

Abstract
Mitochondrial dysfunction is a pathogenic feature of non-alcoholic steatohepatitis (NASH). NASH complicates hepatotropic viral disease. The mitochondrial antiviral signaling protein, (MAVS) is the adapter of helicase receptors involved in sensing dsRNA. We hypothesized that impaired MAVS function may contribute to insufficient anti-viral response and liver damage in steatohepatitis. We identified reduced MAVS protein levels and increased MAVS association with the proteosomal PSMA7 in livers from mice after methionine-choline-deficient (MCD) diet. Decreased association of MAVS with mitochondria and increased cytosolic cytochrome-c indicated mitochondrial damage in steatohepatitis. In vivo administration of the synthetic dsRNA, polyI;C, but not LPS or CpG DNA, resulted in impaired induction of Type-I IFNs and proinflammatory cytokines in steatohepatitis. Consistent with a defect in helicase receptor-induced signaling, there was loss of poly I:C induced translocation of MAVS to the cytosol and decreased IRF3 phosphorylation. Both caspase-1 and -8, that cleave MAVS, were increased in MCD diet-fed mice. At baseline, steatohepatitis was associated with increased serum ALT, apoptosis and caspase-3 activation compared to controls. In contrast to apoptosis in controls, necrosis was induced by poly I:C stimulation in steatohepatitis. Hepatocyte necrosis was indicated by elevated serum HMGB1 and ALT and it correlated with increased expression of RIP3, a master regulator of necrosis. Increased expression of MAVS, PSMA7 and RIP3 mRNA was also present in human NASH livers.
Conclusions
Our novel findings suggest that mitochondrial damage in steatohepatitis extends to MAVS, an adapter of helicase receptors, resulting in inefficient Type-I IFN and inflammatory cytokine response but increased hepatocyte necrosis and RIP3 induction in response to a dsRNA viral challenge. These mechanisms may contribute to progressive liver damage and impaired viral clearance in NASH.
Keywords: IPS, VISA, apoptosis, RIP3, caspase, dsRNA
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most rapidly increasing cause of liver disease in the western world (1). The spectrum of NAFLD spans from steatosis to nonalcoholic steatohepatitis (NASH) that can lead to cirrhosis and hepatocellular cancer (1). While the factors determining progression of NASH are yet to be fully defined, the clinical importance of increased susceptibility of the fatty liver to ischemia (2), bacterial lipopolysaccharide (LPS) (3), viral infections (1) and drug-induced (4) liver damage is emerging.
Co-morbidity of NASH with viral infections caused by RNA viruses, such as hepatitis C and HIV remains a clinical challenge (1). HCV-infected patients with significant steatosis or superimposed NASH have rapid progression of liver disease, increased rate of fibrosis, and a decreased likelihood of sustained virological response (SVR) to standard antiviral therapy (5). In human immunodeficiency virus (HIV) infection highly active anti-retroviral therapy (HAART) induces extensive alterations to liver lipid metabolism, including liver damage and sometimes even liver failure (6). Fatty liver also complicates viral infections, such as hepatitis A virus (HAV)-, cytomegalovirus (CMV)-, or Epstein-Barr virus (EBV) resulting in “acute on chronic” liver failure.
HCV, a single stranded RNA virus undergoes dsRNA phase during viral replication (7). The innate immune response to dsRNA is triggered through TLR3 and the helicase receptors, RIG-I (retinoic acid-inducible gene-I) and Mda5 (melanoma-differentiation-associated gene5) and induces Type-I IFN and pro-inflammatory cytokine production. The adapter, MAVS, (also called VISA (virus-induced signaling adaptor) or IPS-1 (IFNβ promoter stimulator protein-1) is associated with the outer membrane of mitochondria and it is critical in both IFN and inflammatory cytokine induction after engagement of helicases by dsRNA. (8,9) Multiple “hits” have been proposed in the pathogenesis of NASH, including hepatocyte death as a result of toxic lipid metabolites and pathogen-derived factors that contribute to inflammation and liver damage (10). Indeed, necro-inflammation is considered as an indicator of NASH progression (1). There is evidence that mitochondrial damage contributes to apoptotic/necrotic cellular damage in NASH (11), but the role of MAVS in NASH and in response to pathogenic RNA is yet to be evaluated.
In this study, we hypothesized that steatohepatitis results in decreased antiviral immunity and increased susceptibility to liver damage in response to a viral challenge. Using the diet-induced NASH model in mice and poly I:C, a synthetic double-stranded RNA sensed by the helicase receptors, we show that fatty livers fail to mount an efficient antiviral IFN response due to dissociation of MAVS from the mitochondria. We also found increased liver damage in steatohepatitis after poly I:C challenge that was associated with RIP3 over-expression and necrosis. Our data offer novel mechanistic insights into the decreased antiviral immune defense in steatohepatitis and indicate that impaired response to virus-derived factors may promote RNA virus-induced liver injury in NASH.
METHODS
Animal studies
Six-eight week-old female C57Bl/6 wild type (wt) mice were fed with methionine-choline-deficient (MCD) diet for five weeks; controls received a DL-methionine (3 g/kg) and choline bitartrate (2 g/kg) supplemented (MCS) diet (Dyets Inc., Bethlehem, PA, USA). Polyinosinic:polycytidylic acid (Poly I:C) (InvivoGen, San Diego, CA, USA), a synthetic double stranded RNA (5mg/bwkg); or CpG-ODN (InvivoGen, San Diego, CA, USA), (5mg/bwkg); or lipopolysaccharyde (LPS) (Sigma-Aldrich Co., St. Louis, MO, USA) (0.5mg/bwkg) were injected inrtaperitoneally for 2 or 6 hours. This study was approved by Institutional Animal Use and Care Committee at UMass.
Biochemical analysis and cytokine measurements
Serum alanine aminotransferase (ALT) was determined using a kinetic method (D-TEK, Bensalem, PA, USA), liver triglyceride levels assessed using the L-Type Triglyceride H kit (Wako Chemicals USA Inc., VA, USA). Serum cytokine levels were determined by BD™ Cytometric Bead Array (BD Biosciences, Sparks, MD, USA). Liver thiobarbituric acid reactive substances (TBARS) were assayed using whole liver homogenates and Oxi-TEK TBARS assay kit (ZeptoMetrix Corp., Buffalo, NY, USA). Serum HMGB1 protein levels were measured by ELISA (IBL Transatlantic, Toronto, Canada).
Histopathological analysis
Sections of formalin-fixed livers were stained with hematoxylin-eosin; all slides were analyzed by microscopy.
RNA analysis
RNA was purified using the RNeasy kit (Qiagen Sciences, Maryland, USA) and on-column DNA digestion. cDNA was transcribed with the Reverse Transcription System (Promega Corp., Madison, WI). Real-time quantitative polymerase chain reaction was performed using the iCycler (Bio-Rad Laboratories Inc., Hercules, CA), as described previously (12); primer sequences are shown in Table 1.
