

Abstract
Despite the discovery of 5 alpha-reduction as an enzymatic step in steroid metabolism in 1951, and the discovery that dihydrotestosterone is more potent than testosterone in 1968, the significance of 5 alpha-reduced steroids in human diseases was not appreciated until the discovery of 5 alpha-reductase type 2 deficiency in 1974. Affected males are born with ambiguous external genitalia, despite normal internal genitalia. The prostate is hypoplastic, nonpalpable on rectal examination and approximately 1/10th the size of age-matched normal glands. Benign prostate hyperplasia or prostate cancer does not develop in these patients. At puberty, the external genitalia virilize partially, however, secondary sexual hair remains sparse and male pattern baldness and acne develop rarely. Several compounds have been developed to inhibit the 5 alpha-reductase isozymes and they play an important role in the prevention and treatment of many common diseases. This review describes the basic biochemical properties, functions, tissue distribution, chromosomal location, and clinical significance of the 5 alpha-reductase isozyme family.
1. Introduction
Testosterone (T) is the most abundant androgen in serum. Approximately 97% of T is bound to albumen and sex-hormone binding globulin and the remaining 3% is free and biologically active. T is synthesized by the Leydig cells of the testes under the control of the hypothalamus and anterior pituitary gland. In male fetuses, T stimulates the differentiation of the Wolffian duct into male internal genitalia (epididymis, vas deferens, and seminal vesicles) and development of libido, enlargement of the vocal cords, skeletal muscles, penis, and scrotum and the initiation of spermatogenesis at puberty [1, 2]. T is taken from circulation to cells through processes that remain poorly understood. Intracellular T is converted to dihydrotestosterone (DHT), the preferred ligand for androgen receptor (AR) transactivation, by the enzyme 5 alpha-reductase (5α-R). Upon ligand binding and transactivation, the DHT-AR complex translocates from cytoplasm to nucleus and activates the transcription of certain genes (the androgen receptor-regulated genes, ARRG).
DHT is important for in utero differentiation and growth of the prostate gland, male external genitalia (penis and scrotum), and pubertal growth of facial and body hair. DHT plays an important role in several human diseases, which include acne, hirsutism, male pattern baldness, benign prostate hyperplasia (BPH), and prostate cancer (CaP) [3]. The role of DHT was discovered after the description of 5α-R2 deficiency in a group of males from the Dominican Republic [4]. DHT has 2–5 times higher binding affinity for AR than T, and 10-fold higher potency of inducing AR signaling than T [5], which means that their effects are different but complementary [6].
Three isozymes of 5α-R are known to exist (5α-R1-3) [7] and two other proteins exhibit 5-alpha reducing capabilities, glycoprotein synaptic 2 (GPSN2), and glycoprotein synaptic 2-like (GPSN2L) proteins. Only one 5 beta-reductase (5β-R) enzyme has been identified. Its products, 5β-isomers, are labeled as epi-product, such as 5β-DHT (epi-DHT) [8]. Several compounds have been developed to inhibit the 5α-R enzyme system and they play an important role in the prevention and treatment of many common diseases [9]. This review describes the basic biochemical properties, functions, tissue distribution, chromosomal location, and clinical significance of this enzyme family.
2. Background
Steroids are a special type of lipid. The backbone of steroids is the compound “gonane”, a 17-carbon molecule composed of 4 rings. The three cyclohexane rings are labeled A, B, and C. These 3 rings together are called phenanthrene. Ring D is a cyclopentane ring. The carbon atoms are numbered from 1 to 17. Typically, steroids have a methyl group (–CH3) at carbons C-10 and C-13 and an alkyl side chain (R) at C-17 (Table 1). Alkanes are saturated hydrocarbons composed of carbon and hydrogen atoms linked by single bonds. The simplest alkyl group is a methyl group. Steroids vary by the configuration of the alkyl side chain, the number of additional methyl groups, and the functional groups attached to the steroid nucleus. Carbons number 18 and 19 are attached to carbons number 13 and 10, respectively. Additional carbon atoms are usually a part of the R side chain or attached elsewhere to the steroid backbone [22]. Androgens are derivatives of androstane and contain 19 carbons and either a keto group (e.g., dehydroepiandrosterone (DHEA) and androstenedione (ASD)) or a hydroxy group (e.g., T and DHT) at position 17 of the steroid nucleus (Figure 1).
Table 1.
Different steroid families.
| Class | Example | Number of carbon atoms |
|---|---|---|
| Steroid backbone | Gonane | 17 |
| Estranes | Estradiol | 18 |
| Androstanes | Testosterone | 19 |
| Pregnanes | Progesterone | 21 |
| Glucocorticoids | Cortisol | 21 |
| Mineralocorticoids | Aldosterone | 21 |
| Cholanes | Cholic acid | 24 |
| Cholestanes | Cholesterol | 27 |
Figure 1.
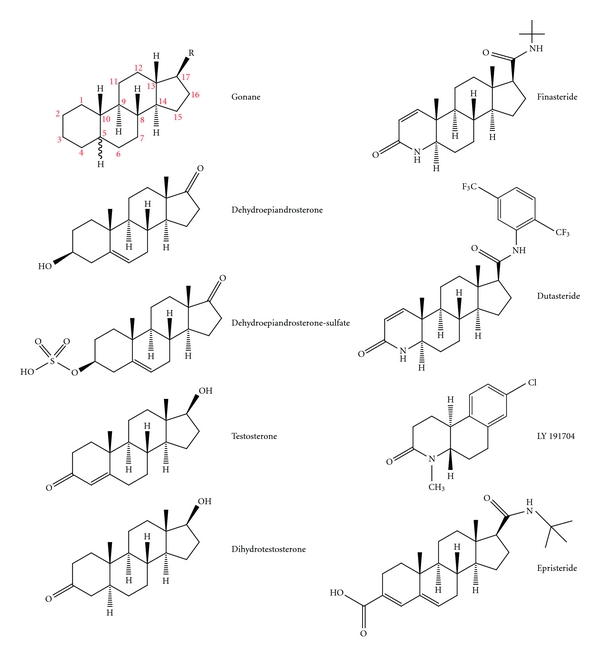
Structure of various steroids.
3. Historical Overview
Steroid-5-reductases (5α-R and 5β-R) were first discovered, purified, and characterized in rat liver homogenates [23]. These early experiments demonstrated that these enzymes were capable of irreversibly reducing the delta 4, 5 bond (double bond between carbons 4 and 5; Δ4,5) of C-19 and C-21 steroids to 5α- and 5β-stereoisomers.
The first androgen isolated was androsterone, a 5α-reduced androstane, which was isolated by Butenant in 1931 from 25,000 liters of urine from adult men. This steroid was assumed to be the male hormone until 1935 when Ernst Laquer and his colleagues isolated T from several tons of bull testes. The 5α-R enzyme was characterized initially in the 1950s in rat liver slices based on its ability to convert deoxycorticosterone to 5α-reduced metabolites [24]. Tomkins and others showed that the enzyme required a reduced pyridine nucleotide cofactor (i.e., NADPH) and could metabolize a variety of steroid substrates [25]. Speculation persisted about whether a single enzyme or multiple enzymes were involved in 5α-reduction of steroids. The 5α-reduction of steroids made them susceptible to further reduction, sulfation, and glucuronidation, modifications that decreased their affinity to bind proteins, made them more hydrophilic and facilitated their excretion. In the 1960s, 5α-reduction was shown to be an irreversible reaction and DHT was found to be a more potent androgen than T in prostate bioassays [26]. The administration of radiolabeled T to rats resulted in a time-dependent accumulation of DHT in the nuclei of ventral prostate cells, which subsequently bound to a specific nuclear (androgen) receptor. These data indicated that 5α-reduction of T is a crucial step in androgen action and focused attention on 5α-R. The central role of 5α-R in mammalian male physiology was obtained from developmental studies of mammalian embryos showing that 5α-reduction activity was highest in the primordia of the prostate and external genitalia prior to their virilization, but very low in Wolffian duct structures [27, 28], and from genetic studies on a rare disorder of male sexual differentiation, originally termed pseudovaginal perineoscrotal hypospadias and subsequently referred to as 5α-R deficiency [4]. Analysis of enzyme activity in skin samples and of urinary and serum steroids revealed a generalized defect in the conversion of T to DHT.
Studying 5α-R was hampered by the insolubility of the protein, a hurdle which was overcome in 1989. The technique of expression cloning in Xenopus laevis oocytes was used to isolate a cDNA from rat liver that encoded 5α-R enzyme, which was used to isolate a human 5α-R by cross-hybridization with a prostate cDNA library. The two expressed proteins had different biochemical properties and different responses to finasteride. These observations suggested the presence of two 5α-R isozymes that were confirmed by studies done in patients with 5α-R deficiency. The coding sequence of the gene specifying the rat liver cDNA was isolated and found to be normal in these patients. Further genetic studies in these patients identified a different mutated gene that encoded a 5α-R in normal individuals with identical biochemical properties to the human prostatic 5α-R. The first cDNA, isolated from rat liver, was named 5α-R1 (SRD5A1) gene, and the second cDNA, which was isolated from human prostate and found defective in 5α-R-deficient patients, was named 5α-R2 (SRD5A2) gene [29].
More recently, with the development of genome-wide gene expression profile analyses, a third 5α-R (SRD5A3) gene was identified. GPSN2 and GPSN2L proteins were identified using sequence searching and NCBI's BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). All primary species (from plant, amoeba, yeast, to vertebrate) in Eukaryota contain all 3 subfamilies [8].
4. Family Members
The 5α-R family is composed of 3 subfamilies and 5 members (isozymes) in total. Isozymes are different proteins that perform the same function:
5α-R1 and 5α-R2,
5α-R3,
GPSN2 and GPSN2L proteins.
5. Functions
5.1. 5 Alpha Reduction: (5α-R1-3) [29, 30]
The substrates for 5α-reductases are 3-oxo (3-keto), Δ4,5 C 19/C21 steroids. The group “3-keto” refers to the oxygen-carbon double bond at carbon 3. Delta 4, 5 refers to the double bond between carbon atoms 4 and 5. The reaction involves a stereospecific, irreversible breakage of the double bond between carbons 4 and 5 (delta 4, 5) with the aid of cofactor NADPH and the insertion of a hydride anion (H−) to the α face at carbon C-5 and a proton to the β face at position C-4. Examples of substrates are T, progesterone, androstenedione, epi-T, cortisol, aldosterone, and deoxycorticosterone. The physiologic role of 5α-reduction of these steroids (other than T) is unknown but probably related to their degradation and excretion or to certain physiologic functions. 5α-dihydroprogesterone (5α-DHP) is a major hormone in the circulation of both normal cycling and pregnant women [31]. 5α-dihydrocortisol is present in the aqueous humor of the eye, is synthesized in the lens of the eye, and may play a role in the regulation of aqueous humor formation [32]. 5α-Dihydroaldosterone is a potent antinatriuretic agent with somewhat different physiologic effects than aldosterone itself; its formation in the kidney is enhanced by restriction of dietary sodium intake, which suggests its importance for the conservation of sodium [33]:
| (1) |
Uemura et al. used small interfering RNA (siRNA) to knock down the expression of 5α-R3 isozyme in 22RV1 and LNCaP-C4-2 CaP cells by transfecting them with several siRNA expression vectors [34]. Subsequently, they studied mRNA expression of 5α-R3 (RT-PCR), cell growth and viability, and the ratio of DHT/T using liquid chromatography-tandem mass spectrometry. Knockdown of 5α-R3 expression caused decreased cell growth and viability and DHT/T ratio. Unpublished work from our group has confirmed the ability of 5α-R3 to 5α-reduce 3-oxo, delta 4,5 C19 and C21 (T, androstenedione and progesterone) steroids in lysates of CHO-K1 cells transfected with 5α-R3 cDNA via an adenovirus vector, CaP cell lines CWR-22 and CWR-22R, and clinical human samples of androgen-stimulated benign prostate (AS-BP), androgen-stimulated (AS-CaP), and castration-recurrent (CR-CaP) CaP.
