

Abstract
Hepatitis C virus infection affects 170 million people worldwide and the majority of individuals exposed to HCV develop chronic hepatitis leading to progressive liver damage, cirrhosis and hepatocellular cancer. The natural history of HCV infection is influenced by genetic and environmental factors of which chronic alcohol use is an independent risk factor for cirrhosis in HCV infected individuals. Both the hepatitis C virus and alcohol damage the liver and result in immune alterations contributing to both decreased viral clearance and liver injury. This review will capture the major components of the interactions between alcohol and HCV infection to provide better understanding for the molecular basis of the dangerous combination of alcohol use and HCV infection. Common targets of HCV and alcohol involve innate immune recognition and dendritic cells, the critical cell type in antigen presentation and antiviral immunity. In addition, both alcohol and HCV affect intracellular processes critical for hepatocyte and immune cell functions including mitochondrial and proteasomal activation. Finally, both chronic alcohol use and hepatitis C virus infection increase the risk of hepatocellular cancer. The common molecular mechanisms underlying the pathological interactions between alcohol and HCV include the modulation of cytokine production, lipopolysaccharide (LPS)-TLR4 signaling, and reactive oxygen species (ROS) production. LPS-induced chronic inflammation is not only a major cause of progressive liver injury and fibrosis but it can also contribute to modification of the tissue environment and stem cells to promote hepatocellular cancer development. Alteration of these processes by alcohol and HCV produces an environment of impaired antiviral immune response, greater hepatocellular injury and activation of cell proliferation and dedifferentiation.
Keywords: antigen presenting cells, CD4+ T cells, CD8+ T cells, cytotoxic T lymphocytes (CTL), cytokines, CYP2E1, dendritic cells (DCs), HCV, HCV core protein, NS5a protein, hepatocytes, hepatocellular carcinoma (HCC), innate and adaptive immunity, Lieber-DeCarli diet, Lipopolysaccharide (LPS), mitochondrial Ca2+, mitochondrial dysfunction, Mn-superoxide dismutase (SOD2), proteasome, reactive oxygen species (ROS), toll-like receptor 4 (TLR4), transgenic mice
Introduction
Hepatitis C virus (HCV) infects an estimated four million Americans and 170 million people worldwide. About 80% of all acute infections develop into chronic infections, which is a cause of progressive liver disease characterized by continuing hepatocellular inflammation and progression from mild to advanced fibrosis and to cirrhosis. Patients with cirrhosis also have a very high risk for developing hepatocellular carcinoma (HCC). Nearly 40% of all chronic liver disease cases in the US and 27% (in the US) and up to 90% (in Japan) of HCC cases have an underlying chronic HCV infection (Singal and Anand, 2007; Blonski and Reddy, 2008). While a pegylated inferferon/ribavirin based anti-HCV therapy is available, it results in a sustained virological response (SVR) in only approximately 55% of the chronic infected patients (Asselah et al., 2009). Study of the underlying mechanisms of HCV infection and pathogenesis has been a very active research area since HCV was first identified (Kuo et al., 1989) and still is in need for better disease prevention and intervention.
HCV is a positive stranded RNA virus and a member of Flaviviridae. Its genome has approximately 9,600 nucleotides containing a large open reading frame coding for a polyprotein. Once made, this polyprotein is processed into ten separate proteins. Among these, core (nucleocapside protein), NS3 (helicase/protease), NS5a, and NS5b (RNA polymerase) have been implicated in HCV related tissue damage and carcinogenesis. Based on phylogenetic studies, HCV variants are classified into 6 major genotypes (Simmonds et al., 2005). The most common variants found in the US are genotype 1. Interestingly, this genotype, as well genotype 4, is more resistant than genotypes 2 and 3 to the standard pegylated inferferon/ribavirin therapy (Lemon et al., 2007).
The natural history of HCV infection and pathogenesis are influenced by both genetic and environmental factors. Among these, alcohol abuse is the most consistent environmental risk factor and independently associated with a much reduced HCV clearance and accelerated disease course (Siu et al., 2009). From a study of a US veteran cohort, patients with alcohol use disorder (AUD, including current and past use) are less than half as likely to have spontaneous viral clearance compared to those without AUD (Piasecki et al., 2004). It was recognized soon after HCV was identified that, in alcoholics, HCV infection positively correlates with clinical severity of liver disease (Mendenhall et al., 1991; reviewed in Hutchinson et al., 2005). Studies since have established that HCV and heavy alcohol use synergistically accelerate the progression of the most severe liver diseases, cirrhosis (Corrao and Arico 1998) and hepatocellular carcinoma (HCC; Hassan et al., 2002; Yuan et al., 2004). The mechanisms by which alcohol affects liver damage and impairs immune elimination of the HCV virus, however, are yet to be fully understood.
HCV and Alcohol separately can cause both immune impairment and tissue damage (reviewed in Szabo and Mandrekar, 2009; Tsukamoto et al., 2009; Rehermann, 2009; Lanford et al., 2009). This review focuses on four recently described aspects of alcohol-HCV interactions that significantly impact on viral clearance and pathogenesis. 1) Dendritic cell (DC) function. DCs are critical to the adaptive immune response because they are the central cell type, via antigen presentation, for activating anti-viral effector CD4+/CD8+ lymphocytes. Previous observations on CD4+/CD+8 cell dysfunction in alcoholics have led to the closer examination of the effect of alcohol on DC cell function; 2) Proteasome activity and reactive oxygen species (ROS).
