infections, the dependency of the parasite on polyamines for survival has been well established [108,117—119].
New generation ODC inhibitors including APA are far more effective than DFMO against P. falciparum and exhibit an IC50 value of ~1 μM (Table 1). However, the effects of these inhibitors are also reversed by putrescine [108]. The apparent recovery of parasites treated with ODC inhibitors is troublesome since it implies that the parasite can overcome ODC inhibition through, for example, efficient polyamine uptake mechanisms (see below), and that additional activities in the biosynthesis or uptake of polyamines may need to be targeted.
Inhibition of AdoMetDC activity of the bifunctional protein with MDL73811 (1000-fold more active than DFMO) also arrests P. falciparum growth in vitro [120], but is not curative in in vivo malaria models (Table 1). In contrast with the trypanosomes [37,42], inhibition of AdoMetDC activity in P. falciparum does not result in accumulation of AdoMet levels and the methylation status of the parasite is unaffected [117].
Co-inhibition of both activities of plasmodial AdoMetDC/ODC results in an additive growth-inhibitory effect and a total block in polyamine biosynthesis, indicating possible applications for therapeutic interventions [117]. The decarboxylase active sites of plasmodial AdoMetDC/ODC are structurally conserved compared with the mammalian orthologues, but the bifunctional nature of AdoMetDC/ODC makes it attractive to target protein–protein interacting sites instead, a strategy that is gaining popularity for selective inhibition of multi-subunit proteins [121,122].
A structural description of the complete AdoMetDC/ODC bifunctional complex is lacking, mostly due to the refractory expression of this large protein complex in heterologous systems. However, homology models have been described for the individual AdoMetDC and ODC domains and were used to predict interaction sites between these domains (Figure 5) [113,123]. The P. falciparum AdoMetDC is predicted to fall within its own structural class [35] due to its presence within the bifunctional protein complex (Figure 5). The homology model of ODC showed a similar topology to those of other ODCs, but also identified important differences in the residues involved in its active site. These differences may be exploited for selective inhibitor design [113,123]. Moreover, the ODC domain is more refractory to change and is dependent on the presence of the so-called linker or hinge region (Figure 5) of the bifunctional complex for activity [111,114].
Inhibition of SpdS in P. falciparum
Investigations of the potential of SpdS as a drug target have not been as extensive as for AdoMetDC/ODC, but global analyses of the functional consequence of its inhibition suggest that it is an important flux-controlling point within the polyamine biosynthetic pathway [119]. Treatment with the SpdS inhibitors CHA, 4MCHA and dicyclohexylamine (Table 1) resulted in a marked reduction in P. falciparum cell growth in vitro [109].
Crystal structures of P. falciparum SpdS containing MTA (5′-methylthioadenosine), spermidine, spermine, AdoDATO, dcAdoMet or 4MCHA have been obtained [124]. These structures explained, among other phenomena, the active-site promiscuity in terms of spermidine binding, enabling the enzyme to synthesize spermine [124,125]. Moreover, these structures have been used in the further development of novel and potent inhibitors of SpdS, identifying several active-site binders with the use of structure-based virtual screening [126]. Clearly, the essential nature of SpdS to the survival of P. falciparum parasites warrants further investigation into the design of new inhibitors.
Inhibition of polyamine function in P. falciparum with polyamine analogues
The treatment of murine malaria (P. berghei) with a combination of DFMO and the N,N′-bis(benzyl)polyamine analogue MDL27695 is curative of the disease. Additionally, these cured mice were resistant to re-infection with the same parasite strain [127]. MDL27695 rapidly inhibits growth of both chloroquine-sensitive and -resistant P. falciparum strains in vitro (Table 1) [127]. A combination of MDL27695 and chloroquine proved to cause an additive effect [128]. This therefore provides evidence of the therapeutic potential of polyamine analogues in perturbing P. falciparum growth.
Several of a series of 1,3,5-triazine-substituted polyamines have shown growth-inhibitory activity against P. falciparum in the low micromolar range, especially against chloroquine-resistant parasites [129]. Chemical library screening of diamine derivatives identified compounds with some activity against plasmodia with IC50 values as low as 0.7 μM, provided these derivatives contained a benzyloxy group [130]. There was no indication of selectivity, but antiparasitic activity was confirmed in Leishmania.
Polyamine-related metabolic pathways as drug targets in P. falciparum
Ornithine, a substrate of ODC, seems to be homoeostatically controlled in P. falciparum. Inhibition of the ODC activity of AdoMetDC/ODC increases production of OAT (ornithine aminotransferase) to catabolize potentially toxic ornithine to glutamate-5′-semialdehyde and ultimately to proline [117]. The three-dimensional structure of P. falciparum OAT was recently solved and it was shown that, even though the overall structure is similar to that of the human protein, there are parasite-specific differences that can be exploited for chemotherapeutic intervention. Specifically, two cysteine residues within the loop involved in substrate binding were found only in the parasite OAT, and were predicted to mediate a unique interaction with thioredoxin [131]. Thioredoxin plays a crucial role in redox regulation in P. falciparum.
The control of ornithine levels links polyamine metabolism to redox control in the parasite. Although there is no evidence for the existence of trypanothione in P. falciparum, a clear relationship between polyamine metabolism and the redox status of the parasites is implied since the transcript levels of 1-Cys peroxiredoxin, thioredoxin, glutathione transferase and glutathione reductase are decreased in polyamine-depleted parasites [117].
In the absence of polyamines, P. falciparum produces an inducible LDC1 (lysine decarboxylase 1), which is able to form cadaverine from lysine [117,119,132]. In addition, it seems as if P. falciparum possesses an LDC2 paralogue that is constitutively expressed in the cytosol [133]. Although aminopropylcadaverine is detectable in the parasite, the functions of the LDCs in P. falciparum remain unclear.
Polyamine transport as a drug target in P. falciparum
Putrescine transport has been characterized in rhesus monkey erythrocytes infected with P. knowlesi and was shown to be dependent on temperature and competed for by spermidine and spermine [134]. P. falciparum-infected erythrocytes exhibited similar putrescine uptake characteristics as did P. knowlesi-infected erythrocytes [135].
Both putrescine and spermidine are taken up across the plasma membrane of isolated viable P. falciparum parasites via a saturable temperature-dependent process that showed competition between different polyamines, as well as the polyamine precursor ornithine and other basic amino acids (J. Niemand, unpublished work). Inhibition of polyamine biosynthesis in the isolated parasites led to increased total uptake of both putrescine and spermidine and the rate of uptake was independent of extracellular Na+ or K+ concentrations, but increased with increasing extracellular pH. Putrescine and spermidine uptake decreased with membrane depolarization and increased with membrane hyperpolarization. Unlike in L. major and T. cruzi, a polyamine transporter has as yet not been identified molecularly in Plasmodium spp. The putrescine and spermidine uptake systems may provide a mechanism for selective delivery of antimalarials by conjugating them to polyamines [136,137]. This approach may prove to be effective in improving the inhibitory capacity of the antimalarials, as well as to decrease their non-specific side-effects.