5.2. N-Glycosylation of Proteins: (5α-R3)
Congenital deficiency of 5α-R3 has been linked to a rare, autosomal recessive disorder in which patients are born with mental retardation, cerebellar, and ophthalmologic defects [35]. The presumed defect involves the reduction of the terminal double bond of polyprenols to dolichols, an important step in protein N-glycosylation. N-linked protein glycosylation involves the addition of a 14-sugar glycan to select asparagine residues on a nascent protein to facilitate proper folding and trafficking of the protein and occurs in the membranes of endoplasmic reticula. This disorder is part of the family of congenital disorders of glycosylation and was described for the first time in a family in the United Arab Emirates by Cantagrel et al. [35].
5.3. Potential Biomarker of Malignancy: (5α-R1-3) [14, 20]
The bulk of published literature indicates that the expression of 5α-R1 increases and 5α-R2 decreases in CaP compared to benign prostate and BPH. Umera et al. confirmed for the first time increased expression of 5α-R3 at the mRNA level in CR-CaP. Godoy et al., confirmed this at the protein level. A validated monoclonal antibody showed that expression of 5α-R3 was increased similarly in AS-CaP and CR-CaP compared to AS-BP. 5α-R3 expression was increased in lung, breast, papillary thyroid, and testicular (seminoma and yolk sac) cancers compared to their benign counterparts.
5.4. Erythropoiesis [36]
5α-C 19 steroids increase the production of erythropoietin hormone in the kidneys. 5β-C 19 steroids are important for heme synthesis in the liver.
5.5. Regulation of Bile Synthesis [37]
Both 5α-R and 5β-R are involved in bile biosynthesis, where they catalyze the conversion of 7α, 12α-dihydroxy-4-cholesten-3-one into 7α,12α-dihydroxy- 5α-cholestan-3-one, and 7α,12α-dihydroxy-5β-cholestan-3-one, respectively. Only the 5β-isomer has been shown to be biologically active and is used for bile synthesis. The 5α-isomer is inactive and suggested to be an inhibitory step in bile biosynthesis regulation in humans.
5.6. GPSN2 Family [38]
While the functions of the GPSN2 subfamily are not understood fully, several reports have shown that GPSN2 members are involved in the fourth reaction of fatty acid elongation by reducing a fatty chain double bond in mammals. Although the substrate (fatty acid) of GPSN2 members is structurally different from that of the other two 5α-R subfamilies, all three subfamilies of 5α-R share a similar biochemical ability of reducing a double bond of the substrate.
6. Protein Structure and Gene Location [8, 29, 39]
5α-R1 and 2 isozymes are NADPH-dependent, membrane-associated (microsomal) enzymes, composed of 259 and 254 amino acids, and have molecular weights of 29.5 and 28.4 kilodaltons, respectively. They contain a high content of hydrophobic amino acids distributed throughout their sequences, which suggests that they are intrinsic membrane proteins deeply embedded in the lipid bilayer.
Even though these two isozymes are intrinsic membrane proteins and catalyze the same reaction, they only share a limited degree of homology in protein sequence, are located on different chromosomes, and possess distinctive biochemical properties. The average sequence identity between these two isozymes within a given species is approximately 47%, while the sequence identity between the same isozyme across species is 60% for 5α-R1 and 77% for 5α-R2. They are encoded by the 5α-R1 and 5α-R2 genes. These genes have similar structures, with five coding exons separated by four introns. The positions of the introns are essentially identical in the two genes. However, SRD5A1 is located on chromosome 5p15 whereas SRD5A2 is on 2p23. Gene polymorphisms exist for the two genes and are more common for 5α-R2. More than 850 and more than 550 single nucleotide polymorphisms (SNPs) have been reported for 5α-R2 and 5α-R1 genes, respectively [40, 41]. Only a few of these gene polymorphisms affect enzyme activity; some decrease (e.g., V89L SRD5A2 variant) and others increase (e.g., A49T SRD5A2 variant) enzyme activity [42]. Molecular epidemiologic studies are inconclusive as to whether altered 5α-R2 isozyme activity due to 5α-R2 gene polymorphism affects CaP risk [43]. A variant of 5α-R1 gene was reported to increase risk of polycystic ovary syndrome (PCOS) and more severe hirsutism in lean women, whereas a variant of 5α-R2 gene was associated with decreased risk of PCOS in the same cohort [44]. More than 300 SNPs have been reported for 5α-R3 gene; however, their clinical significance remains uncertain [45]. 5α-R3 is composed of 318 amino acids and has only 19% homology with 5α-R1 and 20% homology with 5α-R2 [39]. 5α-R3 is encoded by SRD5A3, which is located at 4q12. The genes encoding GPSN2, GPSN2-like, and 5β-R are located at 19p13.12, 4q13.1, and 7q34, respectively. GPSN2 and GPSN2-like proteins are composed of 308 and 363 amino acids, respectively. The amino acid sequence homology for GPSN2 is 15% with 5α-R1, 17% with 5α-R2 and 11% with 5α-R3. GPSN2-like has 6%, 11%, 6%, and 44% sequence homology with 5α-R1, 5α-R2, 5α-R3, and GPSN2, respectively.
7. Biochemical Properties [8, 29]
When examined in lysates of transfected cells, 5α-R1 exhibits a broad pH optimum, which ranges between 6.0 and 8.5, while 5α-R2 shows a narrow acidic pH optimum (pH 5–5.5). However, there is evidence to suggest that inside intact human cells, 5α-R2 isozyme functions optimally at a more neutral pH range (6.0–7.0). 5α-R1 has a larger turnover number, as indicated by its K cat value and a lower substrate affinity for T, K m = 1–5 μM. 5α-R2 has a lower turnover number (K cat) and a higher substrate affinity, as indicated by K m = 0.004–1 μM for T. Under optimal conditions, 5α-R2 has a higher 5α-reducing activity than 5α-R1, as indicated by its high V max/k m ratio. Both isozymes contain an NH2-terminal steroid (ligand) binding domain and a COOH-terminal NADPH binding domain. The apparent dissociation constant for NADPH cofactor is similar for both isozymes (3–10 μM). No such comparisons exist for 5α-R3 except that it appears to be efficient at pH 6.5–6.9 (unpublished work from our group).
8. 5α-Reductase Inhibitors [9, 30, 46]
Goal of development of 5α-reductase inhibitors (5α-RI) was to bind to 5α-R with little or no affinity for the androgen or other steroid receptors. The first inhibitors were steroids that mimicked T and, in many cases, were substrates themselves (i.e., not true inhibitors). The inhibitors can be broadly classified into two categories: steroidal and nonsteroidal. The steroidal class has more inhibitors thus far.
The mechanism of 5α-RI is complex but involves the binding of NADPH to the enzyme followed by the substrate. The Δ4,5 bond is broken and a hydride anion is transferred from NADPH directly to the C-5 carbon on the α face followed by a proton attacking the C-4 carbon on the β face leading to the formation of the product that subsequently leaves the enzyme-NADP+ complex. NADP+ departs last and the enzyme becomes free for further catalysis cycles. Based on this, the mechanism of inhibition of 5α-R isozymes is divided into three types [30]:
competitive with the cofactor (NADPH) and substrate (bi-substrate inhibitors): the inhibitor binds the free enzyme, for example, ONO-3805;
competitive with the substrate: the inhibitor binds the enzyme-NADPH complex for example, 4-, 6-, and 10-azasteroids;
uncompetitive with the enzyme-NADP+ complex: the inhibitor binds the enzyme-NADP+ complex after the product leaves, for example, epristeride.
8.1. Steroidal 5α-RI (Figure 1)
(1) 4-Azasteroids: the 3-oxo, 5-alpha steroids with a nitrogen atom at position 4 have been the most extensively studied. Examples include finasteride (MK-906), dutasteride (GG745), 4-MA, turosteride, MK-386, MK-434, and MK-963.
Finasteride is a synthetic 4-azasteroid and is the first 5α-RI approved for treatment of benign prostatic enlargement (BPE) and subsequently male pattern baldness. Finasteride is a potent (mean inhibitory concentration [IC50], 69 nM) competitive inhibitor of 5α-R2 but inhibits less effectively 5α-R1 (IC50 360 nM) [47]. Finasteride decreases mean serum level of DHT by 71% after 24 weeks of use [48]. Seven-day treatment with finasteride (1 or 5 mg daily) has been reported to suppress intraprostatic DHT in men with lower urinary tract symptoms (LUTSs) attributed to BPE by approximately 85% relative to placebo [49], whereas another study of finasteride 5 mg/d (also in men with LUTS attributed to BPE) demonstrated a reduction of 68% at 6 months [49]. Finasteride was shown in vitro to inhibit 5α-R3 at a similar potency to 5α-R2 (IC50 = 17.4 nM, 14.3 nM, resp.) in transfected HEK-293 cells [21].
Dutasteride is a synthetic 4-azasteroid with a half-life of nearly 5 weeks and is only approved for treatment of BPH. Dutasteride is a dual 5α-RI since it is more effective (more potent) at inhibiting 5α-R1 and 2 than finasteride; IC50 for inhibiting 5α-R1 is 7 nM and 5α-R2 is 6 nM. Dutasteride reduced mean levels of serum DHT at 24 weeks better than finasteride (94.7% versus 70.8% suppression) [50] and caused a 97% reduction in intraprostatic DHT levels in men with CaP treated with 5 mg/d for 6–10 weeks [51]. Another trial of dutasteride 3.5 mg/d for 4 months prior to RP decreased intraprostatic DHT by 99% [52]. The near-maximal suppression of intraprostatic DHT with dutasteride 3.5–5 mg daily and the report that dutasteride inhibits 5α-R3 in vitro (IC50 = 0.33 nM) [21] suggest that the development of a triple 5α-R inhibitor may not be necessary. Table 2 provides a comparison between finasteride and dutasteride.
4-MA was a potent dual inhibitor of 5α-R1 (IC50 = 1.7 nM) and 5α-R2 (IC50 = 1.9 nM). 4-MA had a very low affinity for AR and thus was not expected to produce undesirable antiandrogen effects, such as impotence, impaired muscle growth, or gynecomastia. However, 4-MA was withdrawn from clinical development after it was shown to be an inhibitor of 3β-hydroxysteroid dehydrogenase and to cause hepatotoxicity [9].
Turosteride, MK-434, and MK-963 inhibit mainly 5α-R2. MK-386 is a selective 5α-R1 inhibitor [46].
Table 2.