Intracellular proteasome activity is important for antigen processing and presentation on viral infected cells. This is the mechanism through which these cells become recognizable by antiviral immune cells. Reduced proteasome activity is associated with alcohol-induced oxidative stress. The identification of ROS induction in hepatocytes by HCV core protein has led to the investigation of the combined effect of alcohol and HCV on proteasome activity. 3) Mitochondrial function and ROS in tissue injury. ROS induced mitochondrial dysfunction plays a major role in alcohol induced tissue injury. Mitochondria are also one of the intracellular targets for HCV core protein. The role of mitochondria as targets and effectors of HCV-alcohol induced liver injury is thus an active area of research. 4) Inflammation and hepatocellular cancer (HCC). Both alcohol and chronic HCV infection are associated with an increase of the level of circulating lipopolysaccharide, a well known inflammation inducer. It has been found only recently that TLR4, a receptor for LPS, plays a key role in the generation of liver cancer cells with stem cell characteristics.
Together the combined effects of HCV and alcohol on various host cell types, via modulating reactive oxygen species (ROS) production, LPS signaling, and cytokine production, produce an environment of impaired antiviral immune response, greater hepatocellular injury and activation of cell proliferation and dedifferentiation responsible for the range of diseases seen in patients.
1. Hepatitis C Virus, Alcohol and Dendritic Cell Dysfunction
Chronic alcoholics have a high incidence of hepatitis C virus (HCV) infection (Schiff and Ozden 2003; Oshita et al., 1994; Siu et al., 2009) which may be due, in part, to the action of alcohol on the cellular immune response to epitopes that reside on viral structural and non-structural proteins. Studies have been performed in animal models where genetic immunization has been employed to generate viral specific antibody and CD4+ and CD8+ responses to HCV core protein (Tokushige et al., 1996; Geissler et al., 1997a; Encke et al., 1998; Stylianou and Saklatvala, 1998). Low level expression of HCV proteins encoded by such plasmids either in muscle or fibroblasts following DNA inoculation led to activation of dendritic cells (DCs) at the site of immunization or in distant draining lymph nodes to subsequently prime anti-viral immune responses. Chronic alcohol feeding had a substantial suppressive effect on the generation of viral antigen specific CD4+ and CD8+ cellular immune activity when HCV core was employed as the immunogen. Of interest, this inhibitory effect of alcohol could be partially, or completely, reversed by co-immunization with an interleukin-2 (IL-2) or granulocyte macrophage colony-stimulating factor (GM-CSF) expression plasmid (Encke et al., 1998; Tokushige et al., 1996; Stylianou and Saklatvala, 1998; Geissler et al., 1997a; Encke and Wands, 2000; Geissler et al., 1999). Such experimental results are consistent with the concept that DNA based immunization was enhanced by cytokine production at the site of antigen presentation, and that DCs could be an important cellular target during chronic alcohol consumption (Encke et al., 1998; Tokushige et al., 1996; Stylianou and Saklatvala, 1998; Geissler et al., 1997a; Encke and Wands, 2000; Geissler et al., 1999). Similar results were obtained using a plasmid encoding for the HCV NS5 non-structural protein as well (Encke et al., 1998; Rehermann and Nascimbeni, 2005). Taken together, these investigations reveal that DCs, differentiated in vivo in the context of chronic alcohol feeding, may have intrinsic functional defects which could partially explain the depressed cytotoxic T cell lymphocyte (CTL) activity previously observed after DNA-based immunization using HCV core and NS5 as the immunogens (Encke et al., 1998; Encke and Wands, 2000; Aloman et al., 2007, Geissler et al., 1997b; Geissler et al., 1997c).
DCs are known to play a crucial role in generating immune responses to viral proteins. In this regard, DCs are highly specialized antigen presenting cells (APCs). Such cells are known to take-up, process, and present small peptide fragments (8-10 amino acids) to effector T cells. They generally elicit an adaptive immune response or, paradoxically, a tolerogenic reaction that depends in part, on how the antigen is presented and its concentration. To initiate a successful adaptive immune response, DCs need to express a variety of co-stimulatory molecules on their cell surface (Banchereau and Steinman, 1998). Upregulation of their activity is mediated by several factors including toll-like receptor-ligand interactions, or binding of CD40 to the CD40 ligand on the cell surface. These interactions between DCs and T cells promote sustained stimulation and improve the efficiency of antigen presentation. DCs are ideal for immune-modulatory strategies since their activity is crucial for eliciting a robust immune response to viral peptides. It is important to note that DC-based immunization was recently shown to be a suitable approach to elicit sustained immune responses to HCV proteins of the type necessary for viral eradication from the liver (Encke et al., 2005; Kuzushita et al., 2006). DCs are equipped with receptor mediating antigen-uptake which include members of the C-type lectin family. It is known that the phagocytic process is initiated through the calcium dependent binding of C-type lectins to carbohydrate-bearing pathogen-derived antigens using highly conserved carbohydrate recognition domains. Some of these receptors appear uniquely on phagocytic cells such as macrophages, monocytes, B lymphocytes, neutrophils, and DCs (Figdor et al., 2002).