CONCLUSIONS AND FUTURE PERSPECTIVES
The inhibition of polyamine biosynthesis by DFMO has demonstrated impressive cure rates of T. b. gambiense infections, validating this pathway as an antiparasitic drug target. However, the poor pharmacokinetic properties of DFMO necessitate the discovery of alternative inhibitors targeting polyamine metabolism. The trypanosomatids as well as malaria parasites have various unique properties that can be exploited in future drug-development efforts. As such, within the trypanosomatids, the synthesis of trypanothione and particularly the bifunctional TSA may be targeted. Targeting the polyamine uptake mechanism to either block polyamine uptake or exploit this transport mechanism to deliver cytotoxic cargo [11,136] may be highly effective against the parasites. Such an approach may be particularly useful in the case of T. cruzi, being a polyamine auxotroph, with a highly efficient putrescine/cadaverine transporter. A unique and selective tool to inhibit AdoMetDC in trypanosomatids may be to prevent its essential interaction with prozyme. Such targeting of protein–protein interactions has led to the development of small-molecule inhibitors of interactions that are currently in clinical trials in other systems [121]. The formation of the bifunctional AdoMetDC/ODC in plasmodia may be similarly interfered with. Furthermore, a global understanding of the importance of the polyamines to these parasites may highlight additional activities that can be targeted in antiparasitic strategies [138].
Online data
ACKNOWLEDGEMENTS:
We thank Gordon Wells for his help in creating Figure 5(B).
FUNDING
This work was supported by a collaborative research exchange grant between the South African National Research Foundation (NRF) and the Swedish International Development Cooperation Agency (SIDA, Swedish Research Links Programme). J.N., M.W., A.I.L. and L.B. are members of the South African Malaria Initiative (http://www.sami.org.za). Support was also obtained from the European Cooperation on Science and Technology [grant number COST-CM0801] (to L.P. and O.H.) and the Royal Physiographic Society in Lund (to O.H.).
References
- 1.Gerner E. W., Meyskens F. L., Jr Polyamines and cancer: old molecules, new understanding. Nat. Rev. Cancer. 2004;4:781–792. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 2.Casero R. A., Marton L. J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discovery. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 3.Pegg A. E. Mammalian polyamine metabolism and function. IUBMB Life. 2009;61:880–894. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace H. M. The polyamines: past, present and future. Essays Biochem. 2009;46:1–9. doi: 10.1042/bse0460001. [DOI] [PubMed] [Google Scholar]
- 5.Heby O., Persson L., Rentala M. Targeting the polyamine biosynthetic enzymes: a promising approach to therapy of African sleeping sickness, Chagas' disease, and leishmaniasis. Amino Acids. 2007;33:359–366. doi: 10.1007/s00726-007-0537-9. [DOI] [PubMed] [Google Scholar]
- 6.Pegg A. E. Regulation of ornithine decarboxylase. J. Biol. Chem. 2006;281:14529–14532. doi: 10.1074/jbc.R500031200. [DOI] [PubMed] [Google Scholar]
- 7.Pegg A. E. S-Adenosylmethionine decarboxylase. Essays Biochem. 2009;46:25–45. doi: 10.1042/bse0460003. [DOI] [PubMed] [Google Scholar]
- 8.Bale S., Ealick S. E. Structural biology of S-adenosylmethionine decarboxylase. Amino Acids. 2010;38:451–460. doi: 10.1007/s00726-009-0404-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikeguchi Y., Bewley M. C., Pegg A. E. Aminopropyltransferases: function, structure and genetics. J. Biochem. 2006;139:1–9. doi: 10.1093/jb/mvj019. [DOI] [PubMed] [Google Scholar]
- 10.Casero R. A., Pegg A. E. Polyamine catabolism and disease. Biochem. J. 2009;421:323–338. doi: 10.1042/BJ20090598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer A. J., Wallace H. M. The polyamine transport system as a target for anticancer drug development. Amino Acids. 2010;38:415–422. doi: 10.1007/s00726-009-0400-2. [DOI] [PubMed] [Google Scholar]
- 12.Almrud J. J., Oliveira M. A., Kern A. D., Grishin N. V., Phillips M. A., Hackert M. L. Crystal structure of human ornithine decarboxylase at 2.1 Å resolution: strucural insights to antizyme binding. J. Mol. Biol. 2000;295:7–16. doi: 10.1006/jmbi.1999.3331. [DOI] [PubMed] [Google Scholar]
- 13.Kern A. D., Oliveira M. A., Coffino P., Hackert M. L. Structure of mammalian ornithine decarboxylase at 1.6 Å resolution: stereochemical implications of PLP-dependent amino acid decarboxylases. Structure. 1999;7:567–581. doi: 10.1016/s0969-2126(99)80073-2. [DOI] [PubMed] [Google Scholar]
- 14.Tobias K. E., Kahana C. Intersubunit location of the active site of mammalian ornithine decarboxylase as determined by hybridization of site-directed mutants. Biochemistry. 1993;32:5842–5847. doi: 10.1021/bi00073a017. [DOI] [PubMed] [Google Scholar]
- 15.Wu H., Min J., Ikeguchi Y., Zeng H., Dong A., Loppnau P., Pegg A. E., Plotnikov A. N. Structure and mechanism of spermidine synthases. Biochemistry. 2007;46:8331–8339. doi: 10.1021/bi602498k. [DOI] [PubMed] [Google Scholar]
- 16.Wu H., Min J., Zeng H., McCloskey D. E., Ikeguchi Y., Loppnau P., Michael A. J., Pegg A. E., Plotnikov A. N. Crystal structure of human spermine synthase: implications of substrate binding and catalytic mechanism. J. Biol. Chem. 2008;283:16135–16146. doi: 10.1074/jbc.M710323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuart K., Brun R., Croft S., Fairlamb A., Gurtler R. E., McKerrow J., Reed S., Tarleton R. Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Invest. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simarro P. P., Jannin J., Cattand P. Eliminating human African trypanosomiasis: where do we stand and what comes next? PLoS Med. 2008;5:e55. doi: 10.1371/journal.pmed.0050055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metcalf B. W., Bey P., Danzin C., Jung M. J., Casara P. L., Vevert J. P. Catalytic irreversible inhibition of mammalian ornithine decarboxylase by substrate and product analogues. J. Am. Chem. Soc. 1978;100:2551–2552. [Google Scholar]