Comparison between finasteride and dutasteride (referenced in text).
| Finasteride | Dutasteride | |
|---|---|---|
| Family | Steroidal 5α-RI (4-azasteroid) | Steroidal 5α-RI (4-azasteroid) |
|
| ||
| IC50 for 5α-R1, 2 and 3 (nM) | 360, 69, 17.4 | 7, 6, 0.33 |
|
| ||
| FDA-approved clinical uses | Male androgenic alopecia Benign prostatic enlargement |
Benign prostatic enlargement |
|
| ||
| Clinical dose | 1 mg daily for male androgenic alopecia 5 mg daily for benign prostatic enlargement |
0.5 mg daily |
|
| ||
| Half-life (T 1/2) | 6–8 hours | 5 weeks |
|
| ||
| Suppression of DHT | ↓Serum DHT by 71% | ↓Serum DHT by 95% |
|
| ||
| ↓Intraprostatic DHT by 85% | ↓Intraprostatic DHT by 97–99% | |
(2) 6-Azasteroids (e.g., GIlS7669X) have a heterocyclic B ring (nitrogen atom at position 6) and a Δ4,5 bond in the A ring and are potent competitive inhibitors of 5α-R1 and 2 [46].
(3) 10-azasteroids, for example, AS97004, are competitive 5α-RI with a similar mechanism of action to 6-azasteroids [9].
(4) Androstanecarboxylic acids, such as epristeride, are noncompetitive, specific inhibitors for 5α-R2 [30].
(5) Other steroidal inhibitors include progesterone esters such as 4-bromo-17α-(p-fluorobenzoyloxy)-4-pregnene-3, 20-dione [53], 2-azasteroids, 3-azasteroids, 19-nor-10-azasteroids, and diazasteroids [9].
8.2. Nonsteroidal Inhibitors [9, 30, 46]
Several pharmaceutical and academic groups have pursued the synthesis of nonsteroidal compounds that inhibit human 5α-reductases due to the undesired hormonal side effects of steroidal compounds. Nonsteroidal inhibitors can be classified according to their structure. Most have been derived from azasteroidal inhibitors by removing one or more rings from the azasteroidal structure. Nonsteroidal inhibitors are thought to act as competitive inhibitors with exception of epristeride analogues, which are noncompetitive inhibitors. The most potent and selective inhibitors of human 5α-R1 are found among these classes of compounds and include the following:
-
Benzoquinolines include many subgroups.
- Benzo[f]quinolinones are tricyclic compounds that are derived from 4-azasteroids by removing the D ring and substituting the C ring with an aromatic one. These are selective against 5α-R1. The potency against 5α-R1 increases by substituting a halogen atom at position 8 (F, Br, or specially a Cl) and a methyl group at position 4. LY 191704 is the most potent (IC50 = 8 nM).
- Piperidones lack B and D rings.
- Quinolinones lack C ring.
- Pyridines lack B and C rings.
- Benzo[c]quinolinones tricyclic compounds derived from 6-azasteroids (no D ring, aromatic ring for the C ring) that have selective but weak inhibitory activity against 5α-R1.
- Benzo[c]quinolizinones are tricyclic compounds derived from 10-azasteroids (no D ring, aromatic ring for the C ring) that include some very potent, selective inhibitors of 5α-R1.
(Subgroups (b), (c), and (d)) are very weak 5α-R1I.
-
(2)
Nonsteroidal aryl acids are tricyclic compounds derived from androstanecarboxylic acids that differ from their parent compounds in being selective, noncompetitive 5α-R1I.
-
(3)
Butanoic acid derivatives contain an aromatic ring (generally benzene or indole) that bears a butanoic acid chain and aromatic moieties. Examples include ONO-3805, demonstrated in vitro to be a selective inhibitor of 5α-R1, and FK143, which inhibits 5α-R1 and 5α-R2 equally and noncompetitively.
-
(4)
Polyunsaturated fatty acids, found in vegetable oils, have been found to inhibit human and rat microsomal 5α-R activity. In this group, y-linolenic acid is the most potent compound tested. Since 5α-R isozymes are intrinsic membrane proteins, their activity may depend on the unique environment of the lipid bilayer. Whether and how fatty acids may function as endogenous regulators of 5α-R remain unknown.
-
(5)
Some cations, especially zinc, have been reported to reduce sebum production in vivo and have been used to treat acne. In vitro assays have indicated that zinc specifically inhibits 5α-R1. This inhibition may be mediated both by non-competitive inhibition of T binding to 5α-R and by reduced formation of the NADPH co-factor.
-
(6)
Other nonsteroidal inhibitors include epicatechin-3-gallate and epigallocatechin-3-gallate, which are major constituents of green tea. Also included are 7-hydroxycoumarin derivatives, 2,6-disubstituted 4-hydroxy-4-hydroxymethyl biphenyl derivatives, isoflavonoids, and 3,3-diphenylpentane derivatives [9].
9. Tissue Distribution
Numerous reports exist in the literature on the expression pattern of 5α-R1 and 5α-R2 in human tissue at various stages of development. The results vary due to differences in antibody sensitivity and specificity, mRNA analysis (in situ hybridization versus northern blotting versus reverse transcriptase-polymerase chain reaction), protein analysis (immunohistochemistry versus western blotting), tissue preparation, nature of tissue, evaluation of results, tissue fixation protocols, and control tissue. In addition, normal, benign, and malignant human tissue specimens are heterogeneous with variable expression of proteins among specimens from different individuals and within the same specimen, that is, inter- and intraindividual variability. Therefore, a summary of many studies that discussed the tissue distribution of 5α-R1-3 in different human tissues was tabulated to demonstrate differences in results (Table 3).
Table 3.
Tissue distribution of 5α-reductase 1–3 according to different authors.
| Author isozymes studied | Tissue type | Materials and methods | 5α-R1 | 5α-R2 | Notes |
|---|---|---|---|---|---|
| Eicheler et al. [10] 5α-R1-2 | Epidermis: genital (scrotum) nongenital (Axilla, breast, lip, eyelid) using a semiquantitative visual scale | Protein expression (IHC) using rabbit polyclonal antibodies against synthetic peptides from C-terminus parts of 5α-R1-2 proteins. Antibody sensitivity and specificity confirmed by ELISA and WB on FFPE biopsy or autopsy tissues | Nuclear, in epidermis from all sites: (scrotal> axilla> breast> lip> eye lid): stratum basale (++), stratum spinosum (++), absent in stratum granulosum and stratum corneum, dermal papillae, fibrous and outer epithelial RS (++), inner epithelial RS (+), matrix cells of hair bulb (++), scrotal fibroblast (++), basal and secretory cells of sebaceous glands (++), secretory and myoepithelial cells of sweat glands (++), arrector pili muscles (+), dermal adipocytes (+). No qualitative differences in males and females |
Cytoplasmic, in epidermis from all sites: stratum spinosum (++), stratum basale (+), absent in stratum granulosum and stratum corneum, inner epithelial RS (++), matrix cells of hair bulb (+), absent in dermal papillae, fibrous and outer epithelial RS, basal (+) and glandular (−) cells of sebaceous glands, myoepithelial (+) and secretory cells (−) of sweat glands, dermal adipocytes (+) No qualitative differences in males and females |
5α-R1 more uniformly spread in skin versus 5α-R2 that is mainly found in inner epith RS |
|
| |||||
| Aumüller et al. [11] 5α-R1-2 | Many tissue types, using a semiquantitative visual scale | Protein expression (IHC) using rabbit polyclonal antibodies against synthetic peptides from C-terminus parts of 5α-R1-2 proteins. Antibody sensitivity and specificity confirmed by ELISA and WB. Tissues from surgical pts and autopsies, fixed in Bouin's or formaldehyde |
Mainly nuclear: Prostate: stroma (+), epithelium (+) Seminal vesicle: stroma (+), epithelium (+) Epididymis: stroma (+), epithelium (+) Testis: Leydig cells (+), Sertoli cells (+) Ovary: stroma (++), theca and granulosa cells (−) Uterus: endometrium (+), myometrium (+) Liver: hepatocytes (+/−), bile duct c (+), kupffer cells (++) Pancreas: exocrine (+), islets of Langerhans (−) Kidney: glomerulus (+), PT (−), DT (++), CD (+) Adrenal: cortex (−), medulla (−) Thyroid: thyrocytes (−), C cells (−) Cerebral cortex: pyramidal c (+), glial cells (+/−) Pituitary: prolactin cells (+), others (−) |
Mainly cytoplasmic: Prostate: stroma (+), epithelium (++), specially basal cells Seminal vesicle.: stroma (+), epithelium (++) Epididymis: stroma (+), epithelium (+) Testis: spermatogonia (+), Leydig and Sertoli cell (−) Ovary: stroma (+), theca and granulosa cells (+/−) Uterus: endometrium (+), myometrium (+) Liver: hepatocytes (++), bile duct c (+), kupffer cells (−) Pancreas: exocrine (+), islets of Langerhans (−) Kidney: glomerulus (−), PT (++), DT (+/−), CD (+) Adrenal: ZG (+), ZF (+/−), ZR (+/−), med (−) Thyroid: thyrocytes (−), C cells (−) Cerebral cortex: pyramidal c (++), glial c (−) Pituitary: prolactin cells (+), others (−) |
5α1-2 is ubiquitous |
|
| |||||
| Bayne et al. [12] 5α-R1-2 | Scalp biopsies from bald and non-bald men | Protein expression (IHC) using validated mouse monoclonal antibodies against peptides from N-terminus parts of 5α-R1-2 and enzyme activity using 3H-T | In balding and non-balding scalp: 5α-R1 is expressed only in sebaceous glands No expression was detected in hair follicles or in epidermis |
In balding and nonbalding scalp: 5α-R2 is expressed in infundibula, outer (mainly) and inner epithelial RS of hair follicles. No expression was detected in dermal papillae or in sebaceous glands |
|
|
| |||||
| Thigpen et al. [13] 5α-R1-2 | Autopsy and surgical tissue samples | Messenger RNA (NB) and protein expression (IHC and WB) using rabbit polyclonal antibodies against peptides from C-terminus parts of 5α-R1-2 | 5α-R1 protein is expressed in liver and chest skin and 5α-R1 mRNA was detected in cerebellum, hypothalamus, pons, medulla oblongata, skin, and liver 5α-R1 was not detected by WB in any fetal tissue. 5α-R1 was detected by WB in newborn liver, skin, and scalp.5α-R1 was detected by WB in all scalp samples from balding and nonbalding men (except one). It was not detected in any normal prostate, BPH, or PC sample |
5α-R2 protein is expressed in prostate, seminal vesicles, epididymis, and liver. 5α-R2 mRNA was detected in prostate, SV, epididymis. and liver. 5α-R2 was detected by WB only in fetal genital skin (not detected in fetal liver, adrenal, testis, ovary, scalp, and brain). 5α-R2 was detected by WB in newborn prostate, SV, epididymis, liver, skin. and scalp. 5α-R2 was not detected by WB in any sample of balding and nonbalding scalp from one man. It was detected in all normal prostate, BPH, and PC samples |
5α-R1 protein was not detected in any prostate sample |
|
| |||||