One of the major effects of long-term alcohol consumption on the biology of DCs in vivo appears to be at the level of cytokine production. DCs generated from alcohol-fed mice had a greater ability to produce IL-1β, the initial cytokine that drives lymphocyte proliferation (Aloman et al., 2007). However, secretion of IL-6, another cytokine implicated in the overall proliferative response, is impaired. It is noteworthy that there is a propensity to generate TH2-type cytokine responses because the secretion of TNFα, IFN-γ, and IL-12, which promote the TH1 differentiation pathway of CD4+ and CD8+ into viral specific functional cells, is strikingly impaired by chronic alcohol. Indeed, IL-10 is increased in alcohol fed mice which provides another boost toward the TH2-type immune response. These in vivo findings provide information on cytokine production by DCs from mice fed alcohol long-term followed by LPS and poly-I:C stimulation. These in vivo observations in mice support previous in vitro findings on human DCs expanded in the presence of alcohol (Mandrekar et al., 2004; Dolganiuc et al., 2003). However, chronic alcohol consumption may also affect antigen uptake, degradation, processing and transport of peptides to the cell surface in the context of MHC class I and II molecules to interact with and stimulate viral specific immune responses, and further studies will be required to examine these possibilities.
It was important to further characterize the role of DCs in vivo in animals with an intact immune system. Therefore, the model of DNA-based immunization and syngeneic transfer of non-selective splenic DCs has been employed. Studies were performed with the NS5 viral antigen because previous studies revealed that a DNA construct expressing this protein generates a strong cellular immune response (Encke and Wands, 2000). The strategy involved the concept that syngeneic DC transfer would allow one to determine whether the effects of alcohol on cellular and humoral immune responses could be reversed by simply co-immunizing with DCs isolated from normal animals, and whether these effects could be inhibited by DCs generated from alcohol-fed mice.
In addition, it was critical to evaluate CTL responses after splenic DC transfer to a HCV non-structural protein such as NS5 (Aloman et al., 2007). Surprisingly, it was found that normal DCs can correct a functional defect in the cellular immune response as measured by CTL activity previously altered by long-term alcohol feeding. The correction of CTL activity after transfer of normal DCs to animals on the chronic alcohol diet provided strong evidence that it is the effect of alcohol on DCs rather than a direct effect of alcohol on CD8+ lymphocyte function. In addition, pair-fed control mice receiving DCs from alcohol fed animals were unable to drive an effective CTL response and developed partial tolerance with respect to CTL activity against the HCV NS5 protein (Aloman et al., 2007).
In summary, in vivo generation of DCs combined with DC transfer and DNA-based immunization revealed that alcohol-induced dysfunction of DCs is a major factor responsible for the reduced cellular immune response to HCV-related peptides. Such observations may have relevance to persistent HCV infection in alcoholic patients, and identify a critical cell type that is at-risk for the effects of alcohol. Therefore, one of the major cellular targets in the immune system for long-term activity of alcohol are DCs with subsequent impaired generation of robust CTL activity to viral structural, and non-structural proteins that may be critical for viral clearance (Mandrekar et al., 2004; Dolganiuc et al., 2003). More important, such observations open new strategies to enhance antiviral immune responses in alcoholics and, particularly, in the setting of long term ethanol consumption by improving DC function. Thus, DCs represent one of the major cellular components of the immune system sensitive to chronic alcohol effects, and subsequently impair the generation of CD8+ CTL and CD4+ proliferative activity against epitopes on HCV viral proteins that may be essential for viral resolution (Aloman et al., 2007; Kuzushita et al., 2006; Mandrekar et al., 2004; Dolganiuc et al., 2003; Szabo et al., 2004; Heinz and Waltenbaugh, 2007; Edsen-Moore et al., 2008). Further studies on the biology of DCs and how alcohol affects their function will lead to a better understanding of the acquisition and persistence of HCV infection, and also will provide opportunities to devise vaccine strategies to eradicate persistent HCV infection in chronic alcoholics.
2. Differential regulation of proteasome activity by HCV core protein and ethanol in liver cells
The generation of viral peptides for antigen presentation in viral infected liver cells is another site at which alcohol may modify the immune response to HCV. The proteasome is a multi-catalytic enzyme that degrades about 80% of intracellular proteins, including signal transduction factors. In addition to protein degradation, the proteasome also generates peptides for MHC class I-restricted antigen presentation. The chymotrypsin-like and the trypsin-like activities of the proteasome are related to antigen presentation due to their ability to cleave peptide bonds after hydrophobic and basic amino acids, respectively (Goldberg et al., 2002; Qian et al., 2006).
Proteasome exists in equilibrium of two particles, 20S proteasome and 26S proteasome. For protein degradation by 26S proteasome, proteins are marked by ubiquitin, a small 8.5-kDa protein that is covalently attached to protein substrate. Proteins subjected to degradation have multiple ubiquitin molecules covalently attached to generate polyubiquitin chain binding to an internal lysine (lysine 48) residue. In contrast, 20S proteasome degrades non-ubiquitylated proteins, most of which are oxidatively modified (reviewed by Donohue et al., 2007 and Osna and Donohue, 2007). Usually, oxidative modification (adduction) of proteins makes them more susceptible for degradation (Curry-McCoy et al., 2009; Grune et al., 1998).