- 20.Bacchi C. J., Nathan H. C., Hutner S. H., McCann P. P., Sjoerdsma A. Polyamine metabolism: a potential therapeutic target in trypanosomes. Science. 1980;210:332–334. doi: 10.1126/science.6775372. [DOI] [PubMed] [Google Scholar]
- 21.Van Nieuwenhove S., Schechter P. J., Declercq J., Bone G., Burke J., Sjoerdsma A. Treatment of gambiense sleeping sickness in the Sudan with oral DFMO (DL-α-difluoromethylornithine), an inhibitor of ornithine decarboxylase: first field trial. Trans. R. Soc. Trop. Med. Hyg. 1985;79:692–698. doi: 10.1016/0035-9203(85)90195-6. [DOI] [PubMed] [Google Scholar]
- 22.Wickware P. Resurrecting the resurrection drug. Nat. Med. 2002;8:908–909. doi: 10.1038/nm0902-908b. [DOI] [PubMed] [Google Scholar]
- 23.Phillips M. A., Coffino P., Wang C. C. Cloning and sequencing of the ornithine decarboxylase gene from Trypanosoma brucei. J. Biol. Chem. 1987;262:8721–8727. [PubMed] [Google Scholar]
- 24.Hasne M. P., Ullman B. Identification and characterization of a polyamine permease from the protozoan parasite Leishmania major. J. Biol. Chem. 2005;280:15188–15194. doi: 10.1074/jbc.M411331200. [DOI] [PubMed] [Google Scholar]
- 25.Bitonti A. J., Cross-Doersen D. E., McCann P. P. Effects of α-difluoromethylornithine on protein synthesis and synthesis of the variant-specific glycoprotein (VSG) in Trypanosoma brucei brucei. Biochem. J. 1988;250:295–298. doi: 10.1042/bj2500295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yarlett N., Bacchi C. J. Effect of DL-α-difluoromethylornithine on methionine cycle intermediates in Trypanosoma brucei brucei. Mol. Biochem. Parasitol. 1988;27:1–10. doi: 10.1016/0166-6851(88)90019-9. [DOI] [PubMed] [Google Scholar]
- 27.Giffin B. F., McCann P. P., Bitonti A. J., Bacchi C. J. Polyamine depletion following exposure to DL-α-difluoromethylornithine both in vivo and in vitro initiates morphological alterations and mitochondrial activation in a monomorphic strain of Trypanosoma brucei brucei. J. Protozool. 1986;33:238–243. doi: 10.1111/j.1550-7408.1986.tb05599.x. [DOI] [PubMed] [Google Scholar]
- 28.Grishin N. V., Osterman A. L., Brooks H. B., Phillips M. A., Goldsmith E. J. X-ray structure of ornithine decarboxylase from Trypanosoma brucei: the native structure and the structure in complex with α-difluoromethylornithine. Biochemistry. 1999;38:15174–15184. doi: 10.1021/bi9915115. [DOI] [PubMed] [Google Scholar]
- 29.Jackson L. K., Brooks H. B., Osterman A. L., Goldsmith E. J., Phillips M. A. Altering the reaction specificity of eukaryotic ornithine decarboxylase. Biochemistry. 2000;39:11247–11257. doi: 10.1021/bi001209s. [DOI] [PubMed] [Google Scholar]
- 30.Osterman A., Grishin N. V., Kinch L. N., Phillips M. A. Formation of functional cross-species heterodimers of ornithine decarboxylase. Biochemistry. 1994;33:13662–13667. doi: 10.1021/bi00250a016. [DOI] [PubMed] [Google Scholar]
- 31.Poulin R., Lu L., Ackermann B., Bey P., Pegg A. E. Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by α-difluoromethylornithine. Characterization of sequences at the inhibitor and coenzyme binding sites. J. Biol. Chem. 1992;267:150–158. [PubMed] [Google Scholar]
- 32.Priotto G., Kasparian S., Mutombo W., Ngouama D., Ghorashian S., Arnold U., Ghabri S., Baudin E., Buard V., Kazadi-Kyanza S., et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet. 2009;374:56–64. doi: 10.1016/S0140-6736(09)61117-X. [DOI] [PubMed] [Google Scholar]
- 33.Smithson D. C., Lee J., Shelat A. A., Phillips M. A., Guy R. K. Discovery of potent and selective inhibitors of Trypanosoma brucei ornithine decarboxylase. J. Biol. Chem. 2010;285:16771–16781. doi: 10.1074/jbc.M109.081588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willert E. K., Fitzpatrick R., Phillips M. A. Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proc. Natl. Acad. Sci. U.S.A. 2007;104:8275–8280. doi: 10.1073/pnas.0701111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bale B., Lopez M. M., Makhatadze G. I., Fang Q., Pegg A. E., Ealick S. E. Structural basis for putrescine activation of human S-adenosylmethionine decarboxylase. Biochemistry. 2008;47:13404–13417. doi: 10.1021/bi801732m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bitonti A. J., Byers T. L., Bush T. L., Casara P. J., Bacchi C. J., Clarkson A. B., Jr, McCann P. P., Sjoerdsma A. Cure of Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense infections in mice with an irreversible inhibitor of S-adenosylmethionine decarboxylase. Antimicrob. Agents Chemother. 1990;34:1485–1490. doi: 10.1128/aac.34.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bacchi C. J., Nathan H. C., Yarlett N., Goldber B., McCann P. P., Bitonti A. J., Sjoerdsma A. Cure of murine Trypanosoma brucei rhodesiense infections with an S-adenosylmethionine decarboxylase inhibitor. Antimicrob. Agents Chemother. 1992;36:2736–2740. doi: 10.1128/aac.36.12.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo J., Wu Y. Q., Rattendi D., Bacchi C. J., Woster P. M. S-(5′-deoxy-5′-adenosyl)-1-aminoxy-4-(methylsulfonio)-2-cyclopentene (AdoMao): an irreversible inhibitor of S-adenosylmethionine decarboxylase with potent in vitro antitrypanosomal activity. J. Med. Chem. 1995;38:1770–1777. doi: 10.1021/jm00010a021. [DOI] [PubMed] [Google Scholar]
- 39.Goldberg B., Yarlett N., Sufrin J., Lloyd D., Bacchi C. J. A unique transporter of S-adenosylmethionine in African trypanosomes. FASEB J. 1997;11:256–260. doi: 10.1096/fasebj.11.4.9068614. [DOI] [PubMed] [Google Scholar]