| Thomas et al. [14] 5α-R1-2 | Prostate: BPH (TURP), ASCaP (RP), CaP metastasis in androgen deprivation-treated men (autopsy) | Protein expression (IHC) using rabbit polyclonal antibodies against peptides from N-terminus of 5α-R1-2 proteins. Antibody specificity confirmed with WB on naïve, 5α-R1- and 5α-R2- transfected COS-1 cells and IHC on transfected COS-1 cells-Evaluated by measuring percentage of moderate- and high-intensity immunostaining areas in relation to total epithelial, PIN, or tumor area in PE tissues |
Nuclear in BPH, shifts to cytoplasm in HGPIN and CaP Immunostaining intensity: Metastasis> CR-CaP> AS-CaP = HGPIN> BPH |
Mainly cytoplasmic in all specimens Immunostaining intensity: BPH = Metastasis = CR-CaP> AS-CaP = HGPIN |
All differences are statistically significant |
|
| |||||
| Thomas et al. [14] 5α-R1-2 | Prostate: AS-CaP (RP) with Gleason score <7, 7, >7 | Protein expression (IHC) using validated rabbit polyclonal antibodies, evaluated by visually estimating percent of total tumor area showing low-, moderate, and high-intensity in relation to Gleason score | Mainly nuclear in BPH, nuclear and cytoplasmic in CaP (all grades), and in adjacent benign epithelial tissue Immunostaining intensity: High grade > moderate grade = low grade > PC-adjacent benign tissue > BPH |
Mainly cytoplasmic in all samples (benign and malignant) Immunostaining intensity: BPH> high grade> moderate grade = low grade = adjacent benign tissue |
Staining in CaP-adjacent benign tissue is not significanty different from low- and high-grade CaP for either isozyme |
|
| |||||
| Söderstöm et al. [15] 5α-R1-2 | Prostate: BPH (TURP) and AS-CaP via RP or RC | Messenger RNA expression (sqRT-PCR) and measurement of 5α-R enzyme activity at pH 5.5 and 7 using 14C-T at 37°C in homogenized frozen pulverized prostate tissue |
5α-R1 mRNA expression is similar in BPH and AS-CaP. There was no correlation between enzyme activity at pH (5.5 and 7) and 5α-R1 mRNA expression as expressed on the basis of β-actin |
5α-R2 mRNA and enzyme activity were higher in BPH than in AS-CaP. There was a positive correlation between enzyme activity at pH 5.5 and expression of 5α-R2 mRNA as expressed on the basis of β-actin |
|
|
| |||||
|
Lehle et al. [61] 5α-R 1-2 |
BPH and CaP tissue post prostate biopsy or RP frozen in liquid nitrogen, one human liver sample | Messenger RNA expression (ISH, sqRT-PCR) | ISH showed that 5α-R1 mRNA is expressed in epithelium > stroma mRNA expression levels by sqRT-PCR: liver> CaP > BPH> Normal prostate |
ISH showed that 5α-R2 mRNA is expressed in epithelium > stroma mRNA expression levels by sqRT-PCR: liver = BPH > Normal prostate > CaP |
|
|
| |||||
| Habib et al. [16] 5α-R1-2 | BPH tissue (TURP) frozen in liquid nitrogen or in ice-cold RPMI with FBS and archival PE-BPH tissue | Messenger RNA expression (ISH, RT-PCR) and measurement of enzyme activity at pH 5 and 7.5 using 3H-T at 37°C in homogenized pulverized frozen prostate tissue | 5α-R1 mRNA expressed in epithelium > stroma | 5α-R1 mRNA expressed in epithelium > stroma 5α-R2 mRNA > 5α-R1 mRNA in BPH 5α-R2 enzyme activity ≫ 5α-R1 enzyme activity in homogenized BPH tissue |
|
|
| |||||
| Bonkhoff et al. [17] 5α-R1-2 | BPH (TURP), AS-CaP (RP), CR-CaP (channeling TUR) | Protein expression (IHC) using polyclonal rabbit antibodies against peptides from C-terminus parts of 5α-R1 and 2, validated by ELISA and WB | Mainly nuclear, in normal prostate and BPH tissue (luminal epithelial > basal) and in stroma 5α-R1 immunostaining > 5α-R2 in BPH (in both epithelium and stroma). In CaP, 5α-R1 immunostaining became nuclear and cytoplasmic and more intense in HGPIN and CaP versus adjacent benign tissue (specially CR-CaP) |
Mainly cytoplasmic (weak), in normal prostate and BPH tissue (basal > luminal epithelial) and stroma. In CaP, 5α-R2 immunostaining became nuclear and cytoplasmic and more intense in HGPIN and CaP versus adjacent benign tissue (specially CR-CaP) |
|
|
| |||||
| Shirakawa et al. [18] 5α-R1-2 | BPH (RC) fixed in formaldehyde and paraffin-embedded | Messenger RNA (qRT-PCR), protein (IHC) expression using polyclonal rabbit antibodies against peptides from C-terminus parts of 5α-R1 and 2, validated by ELISA and enzyme activities at pH 5 and 7.5 using 3H-T at 37°C | 5α-R1 mRNA copy numbers> 5α-R2 mRNA in BPH 5α-R1 protein expression is intense in epithelium of BPH (higher than 5α-R2 protein) 5α-R1 enzyme activity at pH 7.5 is similar to 5α-R2 enzyme activity at pH 5.0 |
5α-R2 mRNA < 5α-R1 mRNA in BPH 5α-R2 protein expression is detected in epithelium and stroma of BPH (less intense than 5α-R1) |
|
|
| |||||
| Titus et al. [19] 5α-R1-2 | ASBP, AS-CaP and CR-CaP (RP or channeling TURP) tissue that was FFPE or snap frozen in liquid nitrogen | Protein expression (by IHC in TMAs that are quantified by visual scoring and digital image analysis and by WB) and enzyme activity in homogenized pulverized prostate tissue using 3H-ASD at 37°C | 5α-R1 is nuclear and cytoplasmic in all 3 tissues Nuclear 5α-R1 staining intensity: ASBP = AS-CaP = CR-CaP Cytoplasmic 5α-R1 staining intensity: ASBP = AS-CaP> CR-CaP Not detected in stroma in any of the 3 tissues In WB, 5α-R1 > 5α-R2 in all 3 tissues 5α-R1 enzyme activity > 5α-R2 in CR-CaP (3 folds) |
5α-R2 is mainly cytoplasmic in all 3 tissues Nuclear 5α-R2 staining intensity: ASBP = AS-CaP> CR-CaP Cytoplasmic 5α-R2 staining intensity: ASBP = AS-CaP> CR-CaP Not detected in stroma in any of the 3 tissues In WB, 5α-R2 was undetectable in CR-CaP 5α-R2 activity > 5α-R1 in ASBP and AS-CaP |
In all 3 tissues, expression of 5α-R1 is consistenty more than 5α-R2 (in nucleus) but similar in cytoplasm |
|
| |||||
| Godoy et al. [20] 5α-R3 | Benign and malignant human tissue TMAs | Protein expression (IHC) using validated monoclonal mouse antibody against peptide from N-terminus of 5α-R3 protein and quantified by visual scoring and digital image analysis |
5α-R3 was mainly cytoplasmic Benign tissue immunostaining: Kidney (PT,DT ++), liver (++), exocrine pancreas (++), skeletal muscle (+), skin (strata basale and spinosum ++), gastric epithelial cells (+), myometrium (++) Malignant tissue: colon adenoCA (++), esophageal adenoCA (++), RCC (++), HCC (++), ovarian mucinous CA (++), stomach adenoCA (++), testis seminoma and YS tumor (++), thyroid papillary CA (++), endometrioid CA (++), breast CA (+) ASBP: basal epithelial cells, HGPIN: benign basal and neoplastic epithelial cells, AS-CaP and CR-CaP: in neoplastic cells (5α-R3 immunostaining intensity: AS-CaP = CR-CaP > ASBP) |
5α-R3 protein expression ↑ in the cytoplasm of malignant cells versus benign cells in prostate, testis, thyroid, lung and breast CA | |
|
| |||||
| Yamana et al. [21] 5α-R3 | 20 benign human tissues, CaP, and breast cancer cell lines | 5α-R1-3 mRNA expression (RT-PCR) and measurement of 5α-R 1–3 enzyme activity using 14C-labelled ASD and T in intact cells in culture |
5α-R3 expression at the mRNA level is higher than 5α-R1 and 2 in frontal cortex, heart, colon, stomach, liver, pancreas, lung, BPH, prostate, testis, mammary gland, brain, cervix, ovary, dermis, epidermis, total skin, small intestine, spleen, and kidney 5α-R2 mRNA was the most abundant in BPH and muscle Finasteride inhibits 5α-R2 and 5α-R3 with similar potency (IC50 = 14.3 nM, 17.4 nM, resp.). Dutasteride is a more potent inhibitor of 5α-R3 than finasteride (IC50 = 0.33 nM) |
5α-R3 is ubiquitous Dutasteride is a triple 5α-RI in vitro | |
ELISA: enzyme-linked immunosorbant assay; IHC: immunohistochemistry; WB: western blot; NB: northern blot; FFPE: formalin fixed paraffin embedded; RS: root sheath; RP: radical prostatectomy; sqRT-PCR: semiquantitative reverse transcriptase-polymerase chain reaction; PE: paraffin-embedded; RPMI: Roswell Park Memorial Institute; FBS: fetal bovine serum; PT: proximal tubules; DT: distal tubules; CD: collecting ducts; TURP: transurethral resection of prostate; RC: radical cystectomy; HGPIN: high-grade prostate intraepithelial neoplasia; ISH: in situ hybridization; RCC: renal cell carcinoma; HCC: hepatocellular carcinoma; adenoCA: adenocarcinoma; CA: carcinoma; YS: yolk sac; TMA: tissue microarray.
9.1. According to Age
9.1.1. Fetus
Ellsworth and Harris [54] studied 5α-R activity in fetal scalp, back skin, and prostatic tissues and compared it to 5α-R activity in adult male scalp and prostatic tissues. They studied the conversion of radio-labeled T into DHT in relation to pH and response to selective 5α-R1 and 5α-R2 inhibitors and calculated the k m of T at pH values of 7.0 and 5.5. 5α-R1 is expressed in fetal scalp and nongenital (back) skin at levels that are 5–50 times less than adult skin. 5α-R2 is expressed in the fetal prostate at levels similar to adult prostate. Thigpen et al. [13] studied 5α-R expression in fetal liver, adrenal, testis, ovary, brain, scalp, chest, and genital skin, using immunoblotting. They detected 5α-R2 only in fetal genital skin. Lunacek et al. [55] studied the expression of 5α-R 1 and 5α-R2 at the mRNA (RT-PCR) and protein (immunohistochemistry) levels in fetal and postnatal prostatic tissues until 6 years of age. Both 5α-R1 and 5α-R2 proteins were expressed in prostatic epithelial and stromal components, at consistent levels throughout all age groups. 5α-R1 is expressed mainly in the epithelium and 5α-R2 is expressed mainly in the stroma of the prostate. At the mRNA levels, both are detectable throughout the ages studied and both peak in the second trimester.
9.1.2. Newborn-Onset of Puberty
In newborns, 5α-R1 is expressed at the protein level in the liver, skin, scalp [13] and prostate [55]. 5α-R2 is expressed in prostate, seminal vesicles, epididymis, liver, and to lesser extent in scalp and skin [13]. Hepatic expression of 5α-R1 and 2 is present at the protein level (immunoblotting) throughout postnatal life. At approximately 1.5 years, the expression of both proteins is not detected in the skin and scalp until the onset of puberty. At puberty, only 5α-R1 is reexpressed in the skin and scalp and persists thereafter until 81 years. In the prostate gland, Lunacek et al. reported that both 5α-R1 and 5α-R2 were detectable at the protein level using IHC until approximately 1 year of age. After that, they were detectable at the mRNA level (RT-PCR) until 6 years of age. Thigpen et al. only detected 5α-R2 at the protein level using immunoblotting in prostatic tissue from a 7-year-old male. Since the methods used by this group did not detect 5α-R1 protein in the newborn, juvenile, or adult prostatic tissues, and since other groups detected 5α-R1 at the protein level in fetal and adult benign prostatic tissue, 5α-R1 and 5α-R2 appear to be expressed in the prostate in male fetuses and throughout postnatal life.