Proteasome function is apparently susceptible to oxidative stress that forms adducts with protein carbonyls, 4-hydroxynonenal (4-HNE) and 3-nitrotyrosine derived from peroxynitrite (Bardag-Gorce et al., 2005; Kessova and Cederbaum, 2005; Osna et al., 2004). Other studies have revealed a reduction of proteasome activity as a result of ethanol metabolism in recombinant hepatoma (VL-17A) cells that express both CYP2E1 and ADH (Donohue et al., 2006; Osna et al., 2003; Osna et al., 2007). This inhibition of proteasome function was CYP2E1 dependent (Dey and Cederbaum, 2006; Kessova and Cederbaum, 2005; Osna et al., 2003) and correlated with generation of intracellular oxidants. In the liver, the levels of oxidative stress induced by multiple agents, including ethanol and viral proteins, inhibit proteasome activity (Osna et al., 2004; Osna et al., 2008).
HCV proteins are able to induce oxidative stress in the liver. Specifically, outer mitochondrial membrane associated HCV core protein elevates generation of reactive oxygen species (ROS) by mitochondrial electron transport complex I, resulting in a decrease in mitochondrial GSH and mitochondrial depolarization, which can be augmented by simultaneous ER oxidative stress (Korenaga et al., 2005a; Otani et al., 2005). This effect of core protein is further potentiated by exposure of cells to ethanol (Otani et al., 2005). Thus, the combined action of HCV core protein and ethanol may interfere with proteasome activity, thereby affecting the events that are downstream from proteasome-dependent generation of peptides for MHC class I-restricted antigen presentation in liver cells, a crucial process for recognition of infected hepatocytes by cytotoxic T-lymphocytes. Previously, we have demonstrated the critical role of oxidative stress-modified proteasome activity for the peptide hydrolysis as well as the presentation of peptide-MHC class I complex on liver cell surface (Osna et al., 2009; Osna et al., 2007).
While studying the effects of HCV core protein on proteasome function in CYP2E1-expressing hepatoma cells, we found that HCV core protein enhanced this enzyme activity in intact Huh7 cells, but suppressed proteasome in ethanol-treated cells. These results have been previously presented (Osna et al., 2008). Because glutathione ethyl ester (GSH-EE) and N-acetyl cysteine (NAC), uric acid, diallyl sulfide (DAS, CYP2E1 inhibitor) and catalase reversed the effects of core protein on proteasome activity, the activation of proteasome by core protein was attributed to oxidative stress, which at low levels enhanced proteasome activity. However, when HCV core/CYP2E1-expressing cells were exposed to ethanol, a higher intensity of oxidative stress induced by both HCV core protein and ethanol suppressed proteasome activity, indicating a dual regulation of proteasome activity by differential levels of oxidative stress. In addition to oxidative stress-related proteasome regulation, HCV core protein-proteasome interactions, which involved the proteasome activator, PA28 and were potentiated by mitochondrial and microsomal cell fractions, activated 20S proteasome in cell-free system, in the absence of oxidative stress. Remarkably, when cell fractions were purified from ethanol-exposed cells, the activating effect of HCV core on proteasome was attenuated.
In vivo studies in hepatocytes of HCV core-expressing vs. non-expressing mice fed with ethanol, showed similar pattern of proteasome activation by core protein and suppression by ethanol feeding as observed in vitro. These changes, at least in part, could be attributed to oxidative stress because induction of ROS by ethanol and TBARS levels were higher in ethanol-fed HCV+ mice than in ethanol-fed controls (Osna et al., 2009; manuscript in preparation). Enhanced ROS production by mitochondria of these transgenic mice has been previously demonstrated (Korenaga et al., 2005a; see subsequent section). These data suggest that both HCV core and ethanol induce oxidative stress, which accordingly corresponds to the magnitude of proteasome activity inhibition in hepatocytes induced by either ethanol or ethanol + HCV core protein. Overall, both HCV core protein and ethanol appear to regulate proteasome activity by overlapping mechanisms. HCV core protein activates proteasome activity both via a low level of oxidative stress as well as a direct effect. However, when ethanol is added, either in cells or in mouse models, there is a further increase in oxidative stress and a net decrease in proteosome activation (Osna et al., 2008).
In conclusion, proteasome activity in HCV core protein–expressing cells is regulated by differential levels of oxidative stress: core protein-induced low oxidative stress enhances proteasome activity, while high oxidative stress induced by the combined action of core protein and ethanol suppresses proteasome activity. These core protein-ethanol-mediated changes in proteasome function are confirmed by in vitro and in vivo experiments and may suggest the reduced proteasome-dependent hydrolysis of antigenic peptides, which limits the supply of peptides for MHC class I-restricted antigen presentation, thereby suppressing the display of these peptides in the context of MHC class I on ethanol-metabolizing liver cells.