Mitochondrial extraction
Isolation of mitochondrial and cytosolic fraction from fresh liver tissue was based on the principle of differential centrifugation using Mitochondrial Extraction kit (Imgenex Co., San Diego, CA, USA).
Western blot
Whole liver lysates or mitochondrial fractions were extracted and Western blot performed, as previously described (12). The following antibodies were employed: MAVS (Santa Cruz Biotechnology Inc., sc-6881), cytochrome c (Imgenex, IMG101-A), caspase-1 p10 (Santa Cruz Biotechnology Inc., sc-514), cleaved caspase-8 (Imgenex, IMG5703), RIP3 (Abcam, ab72106), β-actin (Abcam, ab6276), β-tubulin (Abcam, ab6046), Tim23 (BD Biosciences).
Native gel electrophoresis
Native PAGE Novex Bis-Tris Gel System (Invitrogen Life Science, Carlsbad, CA, USA) was used. Liver samples were lysed using 5% Digitonin as mild detergent and separated on Native PAGE Novex 3–12% Bis-Tris Gels. Proteins were transfered to PVDF membrane, fixed with 8% acetic acid and identified with specific primary antibodies followed by HRP labeled secondary antibodies and chemiluminescence assay.
Immunoprecipitation
Whole liver lysates were precleared with anti-rabbit IgG beads followed by overnight incubation with 5ug of the primary antibody (PSMA7 or MAVS) and precipitated with IgG beads. The immunprecipitates were lysed and denatured using β-mercaptoethanol containing buffer and heating. The proteins were separated on polyacrylamid gel, transferred to nitrocellulose membrane and detected by specific antibodies (MAVS, PSMA7).
Human liver samples
Human liver tissue was obtained from biopsies from clinically and biopsy-proven NASH patients without fibrosis and from patients with chronic B hepatitis. Liver samples were frozen immediately and kept in liquid nitrogen before RNA extraction. RNA was extracted as above. The study was approved by the Committee for the Protection of Human Subjects in Research at the University of Massachusetts. Human normal liver and liver tumor total RNA were purchased from OriGene Technologies (Rockville, MD, USA)
Statistical analysis
Statistical significance was determined using the nonparametric Kruskal-Wallis test and Mann-Whitney tests. Data are shown as mean±standard error and were considered statistically significant at p<0.05.
RESULTS
Type-I IFN induction is decreased in steatohepatitis in response to poly I:C stimulation
Polyinosinic-polycytidylic acid (poly I:C), a synthetic double-stranded RNA (dsRNA), is a surrogate for viral infection (13). Double stranded RNA is recognized by TLR3 and helicase receptors and induces robust Type-I IFN response leading to anti-viral immunity (14). Antiviral responses to RNA are important in HCV and HIV infection (6,7). Here we show for the first time that poly I:C-induced Type-I interferon production is significantly decreased in mice with steatohepatitis (Figure 1). We found decreased serum protein (Figure 1A) and liver mRNA levels (Figure 1B) of IFNβ and IFNα4 (Figure 1C) in MCD compared to MCS diet-fed control mice. Consistent with impaired Type-I IFN production after poly I:C stimulation, induction of interferon-inducible genes ISG56 (Figure 1D) and ISG15 (Figure 1E), was also significantly decreased in MCD diet-induced steatohepatitis. These results suggested that steatohepatitis results in impaired Type-I IFN response to dsRNA viral challenge.
Figure 1. MCD diet attenuates Type-I interferon (IFN) production in response to poly I:C.
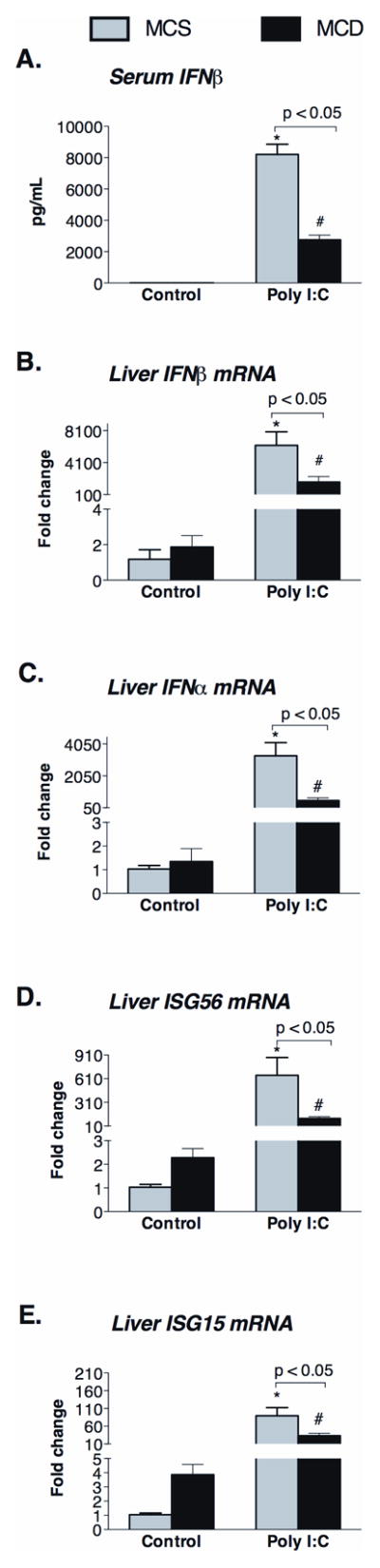
Serum IFNβ (A) and liver mRNA of IFNβ (B) and IFNα (C) and IFN-inducible genes (ISG56 (D), ISG15 (E)) were determined in C57Bl/6 MCD diet-fed mice and compared to control MCS diet-fed mice. Data are shown at baseline and 2 hours after poly I:C challenge. N=4–6 mice/group, (*) indicates p<0.05 vs. MCS baseline, (#) indicates p<0.05 vs. MCD baseline.
Impaired Type-I IFN induction in steatohepatitis is restricted to the RIG-I/Mda5 pathway
To further evaluate the significance of impaired Type-I IFN induction in steatohepatitis, we employed stimulations that induce Type-I IFNs via receptor pathways different from dsRNA recognition by TLR3 and its adapter, TRIF, or RIG-I/Mda5 and their adapter MAVS, respectively (14). LPS is recognized by TLR4 and uses the adapters TRIF and MyD88, while CpG DNA, a ligand for TLR9 solely utilizes the MyD88 adapter in Type-I IFN induction (14).