- 40.Barker R. H., Jr, Liu H., Hirth B., Celatka C. A., Fitzpatrick R., Xiang Y., Willert E. K., Phillips M. A., Kaiser M., Bacchi C. J., et al. Novel S-adenosylmethionine decarboxylase inhibitors for the treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 2009;53:2052–2058. doi: 10.1128/AAC.01674-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willert E. K., Phillips M. A. Regulated expression of an essential allosteric activator of polyamine biosynthesis in African trypanosomes. PLoS Pathog. 2008;4:e1000183. doi: 10.1371/journal.ppat.1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Byers T. L., Bush T., McCann P. P., Bitonti A. J. Antitrypanosomal effects of polyamine biosynthesis inhibitors correlate with increases in Trypanosoma brucei brucei S-adenosyl- l -methionine. Biochem. J. 1991;274:527–533. doi: 10.1042/bj2740527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bitonti A. J., Kelly S. E., McCann P. P. Characterization of spermidine synthase from Trypanosoma brucei brucei. Mol. Biochem. Parasitol. 1984;13:21–28. doi: 10.1016/0166-6851(84)90098-7. [DOI] [PubMed] [Google Scholar]
- 44.Taylor M. C., Kaur H., Blessington B., Kelly J. M., Wilkinson S. R. Validation of spermidine synthase as a drug target in African trypanosomes. Biochem. J. 2008;409:563–569. doi: 10.1042/BJ20071185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao Y., McCloskey D. E., Phillips M. A. RNA interference-mediated silencing of ornithine decarboxylase and spermidine synthase genes in Trypanosoma brucei provides insight into regulation of polyamine biosynthesis. Eukaryot. Cell. 2009;8:747–755. doi: 10.1128/EC.00047-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fairlamb A. H., Cerami A. Metabolism and functions of trypanothione in the Kinetoplastida. Annu. Rev. Microbiol. 1992;46:695–729. doi: 10.1146/annurev.mi.46.100192.003403. [DOI] [PubMed] [Google Scholar]
- 47.Krauth-Siegel R. L., Comini M. A. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim. Biophys. Acta. 2008;1780:1236–1248. doi: 10.1016/j.bbagen.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 48.Wyllie S., Oza S. L., Patterson S., Spinks D., Thompson S., Fairlamb A. H. Dissecting the essentiality of the bifunctional trypanothione synthetase-amidase in Trypanosoma brucei using chemical and genetic methods. Mol. Microbiol. 2009;74:529–540. doi: 10.1111/j.1365-2958.2009.06761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torrie L. S., Wyllie S., Spinks D., Oza S. L., Thompson S., Harrison J. R., Gilbert I. H., Wyatt P. G., Fairlamb A. H., Frearson J. A. Chemical validation of trypanothione synthetase: a potential drug target for human trypanosomiasis. J. Biol. Chem. 2009;284:36137–36145. doi: 10.1074/jbc.M109.045336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krieger S., Schwarz W., Ariyanayagam M. R., Fairlamb A. H., Krauth-Siegel R. L., Clayton C. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol. Microbiol. 2000;35:542–552. doi: 10.1046/j.1365-2958.2000.01721.x. [DOI] [PubMed] [Google Scholar]
- 51.Jones D. C., Ariza A., Chow W. H., Oza S. L., Fairlamb A. H. Comparative structural, kinetic and inhibitor studies of Trypanosoma brucei trypanothione reductase with T. cruzi. Mol. Biochem. Parasitol. 2010;169:12–19. doi: 10.1016/j.molbiopara.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 53.Wilkinson S. R., Horn D., Prathalingam S. R., Kelly J. M. RNA interference identifies two hydroperoxide metabolizing enzymes that are essential to the bloodstream form of the African trypanosome. J. Biol. Chem. 2003;278:31640–31646. doi: 10.1074/jbc.M303035200. [DOI] [PubMed] [Google Scholar]
- 54.Persson K., Åslund L., Grahn B., Hanke J., Heby O. Trypanosoma cruzi has not lost its S-adenosylmethionine decarboxylase: characterization of the gene and the encoded enzyme. Biochem. J. 1998;333:527–537. doi: 10.1042/bj3330527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kinch L. N., Scott J. R., Ullman B., Phillips M. A. Cloning and kinetic characterization of the Trypanosoma cruzi S-adenosylmethionine decarboxylase. Mol. Biochem. Parasitol. 1999;101:1–11. doi: 10.1016/s0166-6851(98)00181-9. [DOI] [PubMed] [Google Scholar]
- 56.Hunter K. J., Le Quesne S. A., Fairlamb A. H. Identification and biosynthesis of N1,N9-bis(glutathionyl)aminopropylcadaverine (homotrypanothione) in Trypanosoma cruzi. Eur. J. Biochem. 1994;226:1019–1027. doi: 10.1111/j.1432-1033.1994.t01-1-01019.x. [DOI] [PubMed] [Google Scholar]
- 57.Yakubu M. A., Majumder S., Kierszenbaum F. Inhibition of S-adenosyl- l -methionine (AdoMet) decarboxylase by the decarboxylated AdoMet analog 5′-([(Z)-4-amino-2-butenyl]methylamino)-5′-deoxyadenosine (MDL73811) decreases the capacities of Trypanosoma cruzi to infect and multiply within a mammalian host cell. J. Parasitol. 1993;79:525–532. [PubMed] [Google Scholar]
- 58.Willert E. K., Phillips M. A. Cross-species activation of trypanosome S-adenosylmethionine decarboxylase by the regulatory subunit prozyme. Mol. Biochem. Parasitol. 2009;168:1–6. doi: 10.1016/j.molbiopara.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brun R., Bühler Y., Sandmeier U., Kaminsky R., Bacchi C. J., Rattendi D., Lane S., Croft S. L., Snowdon D., Yardley V., et al. In vitro trypanocidal activities of new S-adenosylmethionine decarboxylase inhibitors. Antimicrob. Agents Chemother. 1996;40:1442–1447. doi: 10.1128/aac.40.6.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beswick T. C., Willert E. K., Phillips M. A. Mechanisms of allosteric regulation of Trypanosoma cruzi S-adenosylmethionine decarboxylase. Biochemistry. 2006;45:7797—7807. doi: 10.1021/bi0603975. [DOI] [PubMed] [Google Scholar]