3. Mitochondria, oxidative stress and HCV-alcohol induced liver injury
Although non-hepatic cells including lymphoid cells can support HCV replication, hepatocytes are the major target cell type for HCV infection/replication (Lemon et al., 2007). Hepatic oxidative stress and mitochondrial abnormalities are nearly universally observed in patients with chronic Hepatitis C and appear to be an intrinsic component of the disease. They are manifested by elevations of oxidized derivatives of lipids, proteins and nucleic acids (Konishi et al., 2006; Fujita et al., 2007; Paradis et al., 1997), decreases in antioxidant content (Yadav et al., 2002; Vendemiale et al., 2001; Mahmood et al., 2004; Jain et al., 2002; Sumida et al., 2000; Larrea et al., 1998), and ultrastructural abnormalities of mitochondria (Barbaro et al., 1999). While some degree of oxidative stress is a characteristic of any inflammatory disease, it occurs with greater frequency and magnitude for hepatitis C than for other liver diseases (Valgimigli et al., 2002) and it correlates with greater severity of inflammation, the presence of insulin resistance (Vidali et al., 2008b; Mitsuyoshi et al., 2008), more rapid progression of fibrosis and the development of hepatocellular carcinoma (Maki et al., 2007). In addition, successful viral clearance of Hepatitis C results in a decrease of the biomarkers of oxidative stress concomitantly with an improvement of the clinical sequelae (Serejo et al., 2003).
Hepatitis C interacts synergistically with other factors to worsen liver disease. The most well known example of this phenomenon is that heavy alcohol consumption in HCV infected individuals predisposes to more rapid fibrosis progression, a greater incidence of HCC and impaired response to therapy (Siu et al., 2009). In addition, HCV is a risk factor for toxicity of acetaminophen as well as antiretroviral agents in HIV co-infected patients (Nguyen et al., 2008). Mitochondrial effects are prominent in each of these disease processes. There has thus been considerable investigation of how HCV induced mitochondrial changes occur and their contribution to combined HCV-alcoholic liver disease.
Mechanisms of HCV-induced mitochondrial ROS production
An understanding of HCV-induced mitochondrial effects has emerged from studies in HCV model systems including hepatoma cells expressing viral proteins, cells replicating the HCV RNA, cells infected with the JFH1 cell culture strain of HCV, and transgenic mice with constitutive or inducible expression of HCV proteins. Each of these systems has shown evidence of viral protein mediated ROS production, lipid peroxidation and activation of stress kinase pathways (Wang and Weinman, 2006; Piccoli et al., 2009; Machida et al., 2006a). The most important observation is that increased mitochondrial ROS production is a direct consequence of viral proteins and does not require the full process of viral infection. Several transgenic mouse models for hepatitis C reproduce the oxidative stress phenotype, even in the absence of inflammation (Moriya et al., 2001; Chang et al., 2008; Okuda et al., 2002). These findings demonstrate that in the case of oxidative stress, direct effects of viral proteins predominate over specific events in viral RNA replication.
Four different HCV proteins, core, NS3, NS4a, and NS5a, have been shown to cause oxidative stress. Core protein appears to be the primary cause of HCV oxidative stress since it is both sufficient, and in some cases necessary for the phenomenon (Piccoli et al., 2009), and cells expressing HCV core protein have multiple mitochondrial abnormalities (Moriya et al., 2001; Okuda et al., 2002; Piccoli et al., 2007). The effect is entirely Ca2+ dependent and is secondary to both ER stress and direct effects on the mitochondrial Ca2+ uptake mechanism. Core protein, like all the viral proteins is initially produced in the ER and its expression causes ER stress (Benali-Furet et al., 2005). This results in activation of the unfolded protein response, and release of Ca2+ with diminished retention within the ER lumen (Piccoli et al., 2007; Tardif et al., 2005; Qadri et al., 2004).
At the same time, core protein more directly interacts with the mitochondria as well. Core localizes to mitochondria (Schwer et al., 2004; Korenaga et al., 2005b; Suzuki et al., 2005) and a specific sequence in the C terminal portion of the molecule serves as a targeting sequence to the mitochondria-associated membrane (MAM) fraction of the ER. This is a point of close contact between the ER and the mitochondrial outer membrane (Williamson and Colberg-Poley, 2009). Core protein then serves as a modulator of the mitochondrial Ca2+ uniporter causing more rapid Ca2+ entry and increased net mitochondrial Ca2+ accumulation for a given extramitochondrial Ca2+ concentration (Li et al., 2007). This effect occurs in cells expressing core as well as in isolated mitochondria incubated with recombinant core protein. It is thus is a direct mitochondrial effect. The consequence is to make the process of Ca2+ transfer from ER to mitochondria more efficient with a net increase in baseline matrix Ca2+ and a dramatically increased rise in mitochondrial Ca2+ in response to agonists or ER stress. This Ca2+ influx increases mitochondrial superoxide production which then initiates an amplification phenomenon whereby oxidation of GSH leads to inhibition of electron transport and increased superoxide production from complex 1. Under some circumstances, such as in the presence of alcohol and exogenous peroxides, this can be sufficient to trigger mitochondrial inner membrane permeabilization and cell death (Otani et al., 2005; Abdalla et al., 2005).
Other viral proteins increase mitochondrial ROS production as well, although the mechanisms of these are less well understood. NS5a also causes ER stress, ER Ca2+ release and mitochondrial Ca2+ uptake (Gong et al., 2001; Dionisio et al., 2009) although it has not been shown to directly modulate mitochondrial Ca2+ uptake mechanisms. The NS3 /NS4a complex forms a protease that binds to the mitochondrial outer membrane where it cleaves an important innate immune signaling molecule, MAVS (Horner and Gale, 2009). It has been shown to increase mitochondrial ROS production and regulate mitochondrial apoptosis pathways (Nomura-Takigawa et al., 2006; Selimovic and Hassan, 2008). An additional viral protein, the alternate reading frame protein F has recently been shown to localize to mitochondria but the functional significance of this is not yet known (Ratinier et al., 2009).