We found increased TLR3, Mda5, RIG-I, as well as their corresponding adapters, TRIF and MAVS at the mRNA levels in fatty livers compared to controls (Figure 2A). In contrast to polyI:C, challenge with a TLR4-ligand (LPS), which uses TRIF, or a TLR9-ligand (CpG DNA), which uses MyD88, resulted in increased Type-I IFN induction in MCD compared to MCS diet-fed mice (Figure 2B, C, D). TRIF serves as sole adapter for poly I:C-engaged TLR3 and it also mediates TLR4/LPS-induced Type-I IFN production (14). TRIF deficient mice were shown to be defective in both TLR3 and TLR4 mediated IRF3 activation (15). These data suggested a selective impairment of Type-I IFN induction upon dsRNA viral (poly I:C) challenge in a TLR3/TRIF-independent manner; we thus focused on dissecting the role of the helicase RNA-sensing pathways in steatohepatitis.
Figure 2. MCD diet upregulates expression of helicase receptors (Mda5/RIG-I), TLR3 and their adaptors (MAVS and TRIF) in the liver.
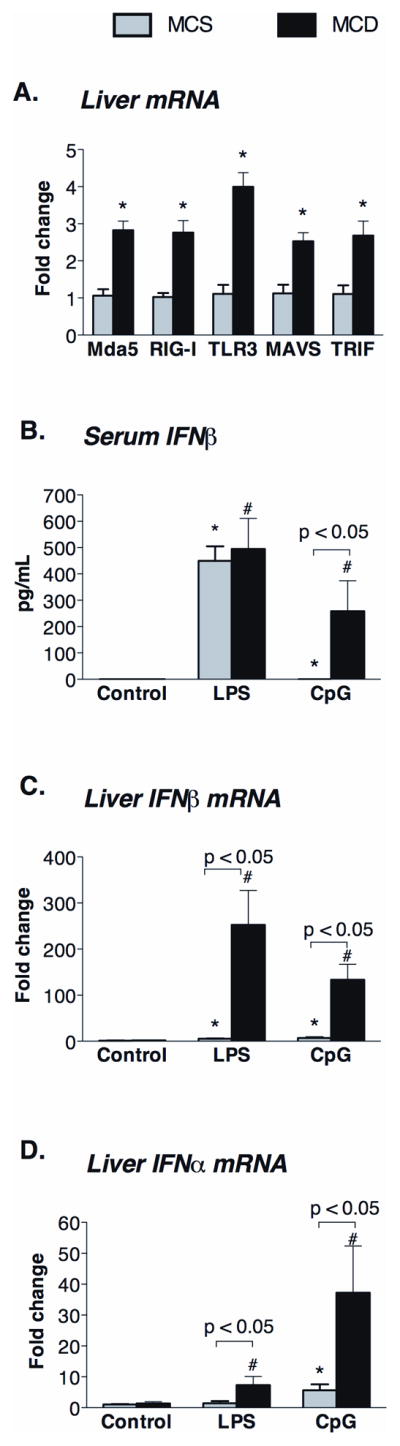
(A) The mRNA expression of poly I:C (dsRNA) sensing receptors, namely Mda5, RIG-I, TLR3 and their adaptor molecules, MAVS and TRIF, respectively, were measured by qPCR in the liver of MCD and MCS diet-fed mice. Serum IFNβ (B) and liver mRNA of IFNβ (C) and IFNα (D) were determined in C57Bl/6 MCD diet-fed mice 2 hours after LPS (0.5mg/bwkg) or CpG (5mg/bwkg) ip. injection and compared to control MCS diet-fed mice. N=4–6 mice/group, (*) indicates p<0.05 vs. MCS baseline, (#) indicates p<0.05 vs. MCD baseline.
Abnormal MAVS function in NASH involves decreased protein levels, dissociation from the mitochondria and impaired oligomerization
The adapter molecule MAVS is critical for the downstream signaling of helicase receptors and its dysfunction impairs proinflammatory cytokine and interferon induction via the NFκB and IRF3 signaling pathways, respectively (8). Consistent with decreased induction of Type-I IFN, we found decreased levels of MAVS protein in whole liver lysates of MCD-diet fed mice compared to controls (Figure 3A). In search of possible mechanisms for decreased MAVS protein levels, we found higher mRNA expression of the PSMA7 subunit of proteasome in MCD steatohepatitis (Figure 3B). PSMA7 can negatively regulate MAVS-mediated immune responses and promotes proteosomal degradation (16). Immunoprecipitation experiments revealed increased association between MAVS and PSMA7 in fatty livers compared to controls (Figure 3C).
Figure 3. Decreased MAVS protein levels and increased MAVS association with proteasome protein PSMA7 in MCD-steatohepatitis.
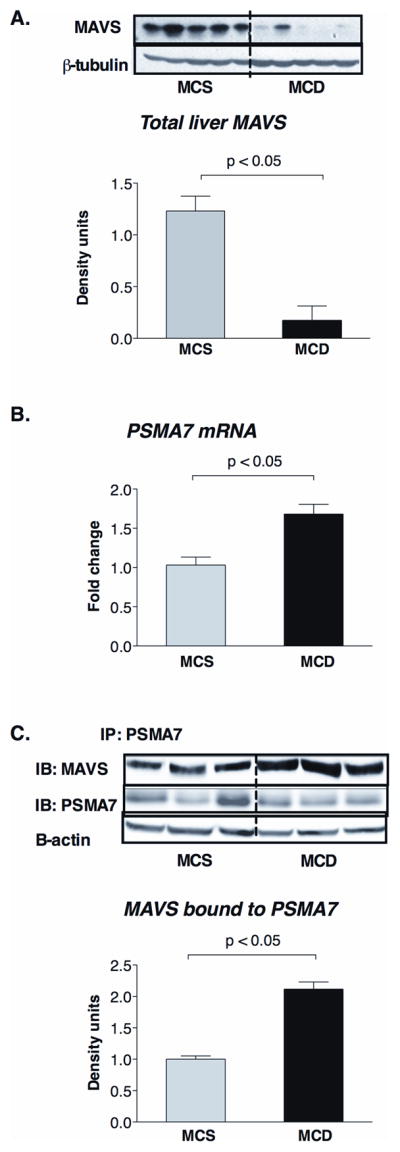
C57Bl/6 mice were fed with methionine-choline-deficient (MCD) or –supplemented (MCS) diet for 5 weeks. MAVS protein expression was analyzed by Western blot in liver whole cell lysates (A); β-tubulin was used as loading control (A). Hepatic mRNA expression of PSMA7 was measured by qPCR (B). PSMA7 associated MAVS expression was evaluated by immunoprecipitation in whole liver lysates using PSMA7 antibody for immunoprecipitating and MAVS antibody for immunoblotting (C). N=4–6 mice/group, (*) indicates p<0.05 vs. MCS.