9.1.3. Adulthood-Old Age
5α-R1-3 is ubiquitously expressed [10, 11, 13, 20, 55]. 5α-R1 and 5α-R2 are expressed differently in liver, genital and nongenital skin, prostate, epididymis, seminal vesicle, testis, ovary, uterus, kidney, exocrine pancreas, and brain (Table 2, Aumuller et al.). Our laboratory described the expression of 5α-R3 using IHC in various benign and malignant tissues. 5α-R3 is overexpressed particularly in lung adenocarcinoma, testicular seminoma and yolk sac tumors, papillary thyroid cancer, and androgen-stimulated and castration-recurrent CaP relative to their benign counterparts [20]. When contrasting these data with the expressed sequence tag (EST) database from NCBI, both sets of data suggest a broad pattern of expression for 5α-R1-3 in human tissues; ESTs for 5α-R1 (271 sequences) have been reported from different human tissues, which include lung, brain, intestine, skin, prostate, testis, and stomach [56]. ESTs for 5α-R2 (39 sequences) have been reported in prostate, lung, liver, kidney, brain, testis, and skin [57]. ESTs for 5α-R3 (149 sequences) have been reported in kidney, testis, intestine, brain, liver, uterus, pancreas, skin, and prostate [58]. Tissue distribution of 5α-R3 protein in several human benign tissues was consistent with the tissue origin of the 5α-R3 EST reported at NCBI [20].
9.2. According to Organs
10. Clinical Role of 5α-R
Alterations in the conversion of T into DHT by the enzyme 5α-R are associated with a number of human disorders:
10.1. 5α-R2 Deficiency (Pseudovaginal Perineoscrotal Hypospadias) [3, 62]
Studies in rabbit, rat, and human fetuses have shown that 5α-R activity is present in the urogenital sinus and external genital anlage prior to prostate and external genital differentiation. However, 5α-R activity is not present in the Wolffian duct, at the time of epididymal, vas deferens, and seminal vesicle differentiation. Thus, T and DHT have selective roles in male sexual differentiation during embryogenesis. T mediates Wolffian ductal differentiation, while DHT mediates male external genital and prostate differentiation.
5α-R2 deficiency is caused by decreased synthesis of DHT due to mutations in the 5α-reductase 2 gene. At least 50 mutations have been reported and is autosomal-recessive in the majority of patients. 5α-R2 deficiency results in a 46,XY disorder of sexual development (formerly male pseudohermaphroditism). Affected males are born with normal male internal reproductive structures (epididymis, seminal vesicles, and vasa deferentia); however, their external genitalia resemble those of females, that is, ambiguous genitalia. These individuals have a small penis that resembles an enlarged clitoris, labioscrotal fusion, and a urogenital sinus in which there are two separate urethral and vaginal openings. The vagina is short and blind ending. The testes are either in the labia, or inguinal canals or intra-abdominal. The vasa terminate at the blind-ending vaginal pouch. The prostate is hypoplastic, nonpalpable on rectal examination and is found to be rudimentary on transrectal ultrasound and MRI. Prostatic volumes are approximately (1/10)th of age-matched normal controls. Prostate biopsy reveals fibrous connective tissue, smooth muscle, and no identifiable epithelial tissue, which suggests atrophic epithelium or lack of epithelial differentiation. Plasma PSA is low or undetectable in these patients. Administration of DHT results in enlargement of the prostate. Neither BPH nor CaP has been reported in these patients. At puberty, these individuals undergo partial virilization of the external genitalia, although their secondary sexual hair remains sparse and they develop less male pattern baldness and acne despite normal sebum production. They undergo an increase in muscle mass, phallic growth, development of male body habitus, and deepening of the voice. Their libido is normal and they are capable of erections. Sperm production and fertility have been reported but depend on testicular location. The mechanism of partial virilization at puberty is through either the androgen receptor binding very high levels of serum T, albeit at lower affinity, or the increased expression of skin 5α-R1 at puberty, which results in peripheral synthesis of DHT from T or via the action of 5α-R3 through unknown mechanisms. 5α-R1 gene is normal in these subjects. No genetic deficiencies of the type 1 enzyme have yet been reported. Inactivation of the 5α-R1 gene in mice adversely affected reproduction in females. In addition, mice deficient in 5α-R1 manifest a parturition defect that is not reversed by the administration of DHT, but is reversed by 3α-androstanediol treatment. The biochemical features of this syndrome include the following.
high normal to elevated levels of plasma T,
low normal to decreased levels of plasma DHT,
increased T to DHT ratio at baseline and following hCG stimulation,
normal metabolic clearance of T and DHT,
decreased levels of urinary 5α-reduced metabolites of C19 and C21 steroids, with increased 5β/5αurinary metabolite ratios,
decreased plasma and urinary 3α-androstanediol glucuronide, a major metabolite of DHT,
increased plasma levels of LH and/or FSH.
Phenotype, development and reproductive function in human females with 5α-R2 deficiency are unaffected.
10.2. LUTS Attributed to BPE
BPE results in a significant morbidity due to urethral obstruction and secondary detrusor dysfunction. Histological evidence of BPH is found in 50% of males by the age of 50 and 90% of males by the age of 80 [63]. The development of BPH depends on androgens, and BPH does not occur in men castrated prior to puberty [64]. 5α-R isozymes play significant roles in BPH development since DHT is the major androgen in the prostate. Patients with decreased DHT production due to 5α-R2 deficiency have a small prostate and BPH has not been reported [4]. In castrated dogs, treatment with either DHT or T results increased intraprostatic DHT and BPH [65]. However, coadministration of T with a 5α-R inhibitor decreased DHT formation and prevented BPH [66]. Finasteride and dutasteride have been shown to decrease circulating and intraprostatic DHT by 60–90%, and 90–98%, respectively [48–52]. Finasteride and dutasteride result in a decrease in prostate size by 20–25% through prostatic epithelial cell apoptosis and a significant improvement in LUTS. Furthermore, they change the natural history of the disease by decreasing the risk of acute urinary retention by 57–79% and decreasing BPH-related surgery by 48–69% [67, 68]. Although both isozymes are overexpressed in prostate tissues from patients with BPH [15], inhibition of 5α-R2 activity is the major contributor in the treatment of BPH as the additional inhibition of 5α-R1 activity by dutasteride does not appear to be of any further benefit in BPH treatment [69].
10.3. Primary Prevention of CaP [67, 68]
Both finasteride and dutasteride have been tested in large, prospective, randomized, placebo-controlled, double-blind studies as primary preventive therapies for CaP. The Prostate Cancer Prevention Trial (PCPT) randomized nearly 19,000 men at low risk for CaP into a treatment group, given finasteride at 5 mg/d and a control group, given placebo, who were followed up for 7 years. At the end of the study, participants were offered a prostate biopsy. For-cause biopsies were done for abnormal DRE and/or PSA > 4.0 ng/mL. PCPT showed that finasteride was effective at reducing the overall risk of biopsy detectable CaP by nearly 25% and this was due mainly to reduction in the risk of low-grade disease (Gleason sum <7). The reduction in the risk of CaP was seen across all subgroups, such as age, race, family history, and baseline PSA. Finasteride users also had improved BPH outcomes. The benefits of finasteride treatment occurred at the expense of a higher rate of diagnosis of moderate- and high-grade CaP (Gleason score ≥7) and more sexual adverse effects. The Reduction by Dutasteride of Prostate Cancer Events trial (REDUCE) studied the effect of dutasteride versus placebo in a large group of men at higher risk of CaP than in PCPT who had at least one negative prostate biopsy at baseline. The study lasted 4 years and participants received mandatory prostate biopsy at 2 and 4 years. Dutasteride decreased the risk of biopsy detectable CaP by nearly 24% and this reduction in risk was evident across all subgroups tested. The frequency of diagnosis of moderate- and high-grade CaP was unchanged over the entire length of the study and beneficial effects were observed on BPH outcomes. However, 12 Gleason score 8–10 cancers were detected in the dutasteride group at years 3-4 versus only one in the placebo group and dutasteride treatment was associated with more sexual adverse effects. The benefit of finasteride or dutasteride in reducing the risk of low-grade CaP is clear. Low-grade CaP is unlikely to be lethal and patients may reduce their risk of overtreatment. However, these drugs may induce high-grade CaP, a concern that has prevented FDA approval of finasteride and dutasteride use for CaP prevention. Several secondary analyses of these two trials have concluded that these drugs actually reduce the risk of moderate- and high-grade disease but these analyses are hypothesis-generating and not definitive. In March 2009, the American Urological Association and the American Cancer Society issued a “cautious” joint statement that accepted these drugs as an option to prevent CaP provided that they are used mainly after a thorough discussion of risks and benefits. In January 2011, the FDA's Oncologic Drug Advisory Committee voted against recommending either dutasteride or finasteride for the specific indication of CaP risk reduction because of the potential increased risk of high-grade disease. Table 4 provides a comparison between PCPT and REDUCE.
Table 4.
Comparison between PCPT and REDUCE.
| PCPT | REDUCE | |
|---|---|---|
| Sponsor | South West Oncology Group | GalxoSmithKline |
| Duration | 7 years | 4 years |
| Risk of CaP in participants | lower | higher |
| No. of participants | 18,882 randomized 9060 included in final analysis |
8122 randomized |
| Age | ≥55 years | 50–75 years |
| Entry serum PSA | ≤3.0 ng/mL | 2.5–10 ng/mL |
| Baseline biopsies | No | Yes (6–12 cores) within 6 months prior to enrollment |
| Study-mandated biopsies | Year 7 | Years 2 and 4 |
| Study-mandated biopsy cores | ≥6 (6 cores in nearly 80%) | 10 (83% had at least 1 biopsy) |
| Overall relative risk reduction in CaP versus placebo | 25% | 23% |
| Incidence of Gleason sum ≥7 CaP | ↑26% (6.4% in finasteride versus 5.1% in placebo), P < 0.05 | Same (6.7% in dutasteride versus 6.8%: in placebo) |
| Incidence of Gleason sum ≥8 CaP | ↑91% (2.1% in finasteride versus 1.1% in placebo), P < 0.05 | Same over 4 years (0.9% in dutasteride versus 0.6% in placebo); however, in years 3-4, there were 12 GS ≥8 CaP in dutasteride group (0.5%) versus 1 in placebo group (<0.1%), P < 0.05 |
10.4. Treatment of CaP
(a) Biochemical Failure after Local Therapy with Curative Intent [70–73] —
Finasteride and dutasteride have been tried, singly and in combination, in patients with biochemical failure after radical prostatectomy or radiotherapy. The most common combination was a 5α-RI and a nonsteroidal antiandrogen. Finasteride and dutasteride monotherapy decreased serum PSA to variable extent. PSA decrease was more frequent and of greater magnitude in patients treated with an antiandrogen and 5α-RI versus 5α-RI alone. However, none of these trials studied the impact on disease-specific or overall survival and none compared 5α-RI mono- or combination therapy against 1st line androgen deprivation treatment in a randomized fashion.
(b) CR-CaP —
CR-CaP was thought for many years to be androgen-independent or hormone-refractory but CR-CaP remains AR-dependent and probably AR-ligand dependent in almost all cases [74]. Despite castrate serum levels of T (<50 ng/dL), CR-CaP tissue levels of T and DHT were similar and 80–90% lower compared to their levels in benign prostatic tissue, respectively [75]. CR-CaP tissue synthesizes testicular androgens (T and DHT) in an intracrine fashion from several substrates such as cholesterol, progesterone, adrenal androgens, and androstandione [76–79]. Other phenomena intrinsically acquired by CR-CaP tissue in response to castration include the continuous expression of AR [80], upregulation of the synthesis of enzymes necessary for steroidogenesis [76], AR hypersensitivity (up to 10,000 times) to low levels of ligands by alteration of its co-activator profile from SRC1 to TIF2 and through its phosphorylation by SRC and Ack1 tyrosine kinases [81–83] and AR functional mutations, which broaden ligand specificity in 5–30% of cases [84].