Two other mechanisms contribute to HCV oxidative stress as well. Plasma membrane associated NADPH oxidase in macrophages and neutrophils is induced by the HCV NS3 (Bureau et al., 2001; Thoren et al., 2004) and this contributes to oxidative stress in the setting of inflammation. An additional contributor is secondary to HCV induced iron overload. HCV infection or HCV polyprotein expression in mouse liver results in a suppression of hepatic hepcidin production. This results from an oxidative stress dependent alteration in transcriptional activity (Nishina et al., 2008; Miura et al., 2008). This iron accumulation that results further increases lipid peroxidation, induces hepatic steatosis and contributes to pathogenesis. Since alcohol has a similar effect it may contribute to the synergistic liver injury (Harrison-Findik et al., 2006).
Consequences of HCV induced oxidative stress
The weight of evidence suggests that oxidative stress in Hepatitis C serves largely as an incomplete host cell response that inhibits viral replication and enhances apoptotic and necrotic clearance of infected cells. In a series of elegant experiments, Choi et al demonstrated that ROS induced increases in Ca2+ inhibits viral replication in replicons or in vitro (Choi et al., 2004; Choi et al., 2006). This conclusion is further supported by data showing a direct antiviral effects of lipid peroxidation (Huang et al., 2007), arsenic related oxidative stress (Kuroki et al., 2009), and oxidant-induced ERK activation (Yano et al., 2009). In contrast, other studies have shown that the oxidative stress produced by alcohol metabolism specifically increases viral replication (McCartney et al., 2008) although it is possible that this may be mediated by other aspects of alcohol metabolism. Oxidative stress does clearly play a role in chronic disease pathogenesis. There is a consistent correlation between greater degrees of oxidative stress markers and more severe disease (Yadav et al., 2002; Vendemiale et al., 2001; Mahmood et al., 2004; Jain et al., 2002; Valgimigli et al., 2002; Cardin et al., 2001) and it is an independent risk factor for fibrosis (Vidali et al., 2008a).
Mitochondrial ROS and HCV-alcohol pathogenesis
It is well established that alcohol also causes hepatic oxidative stress and mitochondrial dysfunction (Mantena et al., 2008), although the characteristics of this differ somewhat from that in HCV. Alcohol appears to cause more global decreases in mitochondrial electron transport complex activities whereas the defect in Hepatitis C is more specific for complex I. Synergistic mitochondrial electron transport inhibition and ROS production has therefore been proposed as a mechanism for liver injury in HCV/alcohol consuming patients. Evidence in support of this hypothesis has largely come from experimental model systems and its strength is dependent on the validity of these models for human disease.
Cell culture models for alcohol-induced liver cell injury are limited because hepatoma cells and even primary hepatocytes fail to express the most important alcohol metabolizing enzymes including ADH, ALDH, and CYP2E1. Nonetheless, hepatoma cells stably transfected with CYP2E1 have been used as a model for alcoholic liver injury and this approach has been tried with Huh7 hepatoma cells expressing HCV core protein (Otani et al., 2005) or replicating HCV RNA (McCartney et al., 2008). These systems show synergistic effects of alcohol and HCV on ROS production, oxidation of the glutathione pool, mitochondrial depolarization, and oxidant-induced cell death. Since cell death due to HCV and alcohol could be prevented by antioxidants it appears that synergistic ROS formation was essential for the cytotoxic effects (Otani et al., 2005).
Transgenic mice expressing HCV proteins are more useful models as they have the potential to better reproduce tissue injury in vivo. The limitation is primarily that alcohol consumption fails to reproduce the full range of alcoholic liver injury seen in humans. The Lieber-DeCarli voluntary feeding model tends to produce steatosis without significant inflammation and higher dose alcohol feeding through continuous intragastric feeding is technically difficult and still does not fully recapitulate human disease. Nonetheless, several groups have reported the results of alcohol feeding via the Lieber-DeCarli method to HCV core protein transgenic mice. These have found that the combination of HCV core and alcohol increased lipid peroxidation, cytokine production, and MAPkinase activation to a greater degree than that seen with either alcohol or core protein alone (Perlemuter et al., 2003; Tsutsumi et al., 2003).
In mice expressing the HCV proteins core, E1, E2 and p7, HCV protein expression mildly sensitizes mice to alcohol-induced hepatic steatosis (Tumurbaatar et al., 2008; and manuscript in preparation). In this transgenic mouse model, it appears that high levels of mitochondrial Mn-superoxide dismutase (SOD2) protect the liver from developing a severe disease in response to alcohol. Decreasing SOD2 levels by heterozygous gene deletion exacerbates the synergy between HCV and alcohol, producing a histological steatohepatitis in some animals (Tumurbaatar et al., 2008). These findings do suggest that HCV and alcohol trigger a superoxide dependent injury cascade in sensitive individuals. In human populations, homozygosity for a frequent polymorphism in the mitochondrial targeting sequence of SOD2 results in up to a 50% decrease in liver mitochondrial SOD2 levels (Martin et al., 2008). The relevance of these mouse models to human disease is thus improving.
In aggregate, the evidence supports the concept that the combined effects of alcohol and HCV on mitochondrial superoxide production initiate an injury cascade in sensitive individuals. Future studies need to examine the downstream effectors of this injury, their role in human disease, and the potential contribution of targeted antioxidant therapy in these diseases.