The localization of MAVS to the outer mitochondrial membrane is crucial for Mda5/RIG-I activation (9). However, we found that steatohepatitis resulted in decreased mitochondria-associated MAVS protein levels compared to controls (Figure 4A). We also observed a corresponding increase in cytosolic MAVS protein levels in MCD compared to the MCS-diet fed livers (Figure 4B). The purity of the mitochondrial and cytosolic preparations was confirmed by the expression of mitochondrial marker Tim23 (Figure 4A) and cytosolic β-tubulin (Figure 4B), respectively. The ratio of the cytoplasmic/mitochondrial MAVS was significantly higher in MCD-steatohepatitis (Figure 4C). These results indicated that displacement of MAVS protein from the mitochondria to the cytosol is likely related to mitochondrial damage in steatohepatitis. The transmembrane domain (TM) of MAVS is crucial for mitochondrial localization and also for dimerization of MAVS that is required for downstream signaling (9,17). We found that in addition to impaired mitochondrial localization, there was decreased oligomerization of MAVS in steatohepatitis compared to controls (Figure 4D).
Figure 4. MAVS is dissociated from the mitochondria in MCD diet induced steatohepatitis.

C57Bl/6 mice were fed with methionine-choline-deficient (MCD) or –supplemented (MCS) diet for 5 weeks and injected with poly I:C (5mg/bwkg) intraperitoneally for 2 hours. MAVS protein expression was analysed by Western blot in liver mitochondrial (A) and cytoplasmic extract (B); tim23 was used as loading control in mitochondrial extract, while β-tubulin was used as loading control in the cytoplasmic extract (A, B, respectively). The ratio of cytoplasmic/mitochondrial MAVS protein levels is shown at panel C. MAVS oligomerization was analyzed on native gel electrophoresis (D). IRF3 activation was evaluated by detection of phosphoIRF3 by Western blot in liver whole cell lysates. N=4–6 mice/group, (*) indicates p<0.05 vs. MCS baseline, (#) indicates p<0.05 vs. MCD baseline.
Given the defects in poly I:C-triggered interferon induction in steatohepatitis (Figure 1), we next explored the function of the MAVS adapter protein. In control mice, poly I:C administration resulted in displacement of MAVS from the mitochondria to the cytosol (Figure 4A, 4B). In contrast, there was no increase in cytoplasmic MAVS translocation after poly I:C stimulation in livers of MCD diet-fed mice (Figure 4A, 4B). PolyI:C-induced engagement of helicases and signaling through MAVS results in downstream activation and phosphorylation of IRF3 (14). In livers of MCD diet-fed mice, impaired MAVS function and decreased mitochondrial association was associated with significantly reduced IRF3 phosphorylation after poly I:C stimulation (Figure 4E). These data suggested that decreased association of MAVS with mitochondria at baseline may impair downstream signaling in stetohepatitis.
Mitochondrial damage occurs in the fatty liver
Mitochondrial dysfunction plays a role in the pathogenesis of NASH (18) and upon mitochondrial damage, its content leaks into the cytosol triggering diverse signaling pathways, including apoptosis (19). Thus, we hypothesized that decreased association of MAVS with mitochondria may be linked to mitochondrial damage in NASH. Indeed, mitochondrial damage was indicated by relocation of cytochrome C from the mitochondria to the cytoplasm (Figure 5A), and by enrichment of the mitochondria with β-actin (Figure 5B) in livers of MCD compared to MCS diet-fed mice. We further identified evidence for increased cellular damage pathways in steatohepatitis as indicated by caspase 8 (Figure 5C) and caspase 1 (Figure 5D) activation. Relevant to our observation of decreased MAVS in steatohepatitis, both caspase 8 and caspase 1 were shown to cleave MAVS from the mirochondria (20,21,22).
Figure 5. MCD steatohepatitis causes mitochondrial damage and caspase-activation.

Cytochrome c (A) and β-actin (B) protein expressions were analyzed by Western blot in liver mitochondrial and cytoplasmic extract of C57Bl/6 MCD diet-fed mice and compared to control MCS diet-fed mice. Lanes run on different gels are separated by vertical white line. The activation of apoptotic caspase-8 (C) and inflammatory caspase-1 (pro-caspase and p10 subunit) (D) were determined by Western blot analysis in liver whole cell lysates using specific antibodies. Tim23 was used as loading control in the mitochondrial fraction (A,B), while β-tubulin in the cytoplasmic fraction (C). Liver thiobarbituric acid (TBARs) levels were analyzed as indirect indicators of ROS production, n=5–6/group (E). (*) indicates p<0.05 vs. MCS baseline, (#) indicates p<0.05 vs. MCD baseline.
Mitochondrial damage in NASH has been linked to excessive levels of reactive oxygen species (ROS) (18). Indeed, we detected significantly increased liver TBARs levels indicating ROS-induced lipid peroxidation at baseline and after poly I:C stimulation in steatohepatitis (Figure 5E). These results indicated that ROS and lipid peroxidation occur in NASH, and their production is exacerbated in response to dsRNA stimulation.
Increased poly I:C-induced liver damage occurs without excessive pro-inflammatory cytokine induction in steatohepatitis
Liver damage, indicated by steatosis and elevated ALT, is a hallmark of steatohepatitis. Here we found that a poly I:C challenge significantly increased liver injury in MCD diet-fed mice indicated by tissue hemorrhage, hepatocyte degeneration (Figure 6A), and significantly increased serum ALT levels compared to MCS controls mice (Figure 6B). Because dsRNA-induced activation of RIG-I and Mda5 leads to Type-I IFN induction as well as activation of NFκB and production of pro-inflammatory cytokines (14), we sought to evaluate whether the increased liver damage was the consequence of enhanced pro-inflammatory cytokine production in steatohepatitis. At baseline, MCD diet-fed mice showed increased serum (Figure 6C) and liver mRNA levels (Figure 6D) of TNFα, IL-6 and IL-1β compared to MCS controls. While poly I:C challenge increased TNFα, IL-6 and IL-1β production both in controls and MCD-diet fed groups (Figure 6C, 6D), the extent of pro-inflammatory cytokine protein (Figure 6C) and mRNA (Figure 6D) induction was significantly lower in MCD compared to MCS diet-fed mice. These data demonstrated that pro-inflammatory cytokine induction was impaired in response to a dsRNA challenge, and thus, it is less likely to account for the increased liver damage in NASH.
Figure 6. Steatohepatitis increases poly I:C-induced liver damage without induction of NK cell activating ligands and attenuates induction of pro-inflammatory cytokines.

C57Bl/6 mice received a methionine-choline-deficient (MCD) or –supplemented (MCS) diet for 5 weeks and then were injected with poly I:C (5mg/bwkg) intraperitoneally for 2 or 6 hours. Representative sections of formalin-fixed, paraffin-embedded livers stained with hematoxilin-eosin (200 fold magnification) are shown at baseline and 6 hrs after poly I:C injection (A). Serum ALT (B), serum protein (C) and liver mRNA (D) of cytokines (TNFα, IL-6, IL-1β) are shown as mean±SEM values at baseline and 2 hours after poly I:C challenge. Liver mRNA expression of NK cell activating ligands was analysed by qPCR (F). N=4–6 mice/group, (*) indicates p<0.05 vs. MCS baseline, (#) indicates p<0.05 vs. MCD baseline.