New second-line hormonal therapeutic agents that have shown better performance in CR-CaP compared to the old generation of second-line hormonal therapies are abiraterone acetate and MDV3100, among others [77, 85]. Abiraterone acetate is a potent, selective, and irreversible inhibitor of CYP17A1 enzyme, which is an important enzyme in the intracrine synthesis of testicular androgens. MDV3100 inhibits ligand binding to the AR and nuclear translocation of AR-ligand complex. The clinical response to these new drugs is indirect proof that CR-CaP remains androgen stimulated. 5α-R isozymes are important in the growth of CR-CaP tissue since they are upregulated in CR-CaP and may contribute to intracrine synthesis of testicular androgens. These enzymes convert progesterone, ASD, and T into pregnanldione, androstandione, and DHT, respectively [74, 79]. Pregnanldione is further converted via several steps into androstandiol which is oxidized by 17β-hydroxysteroid dehydrogenase 2 and 10 (17β-HSD2 and 10) to DHT (the backdoor pathway to DHT synthesis). Androstandione is converted by 17βHSD3 into DHT.
Clinical trials of finasteride [70] and dutasteride [71] as monotherapy in patients with advanced CaP showed no improvement of clinical end points. The presence of 5α-R3 in CR-CaP is a potential explanation, until such time that an inhibitor has been proven effective clinically. Combination therapy of 5α-RI with antiandrogen or ketoconazole and hydrocortisone was tried in CR-CaP as second or third line hormonal therapies [86–89]. PSA decreases of variable magnitudes and durations were achieved in more than half the patients. However, none of the combination trials were designed to test the effect on disease-specific and overall survival. In a phase II, single arm study of 57 patients with CR-CaP, dutasteride added to ketoconazole, and hydrocortisone reduced PSA ≥ 50% of baseline value in 56% of patients, responses that lasted for a median of 20 months. Median time to disease progression was 14.5 months that was better than all prior studies of ketoconazole and hydrocortisone in CR-CaP [89].
10.5. Androgen-Stimulated Skin Disorders (Acne, Androgenic Alopecia and Hirsutism)
Hyperandrogenism, or excessive androgen production, is primarily a disorder of females. Polycystic ovary syndrome (PCOS) is the most common cause of female hyperandrogenism with a prevalence of 6% to 10% in women of childbearing age [90]. Other causes of hyperandrogenism include androgen-secreting ovarian and adrenal tumors, congenital adrenal hyperplasia, and exogenous androgenic hormone administration. Regardless of the cause, overproduction of either T or T precursors leads to exaggerated T action in target tissues such as skin. In skin, T is converted to DHT by the enzyme 5α-R, which acts directly on hair follicles and the sebaceous glands. The most frequent dermatologic manifestations of androgen excess are hirsutism, acne, and androgenic alopecia [91].
Androgen surge can miniaturize hair follicles resulting in male androgenic alopecia at puberty and in the scalp of genetically predisposed individuals. In women, androgen excess plays a role in scalp hair loss; however, this process is different than in men and is referred to as female pattern hair loss. Hirsutism is a disorder of excessive growth of terminal hair in women, in a male-like distribution, which is stimulated by androgen excess [92]. In acne, the pubertal androgen surge increases the stimulation of sebaceous glands resulting in increased sebum production and acne formation in susceptible individuals [93].
In all androgen-stimulated skin disorders, the activity of 5α-R enzyme system is increased such as in the hair follicles of hirsute women [93], in balding scalps [94], and in acne-prone skin [95]. Inhibition of the 5α-R enzyme system appears to be a target for treatment of androgen-stimulated skin disorders, since 5α-R inhibitors may result in fewer side-effects by not blocking the action of T, unlike classical anti-androgens such as cyproterone acetate or spironolactone.
Finasteride and dutasteride reduce scalp DHT levels by 64% and 51%, respectively [96, 97]. In men with androgenic alopecia, finasteride and dutasteride significantly increased hair count after a minimum of 6-month treatment [98, 99]. While finasteride 1 mg daily was superior to 5% topical minoxidil in inducing hair growth [100], finasteride 5 mg daily was inferior to dutasteride 2.5 mg daily in a phase II study as treatment for male androgenic alopecia [97]. Finasteride and topical minoxidil (but not dutasteride) are FDA-approved for male androgenic alopecia. In women with androgenic alopecia, neither finasteride nor dutasteride is FDA-approved treatment options due to teratogenicity. Finasteride has been tested in postmenopausal women at the 1 mg dose without success. However, finasteride was shown to be effective in 4 women with elevated serum T levels [101]. Whether finasteride is only effective in women with hair loss and hyperandrogenism or that higher doses are needed in women remains to be tested. A single case report showed improvement with dutasteride in a woman who had failed to respond to finasteride [102].
5α-RI also is acceptable therapy for hirsutism. Finasteride 5 mg/day was superior to placebo and similar to spironolactone and flutamide in reducing the severity of hirsutism [103]; however, in a different study, finasteride was inferior to flutamide as a treatment for hirsutism [104]. Dutasteride has not been tested as a treatment for hirsutism.
The role of 5α-RI is unclear for treatment of acne. MK-386, a selective 5α-R1 inhibitor, decreased sebum DHT levels in a dose-dependent fashion [105]. MK-386 was examined as a treatment for acne in a placebo-controlled trial and was found to be similar to placebo and inferior to systemic minocycline therapy. Furthermore, MK-386 did not enhance the therapeutic benefit of minocycline when used in combination [106]. In another study, finasteride decreased the severity of acne but was inferior to flutamide and cyproterone acetate with ethinyl estradiol [107]. No studies of dutasteride for treatment of acne have been reported.
11. Conclusions
The 5α-R system is an important player in human physiology and pathology. More work is needed to identify the biochemical characteristics and role of 5α-R3 in several human conditions such as CaP and androgen-stimulated skin diseases. Clinical data are inconclusive regarding the benefit of 5α-RI for CaP prevention. Clinical trials are ongoing to define the role of dutasteride for treatment of CaP, such as REDEEM (dutasteride in low-risk CaP patients on active surveillance), ARTS (biochemical failure after local treatment with curative intent), AVO 108943 (bicalutamide and dutasteride versus bicalutamide and placebo in CR-CaP), and ARI40006 (2-year follow-up study of REDUCE participants who received dutasteride or placebo) [108]. Future trials should focus on blocking multiple steroidogenic enzymes at once, such as in men with biochemical failure after local therapy or men with CR-CaP. Blocking several different steps in steroidogenesis simultaneously may not allow CaP cells time to adjust to loss of androgen stimulation.
References
- 1.Imperato-McGinley J, Zhu YS. Androgens and male physiology the syndrome of 5α-reductase-2 deficiency. Molecular and Cellular Endocrinology. 2002;198(1-2):51–59. doi: 10.1016/s0303-7207(02)00368-4. [DOI] [PubMed] [Google Scholar]
- 2.Siiteri PK, Wilson JD. Testosterone formation and metabolism during male sexual differentiation in the human embryo. Journal of Clinical Endocrinology and Metabolism. 1974;38(1):113–125. doi: 10.1210/jcem-38-1-113. [DOI] [PubMed] [Google Scholar]
- 3.Cilotti A, Danza G, Serio M. Clinical application of 5α-reductase inhibitors. Journal of Endocrinological Investigation. 2001;24(3):199–203. doi: 10.1007/BF03343844. [DOI] [PubMed] [Google Scholar]
- 4.Imperato McGinley J, Guerrero L, Gautier T, Peterson RE. Steroid 5α reductase deficiency in man: an inherited form of male pseudohermaphroditism. Science. 1974;186(4170):1213–1215. doi: 10.1126/science.186.4170.1213. [DOI] [PubMed] [Google Scholar]
- 5.Saartok T, Dahlberg E, Gustafsson JA. Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin. Endocrinology. 1984;114(6):2100–2106. doi: 10.1210/endo-114-6-2100. [DOI] [PubMed] [Google Scholar]
- 6.Beato M. Gene regulation by steroid hormones. Cell. 1989;56(3):335–344. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 7.Stiles AR, Russell DW. SRD5A3: a surprising role in glycosylation. Cell. 2010;142(2):196–198. doi: 10.1016/j.cell.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langlois VS, Zhang D, Cooke GM, Trudeau VL. Evolution of steroid-5α-reductases and comparison of their function with 5β-reductase. General and Comparative Endocrinology. 2010;166(3):489–497. doi: 10.1016/j.ygcen.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Aggarwal S, Thareja S, Verma A, Bhardwaj TR, Kumar M. An overview on 5α-reductase inhibitors. Steroids. 2010;75(2):109–153. doi: 10.1016/j.steroids.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Eicheler W, Dreher M, Hoffmann R, Happle R, Aumuller G. Immunohistochemical evidence for differential distribution of 5α-reductase isoenzymes in human skin. British Journal of Dermatology. 1995;133(3):371–376. doi: 10.1111/j.1365-2133.1995.tb02663.x. [DOI] [PubMed] [Google Scholar]
- 11.Aumüller G, Eicheler W, Renneberg H, Adermann K, Vilja P, Forssmann WG. Immunocytochemical evidence for differential subcellular localization of 5α-reductase isoenzymes in human tissues. Acta Anatomica. 1997;156(4):241–252. doi: 10.1159/000147852. [DOI] [PubMed] [Google Scholar]
- 12.Bayne EK, Flanagan J, Einstein M, et al. Immunohistochemical localization of types 1 and 2 5α-reductase in human scalp. British Journal of Dermatology. 1999;141(3):481–491. doi: 10.1046/j.1365-2133.1999.03042.x. [DOI] [PubMed] [Google Scholar]