- 60a.Fan E., Baker D., Fields S., Gelb M. H., Buckner F. S., Van Voorhis W. C., Phizicky E., Dumont M., Mehlin C., Grayhack E., et al. Structural genomics of pathogenic protozoa: an overview. Methods Mol. Biol. 2008;426:497–513. doi: 10.1007/978-1-60327-058-8_33. [DOI] [PubMed] [Google Scholar]
- 61.Boncher T., Bi X., Varghese S., Casero R. A., Jr, Woster P. M. Polyamine-based analogues as biochemical probes and potential therapeutics. Biochem. Soc. Trans. 2007;35:356–363. doi: 10.1042/BST0350356. [DOI] [PubMed] [Google Scholar]
- 62.Casero R. A., Jr, Woster P. M. Recent advances in the development of polyamine analogues as antitumor agents. J. Med. Chem. 2009;52:4551–4573. doi: 10.1021/jm900187v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baumann R. J., Hanson W. L., McCann P. P., Sjoerdsma A., Bitonti A. J. Suppression of both antimony-susceptible and antimony-resistant Leishmania donovani by a bis(benzyl)polyamine analog. Antimicrob. Agents Chemother. 1990;34:722–727. doi: 10.1128/aac.34.5.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Majumder S., Kierszenbaum F. Inhibition of host cell invasion and intracellular replication of Trypanosoma cruzi by N,N′-bis(benzyl)-substituted polyamine analogs. Antimicrob. Agents Chemother. 1993;37:2235–2238. doi: 10.1128/aac.37.10.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Majumder S., Kierszenbaum F. N,N′-thiophene-substituted polyamine analogs inhibit mammalian host cell invasion and intracellular multiplication of Trypanosoma cruzi. Mol. Biochem. Parasitol. 1993;60:231–239. doi: 10.1016/0166-6851(93)90134-j. [DOI] [PubMed] [Google Scholar]
- 66.Ariyanayagam M. R., Oza S. L., Mehlert A., Fairlamb A. H. Bis(glutathionyl)spermine and other novel trypanothione analogues in Trypanosoma cruzi. J. Biol. Chem. 2003;278:27612–27619. doi: 10.1074/jbc.M302750200. [DOI] [PubMed] [Google Scholar]
- 67.Oza S. L., Tetaud E., Ariyanayagam M. R., Warnon S. S., Fairlamb A. H. A single enzyme catalyses formation of trypanothione from glutathione and spermidine in Trypanosoma cruzi. J. Biol. Chem. 2002;277:35853–35861. doi: 10.1074/jbc.M204403200. [DOI] [PubMed] [Google Scholar]
- 68.Hamana K., Matsuzaki S. Distribution of polyamines in actinomycetes. FEMS Microbiol. Lett. 1987;41:211–215. [Google Scholar]
- 69.Brecher G., Wigglesworth V. B. The transmission of Actinomyces rhodnii Erikson in Rhodnius prolixus stål (hemiptera) and its influence on the growth of the host. Parasitology. 1944;35:220–224. [Google Scholar]
- 70.González N. S., Ceriani C., Algranati I. D. Differential regulation of putrescine uptake in Trypanosoma cruzi and other trypanosomatids. Biochem. Biophys. Res. Commun. 1992;188:120–128. doi: 10.1016/0006-291x(92)92358-5. [DOI] [PubMed] [Google Scholar]
- 71.Le Quesne S. A., Fairlamb A. H. Regulation of a high-affinity diamine transport system in Trypanosoma cruzi epimastigotes. Biochem. J. 1996;316:481–486. doi: 10.1042/bj3160481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bouvier L. A., Silber A. M., Galvao Lopes C., Canepa G. E., Miranda M. R., Tonelli R. R., Colli W., Alves M. J., Pereira C. A. Post genomic analysis of permeases from the amino acid/auxin family in protozoan parasites. Biochem. Biophys. Res. Commun. 2004;321:547–556. doi: 10.1016/j.bbrc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 73.Carrillo C., Canepa G. E., Algranati I. D., Pereira C. A. Molecular and functional characterization of a spermidine transporter (TcPAT12) from Trypanosoma cruzi. Biochem. Biophys. Res. Commun. 2006;344:936–940. doi: 10.1016/j.bbrc.2006.03.215. [DOI] [PubMed] [Google Scholar]
- 74.Hasne M. P., Coppens I., Soysa R., Ullman B. A high-affinity putrescine-cadaverine transporter from Trypanosoma cruzi. Mol. Microbiol. 2010;76:78–91. doi: 10.1111/j.1365-2958.2010.07081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Menezes D., Valentim C., Oliveira M. F., Vannier-Santos M. A. Putrescine analogue cytotoxicity against Trypanosoma cruzi. Parasitol. Res. 2006;98:99–105. doi: 10.1007/s00436-005-0010-1. [DOI] [PubMed] [Google Scholar]
- 76.Chappuis F., Sundar S., Hailu A., Ghalib H., Rijal S., Peeling R. W., Alvar J., Boelaert M. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007;5:873–882. doi: 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- 77.Berman J. Visceral leishmaniasis in the New World & Africa. Indian. J. Med. Res. 2006;123:289–294. [PubMed] [Google Scholar]
- 78.Croft S. L., Sundar S., Fairlamb A. H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006;19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kedzierski L., Sakthianandeswaren A., Curtis J. M., Andrews P. C., Junk P. C., Kedzierska K. Leishmaniasis: current treatment and prospects for new drugs and vaccines. Curr. Med. Chem. 2009;16:599–614. doi: 10.2174/092986709787458489. [DOI] [PubMed] [Google Scholar]
- 80.Moore E. M., Lockwood D. N. Treatment of visceral leishmaniasis. J. Glob. Infect. Dis. 2010;2:151–158. doi: 10.4103/0974-777X.62883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roberts S. C., Jiang Y., Jardim A., Carter N. S., Heby O., Ullman B. Genetic analysis of spermidine synthase from Leishmania donovani. Mol. Biochem. Parasitol. 2001;115:217–226. doi: 10.1016/s0166-6851(01)00293-6. [DOI] [PubMed] [Google Scholar]
- 82.Roberts S. C., Scott J., Gasteier J. E., Jiang Y., Brooks B., Jardim A., Carter N. S., Heby O., Ullman B. S-adenosylmethionine decarboxylase from Leishmania donovani. Molecular, genetic, and biochemical characterization of null mutants and overproducers. J. Biol. Chem. 2002;277:5902–5909. doi: 10.1074/jbc.M110118200. [DOI] [PubMed] [Google Scholar]