Since, previous studies showed a crucial role for NK cells in poly I:C induced liver injury (23), and higher NK cell activating ligand expression has been reported in livers of NASH patients (24), we next investigated the possible role of NK cells. We found increased mRNA expression of the NK-activating ligands, Pan-Rae, Rae-1α and Mult-1 in MCD-steatohepatitis (Figure 6E), but poly I:C did not induce a further increase in the expression of these ligands (Figure 6E). Furthermore, we found that recruitment of NK cells after poly I:C stimulation occurred in control livers and not in steatohepatitis (Supplementary figure 1).
Poly I:C promotes a switch from apoptosis to necrosis and increases RIP3 expression in steatohepatitis
Hepatocyte apoptosis in NASH has been linked to increased susceptibility of the fatty liver to LPS challenge while hepatocyte necrosis is associated with progressive liver damage (3). There is recent evidence that the mitochondria-associated MAVS can regulate apoptosis in viral infection (25). Apoptosis is triggered via intrinsic (involving pro-apoptotic protein Bim, mitochondria, cytochrome c, and caspase 9), or via extrinsic (involving death receptors including TRAIL) pathways that connect at the level of caspase 3 to culminate in cell death. We found increased expression of TRAIL (extrinsic apoptosis) (Supplementary figure 2A) and Bim (intrinsic apoptosis) (Supplementary figure 2B) in livers of MCD diet-fed mice. Expression of caspase-3 was also induced in MCD vs. MCS diet-fed mice (Figure 7A). Here we found that caspase-3 activity was significantly increased by poly I:C in normal (MCS) livers (Figure 7A), but not in steatohepatitis (MCD) (Figure 7A). There were no differences in the extent of poly I:C-induced upregulation of TRAIL and Bim mRNA expression (Supplementary figure 2A, 2B), between MCD and MCS livers indicating that steatotic livers, while exhibit higher apoptosis at baseline, fail to progress to tissue death by apoptosis upon a viral challenge. Notably, the tissue damage was higher in poly I:C-challenged steatotic livers compared to controls (Figure 6B). Thus, we hypothesized that the increased poly I:C-induced liver damage in MCD diet-fed mice was due to necrosis rather than apoptosis. Indeed, we identified increased levels of serum HMGB1 (Figure 7B), a marker of necrosis, in the poly I:C-stimulated MCD group compared to controls.
Figure 7. Poly I:C induces necrosis in MCD-steatohepatitis possibly due to the increased RIP3 expression and ROS production.

Caspase-3 activity was analyzed using a colorimetric assay in the liver of MCD diet-fed mice with or without poly I:C stimulation compared to MCS controls (A). Serum HMGB1 levels were measured by ELISA before and after poly I:C stimulation (B). Liver mRNA (C) and protein (D, top-representative blot and bottom- densitometric analysis) levels of RIP3 were analyzed in whole liver. β-tubulin was used as loading control in the Western blot (D); n=5–6/group. Correlation between HMGB1 and RIP3 mRNA in the liver is shown (E). (*) indicates p<0.05 vs. MCS baseline, (#) indicates p<0.05 vs. MCD baseline.
The balance between apoptosis and necrosis is tightly regulated (26). A recently identified master regulator between apoptosis and necrosis is the protein kinase receptor-interacting protein 3 (RIP3) (26). We found increased levels of RIP3 mRNA (Figure C) and protein (Figure 7D) in livers of MCD- compared to MCS-diet-fed controls. In control mice poly I:C stimulation induced upregulation of RIP3 protein expression at 2 hours post-stimulation which returned to baseline by 6 hours (Figure 7D); in contrast, there was sustained induction of RIP3 in steatohepatitis after poly I:C challenge (Figure 7D). We further identified a positive correlation between RIP3 and liver HMGB1 (Figure 7E) expression. Collectively, these data suggested that pathways that promote necrosis are preferentially upregulated in steatohepatitis after a viral challenge, at least in part due to the regulatory involvement of RIP3.
Altered MAVS and RIP3 mRNA expression in human NASH
To validate our observations in the mouse model of steatohepatitis, we next evaluated human livers. We found an increase of MAVS mRNA levels in livers of NASH patients compared to healthy controls (Figure 8A) and this mirrored MAVS RNA levels in the animal model of steatohepatitis (Figure 2A). MAVS mRNA upregulation was specific to NASH since we did not observe increased MAVS levels in HBV-infection (HBV is a DNA virus) or in liver tumors (no viral infection detected) (Figure 8A). We also found higher expression of PSMA7 mRNA in human NASH livers (Figure 8B) that mirrored findings in the mouse model (Figure 3B). Finally, we detected highly increased RIP3 mRNA levels in NASH patients (Figure 8C) compared to controls; this was parallel to the RIP3 mRNA increase in the mouse model of NASH (Figure 7C).
Figure 8. Altered MAVS and RIP3 mRNA expression in human NASH.

The mRNA expression of MAVS (A), an adaptor of helicase receptors; PSMA7 (B), a proteasome subunit and RIP3 (C), a master regulator of apoptosis-necrosis switch were measured by qPCR in livers of NASH patients (n=6) and were compared to commercially available normal human liver RNA (n=4), HBV-infected patients (n=4) and commercially available RNA from liver tumor (n=1).
DISCUSSION
Steatosis and steatohepatitis are co-factors in the progression of liver diseases, including those of viral etiology, ischemia-reperfusion injury and liver transplantation (2,5). Here we report novel findings related to the impaired capacity of the fatty liver to respond to dsRNA and related viral challenges: First, livers with steatohepatitis failed to activate anti-viral innate immune pathways to produce Type I IFNs in response to a double-stranded RNA challenge. Second, the MAVS adapter, which is required for Type-I IFN induction after recognition of dsRNA by the helicase receptors RIG-I and Mda5, was dissociated from the mitochondria to the cytosol and showed impaired oligomerization and function in steatohepatitis. Third, displacement of MAVS from mitochondria was associated with oxidative stress and instead of upregulation of the apoptosis cascade, poly I:C promoted necrosis via increased expression of RIP3 in steatohepatitis. Fourth, dsRNA challenge resulted in increased liver damage in spite of decreased TNFα and pro-inflammatory cytokine induction in a diet-induced model of NASH.