- 13.Thigpen AE, Silver RI, Guileyardo JM, Casey ML, McConnell JD, Russell DW. Tissue distribution and ontogeny of steroid 5α-reductase isozyme expression. Journal of Clinical Investigation. 1993;92(2):903–910. doi: 10.1172/JCI116665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas LN, Douglas RC, Lazier CB, Too CKL, Rittmaster RS, Tindall DJ. Type 1 and type 2 5α-reductase expression in the development and progression of prostate cancer. European Urology. 2008;53(2):244–252. doi: 10.1016/j.eururo.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 15.Söderström TG, Bjelfman C, Brekkan E, et al. Messenger ribonucleic acid levels of steroid 5α-reductase 2 in human prostate predict the enzyme activity. Journal of Clinical Endocrinology and Metabolism. 2001;86(2):855–858. doi: 10.1210/jcem.86.2.7224. [DOI] [PubMed] [Google Scholar]
- 16.Habib FK, Ross M, Bayne CW, et al. The localisation and expression of 5α-reductase types I and II mRNAs in human hyperplastic prostate and in prostate primary cultures. Journal of Endocrinology. 1998;156(3):509–517. doi: 10.1677/joe.0.1560509. [DOI] [PubMed] [Google Scholar]
- 17.Bonkhoff H, Stein U, Aumüller G, Remberger K. Differential expression of 5α-reductase isoenzymes in the human prostate and prostatic carcinomas. The Prostate. 1996;29(4):261–267. doi: 10.1002/(SICI)1097-0045(199610)29:4<261::AID-PROS7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 18.Shirakawa T, Okada H, Acharya B, et al. Messenger RNA levels and enzyme activities of 5 alpha-reductase types 1 and 2 in human benign prostatic hyperplasia (BPH) tissue. The Prostate. 2004;58(1):33–40. doi: 10.1002/pros.10313. [DOI] [PubMed] [Google Scholar]
- 19.Titus MA, Gregory CW, Ford OH, Schell MJ, Maygarden SJ, Mohler JL. Steroid 5α-reductase isozymes I and II in recurrent prostate cancer. Clinical Cancer Research. 2005;11(12):4365–4371. doi: 10.1158/1078-0432.CCR-04-0738. [DOI] [PubMed] [Google Scholar]
- 20.Godoy A, Kawinski E, Li Y, et al. 5α-reductase type 3 expression in human benign and malignant tissues: a comparative analysis during prostate cancer progression. The Prostate. 2011;71(10):1033–1046. doi: 10.1002/pros.21318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamana K, Labrie F, Luu-The V, et al. Human type 3 5α-reductase is expressed in peripheral tissues at higher levels than types 1 and 2 and its activity is potently inhibited finasteride and dutasteride. Hormone Molecular Biology and Clinical Investigation. 2010;2(3):293–299. doi: 10.1515/HMBCI.2010.035. [DOI] [PubMed] [Google Scholar]
- 22.Moss GP. Nomenclature of steroids (Recommendations 1989) Pure and Applied Chemistry. 1989;61(10):1783–1822. [Google Scholar]
- 23.Dorfman RI, Forchielli E. Separation of delta 4-5 alpha-hydrogenases from rat liver homogenates. The Journal of Biological Chemistry. 1956;223(1):443–448. [PubMed] [Google Scholar]
- 24.Schneider JJ, Horstmann PM. Effects of incubating desoxycorticosterone with various rat tissues. The Journal of Biological Chemistry. 1951;191(1):327–338. [PubMed] [Google Scholar]
- 25.Tomkins GM. The enzymatic reduction of Δ4-3-ketosteroids. The Journal of Biological Chemistry. 1957;225(1):13–24. [PubMed] [Google Scholar]
- 26.Saunders FJ. Some aspects of relation of structure of steroids to their prostate stimulating effects. In: Vollmer EP, editor. Biology of the Prostate and Related Tissue. Washington, DC, USA: U.S. Government Printing Office; 1963. pp. 139–159. [PubMed] [Google Scholar]
- 27.Wilson JD. Recent studies on the mechanism of action of testosterone. The New England Journal of Medicine. 1972;287(25):1284–1291. doi: 10.1056/NEJM197212212872508. [DOI] [PubMed] [Google Scholar]
- 28.Wilson JD, Lasnitzki I. Dihydrotestosterone formation in fetal tissues of the rabbit and rat. Endocrinology. 1971;89:659–668. doi: 10.1210/endo-89-3-659. [DOI] [PubMed] [Google Scholar]
- 29.Russell DW, Wilson JD. Steroid 5α-reductase: two genes/two enzymes. Annual Review of Biochemistry. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- 30.Occhiato EG, Guarna A, Danza G, Serio M. Selective non-steroidal inhibitors of 5α-reductase type 1. Journal of Steroid Biochemistry and Molecular Biology. 2004;88(1):1–16. doi: 10.1016/j.jsbmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 31.Milewich L, Gomez Sanchez C, Crowley G. Progesterone and 5α pregnane 3,20 dione in peripheral blood of normal young women. Daily measurements throughout the menstrual cycle. Journal of Clinical Endocrinology and Metabolism. 1977;45(4):617–622. doi: 10.1210/jcem-45-4-617. [DOI] [PubMed] [Google Scholar]
- 32.Weinstein BI, Kandalaft N, Ritch R, et al. 5α-dihydrocortisol in human aqueous humor and metabolism of cortisol by human lenses in vitro. Investigative Ophthalmology and Visual Science. 1991;32(7):2130–2135. [PubMed] [Google Scholar]
- 33.Kenyon CJ, Brem AS, McDermott MJ. Antinatriuretic and kaliuretic activities of the reduced derivatives of aldosterone. Endocrinology. 1983;112(5):1852–1856. doi: 10.1210/endo-112-5-1852. [DOI] [PubMed] [Google Scholar]
- 34.Uemura M, Tamura K, Chung S, et al. Novel 5α-steroid reductase (SRD5A3, type-3) is overexpressed in hormone-refractory prostate cancer. Cancer Science. 2008;99(1):81–86. doi: 10.1111/j.1349-7006.2007.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantagrel V, Lefeber DJ, Ng BG, et al. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell. 2010;142(2):203–217. doi: 10.1016/j.cell.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gordon AS, Zanjani ED, Levere RD, Kappas A. Stimulation of mammalian erythropoiesis by 5β-H steroid metabolites. Proceedings of the National Academy of Sciences of the United States of America. 1970;65(4):919–924. doi: 10.1073/pnas.65.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kondo K-H, Kai M-H, Setoguchi Y, et al. Cloning and expression of cDNA of human Δ4-3-oxosteroid 5β-reductase and substrate specificity of the expressed enzyme. European Journal of Biochemistry. 1994;219(1-2):357–363. doi: 10.1111/j.1432-1033.1994.tb19947.x. [DOI] [PubMed] [Google Scholar]
- 38.Moon YA, Horton JD. Identification of two mammalian reductases involved in the two-carbon fatty acyl elongation cascade. Journal of Biological Chemistry. 2003;278(9):7335–7343. doi: 10.1074/jbc.M211684200. [DOI] [PubMed] [Google Scholar]
- 39. http://blast.ncbi.nlm.nih.gov/Blast.cgi.
- 40. http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?showRare=on&chooseRs=all&go=Go&locusId=6715.
- 41. http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?showRare=on&chooseRs=all&go=Go&locusId=6716.
- 42.Ntais C, Polycarpou A, Tsatsoulis A. Molecular epidemiology of prostate cancer: androgens and polymorphisms in androgen-related genes. European Journal of Endocrinology. 2003;149(6):469–477. doi: 10.1530/eje.0.1490469. [DOI] [PubMed] [Google Scholar]
- 43.Cussenot O, Azzouzi AR, Nicolaiew N, et al. Low-activity V89L variant in SRD5A2 is associated with aggressive prostate cancer risk: an explanation for the adverse effects observed in chemoprevention trials using 5-alpha-reductase inhibitors. European Urology. 2007;52(4):1082–1089. doi: 10.1016/j.eururo.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 44.Graupp M, Wehr E, Schweighofer N, Pieber TR, Obermayer-Pietsch B. Association of genetic variants in the two isoforms of 5α-reductase, SRD5A1 and SRD5A2, in lean patients with polycystic ovary syndrome. European Journal of Obstetrics Gynecology and Reproductive Biology. 2011;157(2):175–179. doi: 10.1016/j.ejogrb.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 45. http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp&cmd=DetailsSearch&term=nm_024592.3&save_search=false.
- 46.Chen W, Zouboulis CC, Orfanos CE. The 5α-reductase system and its inhibitors. Recent development and its perspective in treating androgen-dependent skin disorders. Dermatology. 1996;193(3):177–184. doi: 10.1159/000246242. [DOI] [PubMed] [Google Scholar]
- 47.Tian G. 17β-(N-tert-butylcarbamoyl)-4-aza-5α-androstan-1-en-3-one is an active site-directed slow time-dependent inhibitor of human steroid 5α-reductase. Biochemistry. 1994;33(8):2291–2296. doi: 10.1021/bi00174a041. [DOI] [PubMed] [Google Scholar]
- 48.McConnell JD, Wilson JD, George FW, Geller J, Pappas F, Stoner E. Finasteride, an inhibitor of 5α-reductase, suppresses prostatic dihydrotestosterone in men with benign prostatic hyperplasia. Journal of Clinical Endocrinology and Metabolism. 1992;74(3):505–508. doi: 10.1210/jcem.74.3.1371291. [DOI] [PubMed] [Google Scholar]
- 49.Span PN, Völler MCW, Smals AGH, et al. Selectivity of finasteride as an in vivo inhibitor of 5α-reductase isozyme enzymatic activity in the human prostate. Journal of Urology. 1999;161(1):332–337. [PubMed] [Google Scholar]
- 50.Clark RV, Hermann DJ, Cunningham GR, Wilson TH, Morrill BB, Hobbs S. Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5α-reductase inhibitor. Journal of Clinical Endocrinology and Metabolism. 2004;89(5):2179–2184. doi: 10.1210/jc.2003-030330. [DOI] [PubMed] [Google Scholar]
- 51.Andriole GL, Humphrey P, Ray P, et al. Effect of the dual 5α-reductase inhibitor dutasteride on markers of tumor regression in prostate cancer. Journal of Urology. 2004;172(3):915–919. doi: 10.1097/01.ju.0000136430.37245.b9. [DOI] [PubMed] [Google Scholar]
- 52.Gleave M, Qian J, Andreou C, et al. The effects of the dual 5α-reductase inhibitor dutasteride on localized prostate cancer—results from a 4-month pre-radical prostatectomy study. The Prostate. 2006;66(15):1674–1685. doi: 10.1002/pros.20499. [DOI] [PubMed] [Google Scholar]
- 53.Cabeza M, Heuze I, Bratoeff E, Murillo E, Ramirez E, Lira A. New progesterone esters as 5α-reductase inhibitors. Chemical and Pharmaceutical Bulletin. 2001;49(9):1081–1084. doi: 10.1248/cpb.49.1081. [DOI] [PubMed] [Google Scholar]
- 54.Ellsworth K, Harris G. Expression of the type 1 and 2 steroid 5α-reductases in human fetal tissues. Biochemical and Biophysical Research Communications. 1995;215(2):774–780. doi: 10.1006/bbrc.1995.2530. [DOI] [PubMed] [Google Scholar]
- 55.Lunacek A, Schwentner C, Oswald J, et al. Fetal distribution of 5α-reductase 1 and 5α-reductase 2, and their input on human prostate development. Journal of Urology. 2007;178(2):716–721. doi: 10.1016/j.juro.2007.03.089. [DOI] [PubMed] [Google Scholar]
- 56. http://www.ncbi.nlm.nih.gov/UniGene/clust.cgi?ORG=Hs&CID=552&MAXEST=271.
- 57. http://www.ncbi.nlm.nih.gov/UniGene/clust.cgi?ORG=Hs&CID=458345&MAXEST=39.
- 58. http://www.ncbi.nlm.nih.gov/UniGene/clust.cgi?UGID=136679&TAXID=9606&SEARCH=SRD5A3.