- 83.Jiang Y., Roberts S. C., Jardim A., Carter N. S., Shih S., Ariyanayagam M., Fairlamb A. H., Ullman B. Ornithine decarboxylase gene deletion mutants of Leishmania donovani. J. Biol. Chem. 1999;274:3781–3788. doi: 10.1074/jbc.274.6.3781. [DOI] [PubMed] [Google Scholar]
- 84.Kaur K., Emmett K., McCann P. P., Sjoerdsma A., Ullman B. Effects of DL-α-difluoromethylornithine on Leishmania donovani promastigotes. J. Protozool. 1986;33:518–521. doi: 10.1111/j.1550-7408.1986.tb05654.x. [DOI] [PubMed] [Google Scholar]
- 85.Balana-Fouce R., Ordonez D., Alunda J. M. Putrescine transport system in Leishmania infantum promastigotes. Mol. Biochem. Parasitol. 1989;35:43–50. doi: 10.1016/0166-6851(89)90140-0. [DOI] [PubMed] [Google Scholar]
- 86.Singh S., Mukherjee A., Khomutov A. R., Persson L., Heby O., Chatterjee M., Madhubala R. Antileishmanial effect of 3-aminooxy-1-aminopropane is due to polyamine depletion. Antimicrob. Agents Chemother. 2007;51:528–534. doi: 10.1128/AAC.01055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dufe V. T., Ingner D., Heby O., Khomutov A. R., Persson L., Al-Karadaghi S. A structural insight into the inhibition of human and Leishmania donovani ornithine decarboxylases by 1-amino-oxy-3-aminopropane. Biochem. J. 2007;405:261–268. doi: 10.1042/BJ20070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mukhopadhyay R., Madhubala R. Effect of a bis(benzyl)polyamine analogue, and DL-α-difluoromethylornithine on parasite suppression and cellular polyamine levels in golden hamster during Leishmania donovani infection. Pharmacol. Res. 1993;28:359–365. doi: 10.1006/phrs.1993.1138. [DOI] [PubMed] [Google Scholar]
- 89.Gradoni L., Iorio M. A., Gramiccia M., Orsini S. In vivo effect of eflornithine (DFMO) and some related compounds on Leishmania infantum preliminary communication. Farmaco. 1989;44:1157–1166. [PubMed] [Google Scholar]
- 90.Burchmore R. J., Barrett M. P. Life in vacuoles-nutrient acquisition by Leishmania amastigotes. Int. J. Parasitol. 2001;31:1311–1320. doi: 10.1016/s0020-7519(01)00259-4. [DOI] [PubMed] [Google Scholar]
- 91.Boitz J. M., Yates P. A., Kline C., Gaur U., Wilson M. E., Ullman B., Roberts S. C. Leishmania donovani ornithine decarboxylase is indispensable for parasite survival in the mammalian host. Infect. Immun. 2009;77:756–763. doi: 10.1128/IAI.01236-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Olenyik T., Gilroy C., Ullman B. Oral putrescine restores virulence of ornithine decarboxylase-deficient Leishmania donovani in mice. Mol. Biochem. Parasitol. 2011;176:109–111. doi: 10.1016/j.molbiopara.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mukhopadhyay R., Kapoor P., Madhubala R. Antileishmanial effect of a potent S-adenosylmethionine decarboxylase inhibitor: CGP 40215A. Pharmacol. Res. 1996;33:67–70. doi: 10.1006/phrs.1996.0011. [DOI] [PubMed] [Google Scholar]
- 94.Goda H., Watanabe T., Takeda N., Kobayashi M., Wada M., Hosoda H., Shirahata A., Samejima K. Mammalian spermidine synthase-identification of cysteine residues and investigation of the putrescine binding site. Biol. Pharm. Bull. 2004;27:1327–1332. doi: 10.1248/bpb.27.1327. [DOI] [PubMed] [Google Scholar]
- 95.Roberts S. C., Jiang Y., Gasteier J., Frydman B., Marton L. J., Heby O., Ullman B. Leishmania donovani polyamine biosynthetic enzyme overproducers as tools to investigate the mode of action of cytotoxic polyamine analogs. Antimicrob. Agents Chemother. 2007;51:438–445. doi: 10.1128/AAC.01193-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fyfe P. K., Oza S. L., Fairlamb A. H., Hunter W. N. Leishmania trypanothione synthetase-amidase structure reveals a basis for regulation of conflicting synthetic and hydrolytic activities. J. Biol. Chem. 2008;283:17672–17680. doi: 10.1074/jbc.M801850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wyllie S., Cunningham M. L., Fairlamb A. H. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J. Biol. Chem. 2004;279:39925–39932. doi: 10.1074/jbc.M405635200. [DOI] [PubMed] [Google Scholar]
- 98.Haimeur A., Guimond C., Pilote S., Mukhopadhyay R., Rosen B. P., Poulin R., Ouellette M. Elevated levels of polyamines and trypanothione resulting from overexpression of the ornithine decarboxylase gene in arsenite-resistant Leishmania. Mol. Microbiol. 1999;34:726–735. doi: 10.1046/j.1365-2958.1999.01634.x. [DOI] [PubMed] [Google Scholar]
- 99.Grondin K., Haimeur A., Mukhopadhyay R., Rosen B. P., Ouellette M. Co-amplification of the γ-glutamylcysteine synthetase gene gsh1 and of the ABC transporter gene pgpA in arsenite-resistant Leishmania tarentolae. EMBO J. 1997;16:3057–3065. doi: 10.1093/emboj/16.11.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wyllie S., Mandal G., Singh N., Sundar S., Fairlamb A. H., Chatterjee M. Elevated levels of tryparedoxin peroxidase in antimony unresponsive Leishmania donovani field isolates. Mol. Biochem. Parasitol. 2010;173:162–164. doi: 10.1016/j.molbiopara.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kandpal M., Tekwani B. L. Polyamine transport systems of Leishmania donovani promastigotes. Life Sci. 1997;60:1793–1801. doi: 10.1016/s0024-3205(97)00139-2. [DOI] [PubMed] [Google Scholar]
- 102.Basselin M., Coombs G. H., Barrett M. P. Putrescine and spermidine transport in Leishmania. Mol. Biochem. Parasitol. 2000;109:37–46. doi: 10.1016/s0166-6851(00)00234-6. [DOI] [PubMed] [Google Scholar]
- 103.Vannier-Santos M. A., Menezes D., Oliveira M. F., de Mello F. G. The putrescine analogue 1,4-diamino-2-butanone affects polyamine synthesis, transport, ultrastructure and intracellular survival in Leishmania amazonensis. Microbiology. 2008;154:3104–3111. doi: 10.1099/mic.0.2007/013896-0. [DOI] [PubMed] [Google Scholar]
- 104.Breman J. G., Egan A., Keusch G. T. The intolerable burden of malaria: a new look at the numbers. Am. J. Trop. Med. Hyg. 2001;64(Suppl):iv–vii. doi: 10.4269/ajtmh.2001.64.iv. [DOI] [PubMed] [Google Scholar]