4. Liver cancer development and stem cells under the influence of HCV and alcohol
Hepatocellular carcinoma (HCC) is highly prevalent in the world, especially in Africa and Asia, does not respond well to conventional therapy, has a high mortality and is one of the most catastrophic consequences of combined HCV and alcohol effects on the liver (Okuda et al., 2000).
Alcohol synergistically enhances the progression of liver disease and the risk for liver cancer caused by hepatitis C virus (HCV). Recent studies have identified effects on toll-like receptor 4 (TLR4) as a primary contributor to HCV-alcohol induced hepatocellular carcinoma. TLR4 is induced by hepatocyte-specific transgenic (Tg) expression of the HCV nonstructural protein NS5A, and this induction mediates synergistic liver damage and tumor development by alcohol-induced endotoxemia (Machida et al., 2009). The stem/progenitor cell marker, Nanog, is up-regulated as a novel downstream gene by TLR4 activation and is responsible for the presence of cancer progenitor cells, (CD133/Nanog-positive cells) in liver tumors of alcohol-fed NS5A Tg mice (Machida et al., 2009). Transplantation of p53-deficient hepatic progenitor cells transduced with TLR4 results in liver tumor development in mice following repetitive lipopolysaccharide (LPS) injection, but concomitant transduction of Nanog short-hairpin RNA abrogates this outcome (Machida et al., 2009). Despite the common understanding that TLR4 is one of the pattern recognition receptors expressed predominantly by innate immune cells such as macrophages and lymphocytes, our study demonstrates that hepatocytes can be the primary cellular site of both TLR4 upregulation and its pathologic consequences in the context of HCV infection. Therefore, the TLR4-dependent mechanism synergizes liver disease by HCV and alcohol and is partly dependent on Nanog, a TLR4 downstream gene.
Although transduction of Nanog expression is required for tumorigenesis, Nanog alone is not as effective as TLR4 activation in production of liver tumors (Machida et al., 2009). It thus appears that TLR4 activation induces other tumor-driver genes which cooperatively work with Nanog to cause liver oncogenesis. Thus, Nanog is essential for TLR4-dependent oncogenesis, but it alone is poorly oncogenic. TLR4 promoter up-regulation by NS5A is mediated by PU.1, Oct-1, and AP-1 elements (Machida et al., 2006b). The similar transcriptional mechanism may underlie TLR4 induction in primary hepatocytes.
The finding that Nanog are transcriptionally upregulated in HCV-alcohol associated liver tumorigenesis suggests that appearance of a stem cell phenotype may have a role in HCC. Recent studies of HCC have centered on cancer stem cells (CSC), including detection of CSC in cancer, identification of CSC markers, and isolation of CSC from human HCC cell lines. The liver has a high regenerative potential, and hepatic small oval progenitor cells around the peripheral branches of the bile ducts, the canals of Hering, can differentiate into biliary epithelial cells and hepatocytes (Roskams et al., 2004). These oval liver progenitor cells share molecular markers with adult hepatocytes (albumin, cytokeratin 7 [CK7], CK19, oval cell markers [OV-6, A6, and OV-1], chromogranin-A, NCAM [neural cell adhesion molecule]) and fetal hepatocytes (α-fetoprotein) (Table 1) (Roskams et al., 2004; Roskams 2006). They are also positive for more common stem cell markers such as CD34+, Thy-1+, c-Kit+, and Flt-3+ (FMS-like tyrosine kinase 3) (Burke et al., 2007). Thus, it currently remains unclear whether these stem cells are derived from the bone marrow and just migrate to this periportal niche or whether they represent true resident liver stem/progenitor cells. Binding of stroma-derived factor-1α (SDF-1α) to its surface receptor CXCR4 activates oval hepatic cells (Hatch et al., 2002). CSC were identified as CD117+/CD133+ hepatic precursors in regenerating liver tissue (Craig et al., 2004) and a CD45−/CD90+ subpopulation of tumor cells in HCC (Yang et al., 2008). The CD90+ cells are not present in the normal liver and, when injected into immunodeficient mice, create tumors repeatedly.
Table 1.
Markers for liver cancer stem cells
| Gene name | Other name | Function | Species | Organ | References |
|---|---|---|---|---|---|
| CD133 | Prominin 1 (PROM1) | Glycoprotein, membrane protrusions | Human, Mouse | Liver, Brain | a |
| CD49f | Integrinα chain α6 (ITGA6) | Cell adhesion, cell signaling | Mouse | Liver | b |
| CD90 | Thy-1 | Glycophosphatidylinositol (GPI) anchor | Mouse | Liver | c |
| CD44 | Hyaluronic acid receptor | Cell adhesion and migration, metastasis | Mouse | Liver, Breast | d |
| CD117 | KIT | C-kit receptor Cytokine receptor | Mouse | Liver | e |
| CK19 | Cytokeratin 19 | Biliary lineage marker | Mouse | Liver | f |
| OV-6 | Oval cell marker | Early progenitor cells | Human | Liver | g |
| CD34 | Glycoprotein | Cell-cell adhesion factor | Mouse | Liver, Leukemia | h |
| AFP | α-fetoprotein | Fetal counterpart of serum albumin | Mouse | Liver | i |
In human HCC and HCC cell lines, specifically CD133+ cells, not CD133− cells, had the ability to self-renew, create differentiated progenies, and form tumors (Ma et al., 2007). This coincided with the expression of genes associated with stem/progenitor status, such as β-catenin, NOTCH, BMI, and OCT3/4. When compared to CD133− cells, the CD133+ cells isolated from the HCC cell lines showed higher expression of CD44 and CD34, but both CD133 subpopulations displayed similar expression for CD29, CD49f (integrin α6), CD90 and CD117 (Ma et al., 2007). The role of cancer stem cells in HCV-alcohol associated liver carcinogenesis is thus an active area of investigation.