- 59.Thomas LN, Lazier CB, Gupta R, et al. Differential alterations in 5α-reductase type 1 and type 2 levels during development and progression of prostate cancer. The Prostate. 2005;63(3):231–239. doi: 10.1002/pros.20188. [DOI] [PubMed] [Google Scholar]
- 60.Thomas LN, Douglas RC, Lazier CB, et al. Levels of 5α-reductase type 1 and type 2 are increased in localized High grade compared to low grade prostate cancer. Journal of Urology. 2008;179(1):147–151. doi: 10.1016/j.juro.2007.08.155. [DOI] [PubMed] [Google Scholar]
- 61.Iehle C, Radvanyi F, Gil Diez de Medina S, et al. Differences in steroid 5alpha-reductase iso-enzymes expression between normal and pathological human prostate tissue. The Journal of Steroid Biochemistry and Molecular Biology. 1999;68:189–195. doi: 10.1016/s0960-0760(99)00030-8. [DOI] [PubMed] [Google Scholar]
- 62.Zhu YS, Sun GH. 5α-reductase isozymes in the prostate. Journal of Medical Sciences. 2005;25(1):1–12. [PMC free article] [PubMed] [Google Scholar]
- 63.Roehrborn CG, Marks L, Harkaway R. Enlarged prostate: a landmark national survey of its prevalence and impact on US men and their partners. Prostate Cancer and Prostatic Diseases. 2006;9(1):30–34. doi: 10.1038/sj.pcan.4500841. [DOI] [PubMed] [Google Scholar]
- 64.Huggins C, Stevens R. The effect of castration on benign hypertrophy of the prostate in man. Journal of Urology. 1940;43:705–714. doi: 10.1016/j.juro.2016.10.075. [DOI] [PubMed] [Google Scholar]
- 65.Moore RJ, Gazak JM, Quebbeman JF, Wilson JD. Concentration of dihydrotestosterone and 3α-androstanediol in naturally occurring and androgen-induced prostatic hyperplasia in the dog. Journal of Clinical Investigation. 1979;64(4):1003–1010. doi: 10.1172/JCI109536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wenderoth UK, George FW, Wilson JD. The effect of a 5α-reductase inhibitor on androgen-mediated growth of the dog prostate. Endocrinology. 1983;113(2):569–573. doi: 10.1210/endo-113-2-569. [DOI] [PubMed] [Google Scholar]
- 67.Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. The New England Journal of Medicine. 2003;349(3):215–224. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 68.Andriole GL, Bostwick DG, Brawley OW, et al. Effect of dutasteride on the risk of prostate cancer. The New England Journal of Medicine. 2010;362(13):1192–1202. doi: 10.1056/NEJMoa0908127. [DOI] [PubMed] [Google Scholar]
- 69.Nickel J, Gilling P, Tammela T, et al. Comparison of dutasteride and finasteride for treating benign prostate hyperplasia: the enlarged prostate international comparator study (EPICS) BJU International. 2011;108(3):388–394. doi: 10.1111/j.1464-410X.2011.10195.x. [DOI] [PubMed] [Google Scholar]
- 70.Andriole G, Lieber M, Smith J, et al. Treatment with finasteride following radical prostatectomy for prostate cancer. Urology. 1995;45(3):491–497. doi: 10.1016/s0090-4295(99)80021-1. [DOI] [PubMed] [Google Scholar]
- 71.Perotti M, Jain R, Abriel L, et al. Dutasteride momotherapy in men with serologic relapse following radical therapy for adenocarcinoma of the prostate: a pilot study. doi: 10.1016/j.urolonc.2010.01.004. Urologic Oncology: Seminars and Original Investigations. In press. [DOI] [PubMed] [Google Scholar]
- 72.Barqawi AB, Moul JW, Ziada A, Handel L, Crawford ED. Combination of low-dose flutamide and finasteride for PSA-only recurrent prostate cancer after primary therapy. Urology. 2003;62(5):872–876. doi: 10.1016/s0090-4295(03)00667-8. [DOI] [PubMed] [Google Scholar]
- 73.Bañez LL, Blake GW, McLeod DG, Crawford ED, Moul JW. Combined low-dose flutamide plus finasteride vs low-dose flutamide monotherapy for recurrent prostate cancer: a comparative analysis of two phase II trials with a long-term follow-up. BJU International. 2009;104(3):310–314. doi: 10.1111/j.1464-410X.2009.08400.x. [DOI] [PubMed] [Google Scholar]
- 74.Mohler JL, Gregory CW, Ford H, et al. The androgen axis in recurrent prostate cancer. Clinical Cancer Research. 2004;10(2):440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 75.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clinical Cancer Research. 2005;11(13):4653–4657. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 76.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Research. 2006;66(5):2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 77.Attard G, Reid AHM, Yap TA, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. Journal of Clinical Oncology. 2008;26(28):4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 78.Mohler J, Titus M, Bai S, et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Research. 2011;71(4):1486–1496. doi: 10.1158/0008-5472.CAN-10-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Locke JA, Guns ES, Lubik AA, et al. Androgen Levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Research. 2008;68(15):6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 80.Ford OH, III, Gregory CW, Kim D, Smitherman AB, Mohler JL. Androgen receptor gene amplification and protein expression in recurrent prostate cancer. Journal of Urology. 2003;170(5):1817–1821. doi: 10.1097/01.ju.0000091873.09677.f4. [DOI] [PubMed] [Google Scholar]
- 81.Gregory CW, Johnson RT, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Research. 2001;61(7):2892–2898. [PubMed] [Google Scholar]
- 82.Agoulnik IU, Vaid A, Nakka M, et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Research. 2006;66(21):10594–10602. doi: 10.1158/0008-5472.CAN-06-1023. [DOI] [PubMed] [Google Scholar]
- 83.Guo Z, Dai B, Jiang T, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10(4):309–319. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 84.Taplin ME, Bubley GJ, Ko YJ, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Research. 1999;59(11):2511–2515. [PubMed] [Google Scholar]
- 85.Scher HI, Anand A, Rathkopf D, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. The Lancet. 2010;375(9724):1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shah SK, Trump DL, Sartor O, Tan W, Wilding GE, Mohler JL. Phase II study of dutasteride for recurrent prostate cancer during androgen deprivation therapy. Journal of Urology. 2009;181(2):621–626. doi: 10.1016/j.juro.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tay MH, Kaufman DS, Regan MM, et al. Finasteride and bicalutamide as primary hormonal therapy in patients with advanced adenocarcinoma of the prostate. Annals of Oncology. 2004;15(6):974–978. doi: 10.1093/annonc/mdh221. [DOI] [PubMed] [Google Scholar]
- 88.Sartor O, Nakabayashi M, Taplin ME, et al. Activity of dutasteride plus ketoconazole in castration-refractory prostate cancer after progression on ketoconazole alone. Clinical Genitourinary Cancer. 2009;7(3):E90–E92. doi: 10.3816/CGC.2009.n.030. [DOI] [PubMed] [Google Scholar]
- 89.Taplin ME, Regan MM, Ko YJ, et al. Phase II study of androgen synthesis inhibition with ketoconazole, hydrocortisone, and dutasteride in asymptomatic castration-resistant prostate cancer. Clinical Cancer Research. 2009;15(22):7099–7105. doi: 10.1158/1078-0432.CCR-09-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee AT, Zane LT. Dermatologic manifestations of polycystic ovary syndrome. American Journal of Clinical Dermatology. 2007;8(4):201–219. doi: 10.2165/00128071-200708040-00003. [DOI] [PubMed] [Google Scholar]
- 91.Falsetti L, Gambera A, Andrico S, et al. Acne and hirsutism in polycystic ovary syndrome: clinical, endocrine-metabolic and ultrasonographic differences. Gynecological Endocrinology. 2002;16(275):27–84. doi: 10.1080/gye.16.4.275.284. [DOI] [PubMed] [Google Scholar]
- 92.Somani N, Harrison S, Bergfeld WF. The clinical evaluation of hirsutism. Dermatologic Therapy. 2008;21(5):376–391. doi: 10.1111/j.1529-8019.2008.00219.x. [DOI] [PubMed] [Google Scholar]
- 93.Essah PA, Wickham EP, Nunley JR, Nestler JE. Dermatology of androgen-related disorders. Clinics in Dermatology. 2006;24(4):289–298. doi: 10.1016/j.clindermatol.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 94.Rathnayake D, Sinclair R. Male androgenetic alopecia. Expert Opinion on Pharmacotherapy. 2010;11(8):1295–1304. doi: 10.1517/14656561003752730. [DOI] [PubMed] [Google Scholar]
- 95.Thiboutot D. Acne: hormonal concepts and therapy. Clinics in Dermatology. 2004;22(5):419–428. doi: 10.1016/j.clindermatol.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 96.Drake L, Hordinsky M, Fiedler V, et al. The effects of finasteride on scalp skin and serum androgen levels in men with androgenetic alopecia. Journal of the American Academy of Dermatology. 1999;41(4):550–554. [PubMed] [Google Scholar]
- 97.Olsen EA, Hordinsky M, Whiting D, et al. The importance of dual 5α-reductase inhibition in the treatment of male pattern hair loss: results of a randomized placebo-controlled study of dutasteride versus finasteride. Journal of the American Academy of Dermatology. 2006;55(6):1014–1023. doi: 10.1016/j.jaad.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 98.Kaufman KD, Olsen EA, Whiting D, et al. Finasteride in the treatment of men with androgenetic alopecia. Journal of the American Academy of Dermatology. 1998;39(4):578–589. doi: 10.1016/s0190-9622(98)70007-6. [DOI] [PubMed] [Google Scholar]
- 99.Eun HC, Kwon OS, Yeon JH, et al. Efficacy, safety, and tolerability of dutasteride 0.5 mg once daily in male patients with male pattern hair loss: a randomized, double-blind, placebo-controlled, phase III study. Journal of the American Academy of Dermatology. 2010;63(2):252–258. doi: 10.1016/j.jaad.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 100.Arca E, Acikgoz G, Tastan HB, et al. An open, randomized, comparative study of oral finasteride and 5% topical minoxidil in male androgenetic alopecia. Dermatology. 2004;209(2):117–125. doi: 10.1159/000079595. [DOI] [PubMed] [Google Scholar]
- 101.Shum KW, Cullen DR, Messenger AG. Hair loss in women with hyperandrogenism: four cases responding to finasteride. Journal of the American Academy of Dermatology. 2002;47(5):733–739. doi: 10.1067/mjd.2002.124608. [DOI] [PubMed] [Google Scholar]
- 102.Olszewska M, Rudnicka L. Effective treatment of female androgenic alopecia with dutasteride. Journal of Drugs in Dermatology. 2005;4(5):637–640. [PubMed] [Google Scholar]
- 103.Moghetti P, Tosi F, Tosti A, et al. Comparison of spironolactone, flutamide, and finasteride efficacy in the treatment of hirsutism: a randomized, double blind, placebo-controlled trial. Journal of Clinical Endocrinology and Metabolism. 2000;85(1):89–94. doi: 10.1210/jcem.85.1.6245. [DOI] [PubMed] [Google Scholar]
- 104.Falsetti L, Gambera A, Legrenzi L, Iacobello C, Bugari G. Comparison of finasteride versus flutamide in the treatment of hirsutism. European Journal of Endocrinology. 1999;141(4):361–367. doi: 10.1530/eje.0.1410361. [DOI] [PubMed] [Google Scholar]
- 105.Schwartz JI, Tanaka WK, Wang DZ, et al. MK-386, an inhibitor of 5α-reductase type 1, reduces dihydrotestosterone concentrations in serum and sebum without affecting dihydrotestosterone concentrations in semen. Journal of Clinical Endocrinology and Metabolism. 1997;82(5):1373–1377. doi: 10.1210/jcem.82.5.3912. [DOI] [PubMed] [Google Scholar]
- 106.Leyden J, Bergfeld W, Drake L, et al. A systemic type I 5 α-reductase inhibitor is ineffective in the treatment of acne vulgaris. Journal of the American Academy of Dermatology. 2004;50(3):443–447. doi: 10.1016/j.jaad.2003.07.021. [DOI] [PubMed] [Google Scholar]
- 107.Carmina E, Lobo RA. A comparison of the relative efficacy of antiandrogens for the treatment of acne in hyperandrogenic women. Clinical Endocrinology. 2002;57(2):231–234. doi: 10.1046/j.1365-2265.2002.01594.x. [DOI] [PubMed] [Google Scholar]
- 108. http://www.clinicaltrials.gov/