- 105.Yamey G. Roll Back Malaria: a failing global health campaign. BMJ. 2004;328:1086–1087. doi: 10.1136/bmj.328.7448.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bronner U., Divis P. C. S., Färnert A., Singh B. Swedish traveller with Plasmodium knowlesi malaria after visiting Malaysian Borneo. Malaria J. 2009;8:15. doi: 10.1186/1475-2875-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Teng R., Junankar P. R., Bubb W. A., Rae C., Mercier P., Kirk K. Metabolite profiling of the intraerythrocytic malaria parasite Plasmodium falciparum by (1)H NMR spectroscopy. NMR Biomed. 2009;22:292–302. doi: 10.1002/nbm.1323. [DOI] [PubMed] [Google Scholar]
- 108.Das Gupta R., Krause-Ihle T., Bergmann B., Müller I. B., Khomutov A., Müller S., Walter R. D., Lüersen K. 3-Aminooxy-1-aminopropane and derivatives have an antiproliferative effect on cultured Plasmodium falciparum by decreasing intracellular polyamine concentrations. Antimicrob. Agents Chemother. 2005;49:2857–2864. doi: 10.1128/AAC.49.7.2857-2864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Haider N., Eschbach M. L., Dias Sde S., Gilberger T. W., Walter R. D., Lüersen K. The spermidine synthase of the malaria parasite Plasmodium falciparum: molecular and biochemical characterisation of the polyamine synthesis enzyme. Mol. Biochem. Parasitol. 2005;142:224–236. doi: 10.1016/j.molbiopara.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 110.Müller S., Da'dara A., Lüersen K., Wrenger C., Das Gupta R., Madhubala R., Walter R. D. In the human malaria parasite Plasmodium falciparum, polyamines are synthesized by a bifunctional ornithine decarboxylase, S-adenosylmethionine decarboxylase. J. Biol. Chem. 2000;275:8097–8102. doi: 10.1074/jbc.275.11.8097. [DOI] [PubMed] [Google Scholar]
- 111.Birkholtz L., Wrenger C., Joubert F., Wells G. A., Walter R. D., Louw A. I. Parasite-specific inserts in the bifunctional S-adenosylmethionine decarboxylase/ornithine decarboxylase of Plasmodium falciparum modulate catalytic activities and domain interactions. Biochem. J. 2004;377:439–448. doi: 10.1042/BJ20030614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wrenger C., Lüersen K., Krause T., Müller S., Walter R. D. The Plasmodium falciparum bifunctional ornithine decarboxylase, S-adenosyl- l -methionine decarboxylase, enables a well balanced polyamine synthesis without domain–domain interaction. J. Biol. Chem. 2001;276:29651–29656. doi: 10.1074/jbc.M100578200. [DOI] [PubMed] [Google Scholar]
- 113.Wells G. A., Birkholtz L., Joubert F., Walter R. D., Louw A. I. Novel properties of malarial S-adenosylmethionine decarboxylase as revealed by structural modelling. J. Mol. Graph. Model. 2006;24:307–318. doi: 10.1016/j.jmgm.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 114.Krause T., Lüersen K., Wrenger C., Gilberger T., Müller S., Walter R. D. The ornithine decarboxylase domain of the bifunctional ornithine decarboxylase/S-adenosylmethionine decarboxylase of Plasmodium falciparum: recombinant expression of two different constructs. Biochem. J. 2000;352:287–292. [PMC free article] [PubMed] [Google Scholar]
- 115.Assaraf Y. G., Golenser J., Spira D. T., Messer G., Bachrach U. Cytostatic effect of DL-α-difluoromethylornithine against Plasmodium falciparum and its reversal by diamines and spermidine. Parasitol. Res. 1987;73:313–318. doi: 10.1007/BF00531084. [DOI] [PubMed] [Google Scholar]
- 116.Bitonti A. J., McCann P. P., Sjoerdsma A. Plasmodium falciparum and Plasmodium berghei: effects of ornithine decarboxylase inhibitors on erythrocytic schizogony. Exp. Parasitol. 1987;64:237–243. doi: 10.1016/0014-4894(87)90148-2. [DOI] [PubMed] [Google Scholar]
- 117.van Brummelen A. C., Olszewski K., Wilinski D., Llinas M., Louw A., Birkholtz L. Co-inhibition of the Plasmodium falciparum S-adenosylmethionine decarboxylase/ornithine decarboxylase reveals perturbation-specific compensatory mechanisms by transcriptome, proteome and metabolome analyses. J. Biol. Chem. 2009;284:4635–4646. doi: 10.1074/jbc.M807085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Clark K., Dhoogra M., Louw A. I., Birkholtz L. M. Transcriptional responses of Plasmodium falciparum to α-difluoromethylornithine-induced polyamine depletion. Biol. Chem. 2008;389:111–125. doi: 10.1515/BC.2008.014. [DOI] [PubMed] [Google Scholar]
- 119.Becker J. V., Mtwisha L., Crampton B. G., Stoychev S., van Brummelen A. C., Reeksting S., Louw A. I., Birkholtz L. M., Mancama D. T. Plasmodium falciparum spermidine synthase inhibition results in unique perturbation-specific effects observed on transcript, protein and metabolite levels. BMC Genomics. 2010;11:235. doi: 10.1186/1471-2164-11-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wright P. S., Byers T. L., Cross-Doersen D. E., McCann P. P., Bitonti A. J. Irreversible inhibition of S-adenosylmethionine decarboxylase in Plasmodium falciparum-infected erythrocytes: growth inhibition in vitro. Biochem. Pharmacol. 1991;41:1713–1718. doi: 10.1016/0006-2952(91)90174-4. [DOI] [PubMed] [Google Scholar]
- 121.Wells J. A., McClendon C. L. Reaching for high-hanging fruit in drug discovery at protein/protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 122.Fuller J. C., Burgoyne N. J., Jackson R. M. Predicting druggable binding sites at the protein/protein interface. Drug Discovery Today. 2009;14:155–161. doi: 10.1016/j.drudis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 123.Birkholtz L., Joubert F., Neitz A. W.H., Louw A. I. Comparative properties of a three-dimensional model of Plasmodium falciparum ornithine decarboxylase. Proteins: Struct. Funct. Genet. 2003;50:464–473. doi: 10.1002/prot.10274. [DOI] [PubMed] [Google Scholar]