Viral-sensing receptors include toll-like receptor (TLR) 3 and the cytoplasmic helicase receptors RIG-I and Mda5 for dsRNA recognition, TLR7/8 for ssRNA and TLR9 for sensing viral DNA (14). Here we identified a selective defect in signaling from viral dsRNA in steatohepatitis that altered both of pro-inflammatory cytokines and Type-I IFNs and was associated with increased liver damage. Although TLR3, Mda5 and RIG-I all sense poly I:C, their signaling pathways are different. Mda5 plays a key role in poly I:C-induced IFNβ production even in the absence of TLR3 or RIG-I (14). Ligand engagement of the helicase receptors catalyzes the phosphorylation of IκB proteins by IKK complex and leads to NFκB activation, along with the phosphorylation and activation of IRF3 (14). NFκB activation triggers the production of pro-inflammatory cytokines, while IRF3 phosphorylation leads to production of Type-I interferons (14).
The cellular source of the Type-I IFNs and inflammatory cytokines remains to be evaluated. Helicase receptors are expressed in several cell types in the liver, including hepatocytes, conventional dendritic cells, Kupffer-cells, NK cells (27,28). RLR expression is enhanced by poly I:C (28). We found that hepatocytes that represent the majority of cells in the liver produce IFNβ after intracellular poly I:C stimulation in vitro (data not shown). The RIG-I/Mda5 pathway is also important in the conventional dendritic cells (27) and NK cells (29), but less prominent in plasmacytoid dendritic cells. Thus, we speculate that hepatocytes and conventional DCs are the likely sources of Type-I IFN production after dsRNA challenge in the liver. Previous studies demonstrated a role of NK cells in NASH (24). Here we found evidence for increased expression of NK cell activating ligands, PanRae, Rae1α, Mult-1 in livers with steatohepatitis without a further increase after dsRNA stimulation. We also determined that NK cell recruitment was not triggered in livers with NASH suggesting that the liver damage was unlikely to be NK cell mediated after poly I:C challenge.
Here we demonstrated that both Type-I IFNs and pro-inflammatory cytokine induction were selectively disturbed in response to dsRNA while TLR4- or TLR9-mediated pathways remained intact in steatohepatitis. This suggested that the signaling defects in fatty livers occurred upstream from the branching of the NFκB and IRF3 signaling pathways and involved a protein that is common to both pathways upon dsRNA stimulation. The mitochondrial antiviral signaling protein (MAVS) mediates the activation of both NFκB and IRF3 in response to viral infection (8). Here we show for the first time that total liver MAVS protein levels are decreased in steatohepatitis. Our data showed increased association of MAVS with the proteasome subunit PSMA7 in MCD-steatohepatitis suggesting that proteosomal degradation could contribute to low MAVS levels. In this context, the apparent discrepancy between our finding of decreased MAVS protein and increased liver MAVS RNA could represent a compensatory feed-back loop mechanism. Increased mRNA levels of MAVS and PSMA7 were also present in human livers with NASH.
Impaired MAVS function was suggested by three of our novel observations. First, MAVS levels were decreased in the mitochondria with a complementary increase in the cytosol in the mouse model of steatohepatitis compared to controls. Second, in parallel with the MAVS dissociation from the mitochondria, we found decreased MAVS oligomerization in livers of MCD diet-fed mice compared to controls. Third, we found impaired induction of IRF3 phosphorylation by poly I:C in livers with steatohepatitis.
The transmembrane domain (TM) of MAVS is crucial to the mitochondrial localization of MAVS, but also required for the dimerization of the protein that is a crucial step during MAVS induced immune responses. (9,17) Our novel finding on the reduced MAVS oligomerization is in accordance with the impaired function of the helicase receptor-MAVS signaling pathway.
Mitochondrial dysfunction is a key component of fat accumulation, ROS generation and the progression of inflammation in NASH (18). Thus, it is plausible that translocation of MAVS from the mitochondria to the cytosol could be a consequence of mitochondrial damage in steatohepatitis. In addition to MAVS redistribution, we found other indications of mitochondrial damage, such as cytochrome C leak from the mitochondria to cytoplasm, enrichment of mitochondria with β-actin, and increased activation of cellular damage pathways. Translocation of β-actin to the mitochondria leading to disruption of mitochondrial membrane was shown in influenza virus-stimulated macrophages (30). We found markedly elevated β-actin protein levels in mitochondrial fractions in steatohepatitis providing evidence for mitochondrial damage in NASH. In normal hepatocytes, MAVS is localized in the outer mitochondrial membrane (9). Our novel data indicate increased activation of multiple caspases, including caspase 1 and caspase-8, in MCD diet-induced steatohepatitis suggesting a possible link between MAVS cleavage and caspase activation. Several viruses, including hepatitis C (NS3/4A protease) and hepatitis A (3ABC protease), disrupt the host antiviral response by cleaving MAVS from mitochondria (20,21). An apoptotic cleavage of MAVS has also been described (21). In NASH, both the death-receptor induced, and cellular-stress induced, apoptotic pathways are involved and apoptosis is indicated by increased caspase-3 activity and plasma cytokeratin-18 fragments (31,32). Studies showed that the pancaspase inhibitor, zVAD, prevents the cleavage of MAVS, while selective blockade of caspase-8, -9 or -3 was not sufficient to prevent MAVS cleavage (21). Relevant to our data, the pan-caspase inhibitor blocks both apoptotic caspases and caspase-1 (21,22). Thus, MAVS cleavage from the mitochondria in NASH is likely to be related to the increased caspase-8 and caspase-1 observed in our experiments.
Damaged proteins are degraded by proteasomes in the cytoplasm or nucleus (33). We show here first time that MAVS protein preferentially binds to the proteasomal protein PSMA7 in fatty livers, suggesting that the damaged, cleaved MAVS protein from the mitochondria accumulates in the cytoplasm, and likely degraded by the proteasomes.
Virus-induced apoptosis requires MAVS in primary mouse fibroblasts (25) and MAVS itself can induce caspase-dependent apoptosis. It has been shown that poly I:C initiates apoptosis via MAVS (34). However, MAVS levels were decreased in MCD diet-induced steatohepatitis in our experiments. Furthermore, we found that while caspase-3 was activated by dsRNA stimulation in normal liver suggesting apoptosis, there was no increase over the elevated baseline apoptosis (caspase-3 activity) in steatohepatitis. Instead, poly I:C induced liver necrosis and increased serum HMGB1 levels in MCD diet-fed mice. We speculate that decreased mitochondrial MAVS levels may result in impaired MAVS-dependent apoptosis following dsRNA challenge in MCD-steatohepatitis.