- 124.Dufe V. T., Qui W., Müller I. B., Hui R., Walter R. D., Al-Karadaghi S. Crystal structure of Plasmodium falciparum spermidine synthase in complex with the substrate decarboxylated S-adenosylmethionine and the potent inhibitors 4MCHA and AdoDATO. J. Mol. Biol. 2007;373:167–177. doi: 10.1016/j.jmb.2007.07.053. [DOI] [PubMed] [Google Scholar]
- 125.Burger P. B., Birkholtz L., Joubert F., Haider N., Walter R. D., Louw A. I. Structural and mechanistic insights into the action of Plasmodium falciparum spermidine synthase. Bioorg. Med. Chem. 2007;15:1628–1637. doi: 10.1016/j.bmc.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 126.Jacobsson M., Gäredal M., Schultz J., Karlén A. Identification of Plasmodium falciparum spermidine synthase active site binders through structure-based virtual screening. J. Med. Chem. 2008;51:2777–2786. doi: 10.1021/jm7016144. [DOI] [PubMed] [Google Scholar]
- 127.Bitonti A. J., Dumont J. A., Bush T. L., Edwards M. L., Stemerick D. M., McCann P. P., Sjoerdsma A. Bis(benzyl)polyamine analogs inhibit the growth of chloroquine-resistant human malaria parasites (Plasmodium falciparum) in vitro and in combination with α-difluoromethylornithine cure murine malaria. Proc. Natl. Acad. Sci. U.S.A. 1989;86:651–655. doi: 10.1073/pnas.86.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Das B., Gupta R., Madhubala R. Combined action of inhibitors of polyamine biosynthetic pathway with a known antimalarial drug, chloroquine, on Plasmodium falciparum. Pharm. Res. 1995;31:189–193. doi: 10.1016/1043-6618(95)80017-4. [DOI] [PubMed] [Google Scholar]
- 129.Klenke B., Barrett M. P., Brun R., Gilbert I. H. Antiplasmodial activity of a series of 1,3,5-triazine substituted polyamines. J. Antimicrob. Chemother. 2003;52:290–293. doi: 10.1093/jac/dkg307. [DOI] [PubMed] [Google Scholar]
- 130.Labadie G. R., Choi S. R., Avery M. A. Diamine derivatives with antiparasitic activities. Bioorg. Med. Chem. Lett. 2004;14:615–619. doi: 10.1016/j.bmcl.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 131.Jortzik E., Fritz-Wolf K., Sturm N., Hipp M., Rahlfs S., Becker K. Redox regulation of Plasmodium falciparum ornithine δ-aminotransferase. J. Mol. Biol. 2010;402:445–459. doi: 10.1016/j.jmb.2010.07.039. [DOI] [PubMed] [Google Scholar]
- 132.Reeksting S. B. MSc Dissertation. Hatfield, Pretoria, South Africa: University of Pretoria; 2009. Metabolomic analyses of the malaria parasite after inhibition of polyamine biosynthesis. [Google Scholar]
- 133.Müller I. B. M., das Gupta R., Lüersen K. L., Wrenger C., Walter R. D. Assessing the polyamine metabolism of Plasmodium falciparum as chemotherapeutic target. Mol. Biochem. Parasitol. 2008;160:1–7. doi: 10.1016/j.molbiopara.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 134.Singh S., Puri S. K., Singh S. K., Srivastava R., Gupta R. C., Pandey V. C. Characterization of simian malarial parasite (Plasmodium knowlesi)-induced putrescine transport in rhesus monkey erythrocytes. A novel putrescine conjugate arrests in vitro growth of simian malarial parasite (Plasmodium knowlesi) and cures multidrug resistant murine malaria (Plasmodium yoelii) infection in vivo. J. Biol. Chem. 1997;272:13506–13511. doi: 10.1074/jbc.272.21.13506. [DOI] [PubMed] [Google Scholar]
- 135.Ramya T. N., Surolia N., Surolia A. Polyamine synthesis and salvage pathways in the malaria parasite Plasmodium falciparum. Biochem. Biophys. Res. Commun. 2006;348:579–584. doi: 10.1016/j.bbrc.2006.07.127. [DOI] [PubMed] [Google Scholar]
- 136.Palmer A. J., Ghani R. A., Kaur N., Phanstiel O., Wallace H. M. A putrescine-anthracene conjugate: a paradigm for selective drug delivery. Biochem. J. 2009;424:431–438. doi: 10.1042/BJ20090815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chadwick J., Jones M., Mercer A. E., Stocks P. A., Ward S. A., Park B. K., O'Neill P. M. Design, synthesis and antimalarial/anticancer evaluation of spermidine linked artemisinin conjugates designed to exploit polyamine transporters in Plasmodium falciparum and HL-60 cancer cell lines. Bioorg. Med. Chem. 2010;18:2586–2597. doi: 10.1016/j.bmc.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 138.Clark K., Niemand J., Reeksting S., Smit S., van Brummelen A. C., Williams M., Louw A. I., Birkholtz L. Functional consequences of perturbing polyamine metabolism in the malaria parasite, Plasmodium falciparum. Amino Acids. 2010;38:633–644. doi: 10.1007/s00726-009-0424-7. [DOI] [PubMed] [Google Scholar]
- 139.Bitonti A. J., Dumont J. A., McCann P. P. Characterization of Trypanosoma brucei brucei S-adenosyl-L-methionine decarboxylase and its inhibition by Berenil, pentamidine and methylglyoxal bis(guanylhydrazone) Biochem. J. 1986;237:685–689. doi: 10.1042/bj2370685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Da'dara A. A., Mett H., Walter R. D. MGBG analogues as potent inhibitors of S-adenosylmethionine decarboxylase of Onchocerca volvulus. Mol. Biochem. Parasitol. 1998;97:13–19. doi: 10.1016/s0166-6851(98)00124-8. [DOI] [PubMed] [Google Scholar]
- 141.Mukhopadhyay R., Madhubala R. Antileishmanial activity of Berenil and methylglyoxal bis (guanylhydrazone) and its correlation with S-adenosylmethionine decarboxylase and polyamines. Int. J. Biochem. Cell Biol. 1995;27:55–59. doi: 10.1016/1357-2725(95)93432-p. [DOI] [PubMed] [Google Scholar]
- 142.Mukhopadhyay R., Madhubala R. Effects of bis(benzyl)polyamine analogs on Leismania donovani promastigotes. Exp. Parasitol. 1995;81:39–45. doi: 10.1006/expr.1995.1090. [DOI] [PubMed] [Google Scholar]
- 143.Wells G. A. Ph.D. Thesis. Hatfield, Pretoria, South Africa: University of Pretoria; 2010. Molecular modeling elucidates parasite-specific features of polyamine pathway enzymes of Plasmodium falciparum. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.