MAVS interacts with protein kinase receptor-interacting protein 1 (RIP1) and facilitates NFκB activation (35). RIP1 and the protein kinase receptor-interacting protein 3 (RIP3) may form a complex with TRADD, FADD and caspase-8 that leads to RIP3 cleavage and proteolytic inactivation (36,37). Studies showed that RIP3 overexpression results in TNFα and NO-mediated necrosis (37,38). RIP3 has been identified as a molecular switch between apoptosis and necrosis (26). Here we show for the first time that increased expression of RIP3 in MCD-diet fed mice occurs both at the mRNA and protein levels. Increased RIP3 mRNA was also present in human livers with NASH. We found a sustained increase in RIP3 expression that correlated with increased necrosis and increased serum HMGB1 levels after poly I:C challenge in steatohepatitis in mice. It is tempting to speculate that increased RIP3 may result in an apoptosis-to-necrosis switch after a dsRNA challenge in steatohepatitis. Recent studies suggest RIP3 association with the mitochondria and its regulation by ROS (38) and RIP3-induced promotion of necrosis is regulated by ROS (26). Our observations confirmed previous findings of increased ROS generation in diet-induced NASH (18). More importantly, we identified that poly I:C augmented ROS generation as well as RIP3 induction and necrosis in MCD-induced steatohepatitis.
In conclusion, our data demonstrate an important role for mitochondrial damage and MAVS dissociation from the mitochondria in the increased susceptibility of steatohepatitis to a dsRNA viral challenge. We report for the first time that livers with steatohepatitis fail to induce Type-I IFNs in response to dsRNA challenge due to dissociation of MAVS from the mitochondria and impaired oligomerization. The MAVS dissociation also leads to impaired induction of apoptosis and promote necrosis together with increased RIP3 expression, impaired anti-viral interferon response and increased liver damage in non-alcoholic steatohepatitis. These key findings were also reproducible in human NASH.
Supplementary Material
C57BL/6 mice received a mthionine-choline-deficient (MCD) or – supplemented (MCS) diet for 5 weeks and were injected with poly I:C (5mg/bwkg) intraperitoneally for 2 hours. Liver mononuclear cells were isolated and stained for NK cell marker NK1.1 and T-cell marker TCRβ. FACS analysis of changes in frequency of NK1.1 positive, TCRβ negative NK cell population compared to MCS diet-fed controls before and after poly I:C challenge is shown as mean±SEM. N=3 mice/group (*) indicates p<0.05 MCS vs. MCS poly I:C.
C57Bl/6 mice were fed with methionine-choline-deficient (MCD) or –supplemented (MCS) diet for 5 weeks and injected with poly I:C (5mg/bwkg) intraperitoneally for 2 or 6 hours.
The mRNA expression of TRAIL (A) and Bim (B), as markers of apoptosis, were analyzed by qPCR in the liver of MCD diet-fed mice with or without poly I:C stimulation and compared to MCS controls. N=4–6 mice/group, (*) indicates p<0.05 vs. MCS baseline, (#) indicates p<0.05 vs. MCD baseline.
The sequence of primers employed for quantitative real-time.
Acknowledgments
This work was supported by grant DK075635 (GS) and the core resources supported by the Diabetes Endocrinology Research Center grant DK32520 were also used (GS is a member of the UMass DERC).
List of abbreviations
- NASH
non-alcoholic steatohepatitis
- TLR
toll-like receptor
- dsRNA
double stranded RNA
- IFN
interferon
- MAVS
mitochondrial antiviral signaling protein
- Poly I
C, polyinosinic-polycytidylic acid
- RIP3
receptor interacting protein-3
- HMGB1
high mobility group box protein-1
- NAFLD
non-alcoholic fatty liver disease
- LPS
lipopolysaccharide
- VISA
virus-induced signaling adaptor
- IPS-1
IFNβ promoter stimulator protein-1 MCD, methionine-choline deficient
- MCS
methionine-choline supplemented
- CpG
cytidine-phosphate-guanosine-rich DNA
- ALT
alanine aminotransferase
- TNF
tumor necrosis factor
- TBARs
thiobarbituric acid reactive substances
- ISG
interferon-inducible gene
- Mda5
melanoma differentiation-associated gene 5
- RIG-I
retinoic acid-inducible gene-I
- MyD88
Myeloid differentiation factor 88
- TRIF
TIR domain-containing adaptor inducing IFN-beta
- ROS
reactive oxygen species
- TRAIL
TNF-related apoptosis inducing ligand
- IKK
IkB kinase
- NFkB
Nuclear factor kB
- zVAD
(carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone)
- PSMA7
proteasome subunit alpha type 7
- Mult1
murine UL16-binding proteinlike transcript 1
- Rae-1α
retinoic acid early inducible-1α
- RLR
RIG-I-like receptor
Footnotes
Disclosures: nothing to disclose
Contributor Information
Timea Csak, Email: Timea.Csak@umassmed.edu.
Angela Dolganiuc, Email: Angela.Dolganiuc@umassmed.edu.
Karen Kodys, Email: Karen.Kodys@umassmed.edu.
Bharath Nath, Email: Bharath.Nath@umassmed.edu.
Jan Petrasek, Email: Jan.Petrasek@umassmed.edu.
Shashi Bala, Email: Shashi.Bala@umassmed.edu.
Dora Lippai, Email: Dora.Lippai@umassmed.edu.
Gyongyi Szabo, Email: Gyongyi.Szabo@umassmed.edu.
References
- 1.Bellentani S, Marino M. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD) Ann Hepatol. 2009;8 (Suppl 1):S4–8. [PubMed] [Google Scholar]
- 2.Selzner M, Rudiger HA, Sindram D, Maddan J, Clavien PA. Mechanisms of ischemic injury are different in the steatotic and normal rat liver. Hepatology. 2000;32:1280–1288. doi: 10.1053/jhep.2000.20528. [DOI] [PubMed] [Google Scholar]
- 3.Szabo G, Velayudham A, Romics L, Jr, Mandrekar P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of toll-like receptors 2 and 4. Alcohol Clin Exp Res. 2005;29:140S–145S. doi: 10.1097/01.alc.0000189287.83544.33. [DOI] [PubMed] [Google Scholar]
- 4.Donthamsetty S, Bhave VS, Mitra MS, Latendresse JR, Mehendale HM. Nonalcoholic steatohepatitic (NASH) mice are protected from higher hepatotoxicity of acetaminophen upon induction of PPARalpha with clofibrate. Toxicol Appl Pharmacol. 2008;230:327–337. doi: 10.1016/j.taap.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 5.Younossi ZM, McCullough AJ. Metabolic syndrome, non-alcoholic fatty liver disease and hepatitis C virus: impact on disease progression and treatment response. Liver Int. 2009;29 (Suppl 2):3–12. doi: 10.1111/j.1478-3231.2008.01949.x. [DOI] [PubMed] [Google Scholar]
- 6.Kahraman A, Miller M, Gieseler RK, Gerken G, Scolaro MJ, Canbay A. Non-alcoholic fatty liver disease in HIV-positive patients predisposes for acute-on-chronic liver failure: two cases. Eur J Gastroenterol Hepatol. 2006;18:101–105. doi: 10.1097/00042737-200601000-00018. [DOI] [PubMed] [Google Scholar]
- 7.Wieland SF, Chisari FV. Stealth and cunning: Hepatitis B and Hepatitis C viruses. J Virol. 2005;79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Tang ED, Wang CY. MAVS self-association mediates antiviral innate immune signaling. J Virol. 2009;83:3420–3428. doi: 10.1128/JVI.02623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 11.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Romics L, Jr, Dolganiuc A, Velayudham A, Kodys K, Mandrekar P, Golenbock D, et al. Toll-like receptor 2 mediates inflammatory cytokine induction but not sensitization for liver injury by Propioni- bacterium acnes. J Leukoc Biol. 2005;78:1255–1264. doi: 10.1189/jlb.0804448. [DOI] [PubMed] [Google Scholar]
- 13.Jacobs BL, Langland JO. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology. 1996;219:339–349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- 14.Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann NY Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 16.Jia Y, Song T, Wei C, Ni C, Zheng Z, Xu Q, et al. Negative regulation of MAVS-mediated immune response by PSMA7. J Immunol. 2009;183:4241–4248. doi: 10.4049/jimmunol.0901646. [DOI] [PubMed] [Google Scholar]
- 17.Baril M, Racine ME, Penin F, Lamarre D. MAVS dimer is crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J Virol. 2009;83:1299–1311. doi: 10.1128/JVI.01659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei Y, Rector RS, Thyfault JP, Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol. 2008;14:193–199. doi: 10.3748/wjg.14.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caroppi P, Sinibaldi F, Fiorucci L, Santucci R. Apoptosis and human diseases: mitochondrion damage and lethal role of released cytochrome C as proapoptotic protein. Curr Med Chem. 2009;16:4058–4065. doi: 10.2174/092986709789378206. [DOI] [PubMed] [Google Scholar]
- 20.Rebsamen M, Meylan E, Curran J, Tschopp J. The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ. 2008;15:1804–1811. doi: 10.1038/cdd.2008.119. [DOI] [PubMed] [Google Scholar]
- 21.Scott I, Norris KL. The mitochondrial antiviral signaling protein, MAVS, is cleaved during apoptosis. Biochem Biophys Res Commun. 2008;375:101–106. doi: 10.1016/j.bbrc.2008.07.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu CY, Chiang RL, Chang TH, Liao CL, Lin YL. The interferon stimulator mitochondrial antiviral signaling protein facilitates cell death by disrupting mitochondrial membrane potential and by activating caspases. J Virol. 2010;84:2421–31. doi: 10.1128/JVI.02174-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong Z, Wei H, Sun R, Hu Z, Gao B, Tian Z. Involvement of natural killer cells in PolyI:C-induced liver injury. J Hepatol. 2004;41:966–973. doi: 10.1016/j.jhep.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 24.Kahraman A, Schlattjan M, Kocabayoglu P, Yildiz-Meziletoglu S, Schlensak M, Fingas CD, et al. Major histocompatibility complex class I-related chains A and B (MIC A/B): A novel role in nonalcoholic steatohepatitis. Hepatology. 2010;51:92–102. doi: 10.1002/hep.23253. [DOI] [PubMed] [Google Scholar]
- 25.Lei Y, Moore CB, Liesman RM, O’Connor BP, Bergstralh DT, Chen ZT, et al. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One. 2009;4:e5466. doi: 10.1371/journal.pone.0005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 27.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, et al. Cell-type specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 28.McCartney S, Vermi W, Gilfillan S, Cella M, Murphy TL, Schreiber RD, et al. Distinct and complementary functions of Mda5 and TLR3 in poly(I:C)-mediated activation of mouse NK cells. J Exp Med. 2009;206:2967–2976. doi: 10.1084/jem.20091181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duluc D, Tan F, Scotet M, Blanchard S, Fremaux I, Garo E, et al. Poly I:C plus IL-2 or IL-12 induce IFNγ production by human NK cells via autocrine IFN-β. Eur J Immunol. 2009;39:2877–2884. doi: 10.1002/eji.200838610. [DOI] [PubMed] [Google Scholar]
- 30.Ohman T, Rintahaka J, Kalkkinen N, Matikainen S, Nyman TA. Actin and RIG-I/MAVS signaling components translocate to mitochondria upon influenza A virus infection of human primary macrophages. J Immunol. 2009;182:5682–5692. doi: 10.4049/jimmunol.0803093. [DOI] [PubMed] [Google Scholar]
- 31.Wieckowska A, Zein NN, Yerian LM, Lopez AR, McCullough AJ, Feldstein AE. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 32.Farrell GC, Larter CZ, Hou JY, Zhang RH, Yeh MM, Williams J, et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J Gastroenterol Hepatol. 2009;24:443–452. doi: 10.1111/j.1440-1746.2009.05785.x. [DOI] [PubMed] [Google Scholar]
- 33.Peters JM, Franke WW, Kleinschmidt JA. Distinct 19S and 20S subcomplexes of the 26S proteasome and their distribution in the nucleusand cytoplasm. J Biol Chem. 1994;269:7709–7718. [PubMed] [Google Scholar]
- 34.Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C, et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type-I interferon-independent apoptosis in human melanoma cells. J Clin Invest. 2009;119:2399–2411. doi: 10.1172/JCI37155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 36.Vandenbeele P, Declerq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 37.He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor-interacting protein kinase 3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 38.Davis CW, Hawkins BJ, Ramasamy S, Irrinki KM, Cameron BA, Islam K, et al. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med. 2010;48:306–317. doi: 10.1016/j.freeradbiomed.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
C57BL/6 mice received a mthionine-choline-deficient (MCD) or – supplemented (MCS) diet for 5 weeks and were injected with poly I:C (5mg/bwkg) intraperitoneally for 2 hours. Liver mononuclear cells were isolated and stained for NK cell marker NK1.1 and T-cell marker TCRβ. FACS analysis of changes in frequency of NK1.1 positive, TCRβ negative NK cell population compared to MCS diet-fed controls before and after poly I:C challenge is shown as mean±SEM. N=3 mice/group (*) indicates p<0.05 MCS vs. MCS poly I:C.
C57Bl/6 mice were fed with methionine-choline-deficient (MCD) or –supplemented (MCS) diet for 5 weeks and injected with poly I:C (5mg/bwkg) intraperitoneally for 2 or 6 hours.
The mRNA expression of TRAIL (A) and Bim (B), as markers of apoptosis, were analyzed by qPCR in the liver of MCD diet-fed mice with or without poly I:C stimulation and compared to MCS controls. N=4–6 mice/group, (*) indicates p<0.05 vs. MCS baseline, (#) indicates p<0.05 vs. MCD baseline.
The sequence of primers employed for quantitative real-time.