In summary, alcohol and HCV NS5A synergistically induce liver tumor development via induction and activation of TLR4 in mice. The importance of Nanog as a direct downstream gene of TLR4 in this oncogenesis has also been identified. Pharmacologic inhibition of TLR4 signaling may become a novel therapeutic strategy for alcohol/HCV-associated liver tumors.
Summary and Future Directions
Research studies discussed in this review demonstrate that alcohol and HCV independently and jointly alter immune and cellular pathways to result in liver damage (Figure 1). Both alcohol and HCV activate pathways of ROS production, cytokine production, induction of TLR expression and proteasome activity. Together, these effects modulate antigen presentation, dendritic cell function and development of a liver stem cell phenotype. Experimental evidence support the hypothesis that alcohol and hepatitis C virus can have additive negative effects on functions of multiple cell types in the liver including hepatocytes, immune cells and stem cells thereby affecting antiviral immunity, hepatocyte survival, liver generation and oncogenesis in favor of HCV survival and replication. While HCV has various ways to affect cell functions, the core protein of the HCV virus appears to be a key modulator of many of these cellular functions.
Figure 1. A schematic of the interactions between alcohol and HCV and their impact on immune cells and liver cells.
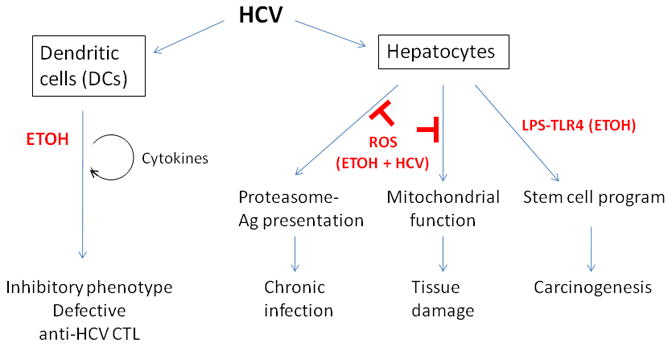
Abbreviation. Ag: antigen.
It is important to note that none of the studies described here were conducted in the context of replicating HCV or the entire HCV genome. While this does not necessarily diminish the significance of the outlined studies, confirmation in an authentic replication/infection system of the modulating effects of HCV proteins and alcohol on cellular and molecular function awaits further studies. It is useful to note, however, that replication of the JFH1 strain of HCV in Huh7.5 cells has nearly identical effects as core protein on mitochondrial ROS production (Wang et al., J. Viral Hepatitis Epub ahead of print) and proteasome inactivation (Osna, unpublished observations).
Increasing evidence suggests that the combination of alcohol use and HCV amplify abnormalities of the functions of dendritic cells that otherwise are induced by the individual insults. In response to LPS stimulation, DCs isolated from alcohol-fed mice had decreased IFNγ and IL-12 but increased IL-10 production (Aloman et al., 2007). This observation is remarkably in parallel to the effect of HCV core protein on human myeloid dendritic cells (Dolganiuc et al., 2004; Dolganiuc et al., 2006). The role of HCV core protein in induction of dendritic cell defects appears to be central as impaired CTL responses in mice immunized with a DNA construct expressing HCV core protein showed decreased activity that could be corrected by transfer of dendritic cells from control but not from alcohol-treated mice. A similar inhibitory effect of HCV core protein has been shown in human monocyte-derived dendritic cells along with an amplified inhibition of DC T cell activating capacity with the combination of alcohol and HCV infection (Dolganiuc et al., 2004).
The pathogenic role of HCV core protein is also evident in hepatocytes. While mitochondrial oxidative stress is primarily induced by the HCV core protein, NS3, NS4a and NS5a proteins as well as the replicating JFH1 culture strain of HCV can induce ROS and mitochondrial injury (Li et al., 2007). Studies by the Weinman group demonstrated that through disturbing mitochondrial Ca2+ transport from the ER, HCV core protein can amplify mitochondrial superoxide production.
While alteration of the mitochondrial electron transport by chronic alcohol has been demonstrated in several previous studies, recent evidence suggest a combined effect of alcohol and HCV core protein on mitochondria. The increased cell death due to HCV and alcohol could be prevented by antioxidants suggesting an important role for ROS (Korenga et al., 2005a).
Recent interesting findings link proteasome function and oxidative stress in alcoholic liver disease and HCV infection. Data presented by Osna indicate that the combined action of HCV core protein and ethanol may interfere with proteasome activity (Osna et al., 2008). Such change in proteasome activity was suggested to contribute to alterations in proteasome-dependent generation of peptides for MHC class-I restricted antigen presentation in hepatocytes.
While clinical evidence for the combined effects of HCV infection and alcohol in hepatocellular cancer development is well established, only recent developments provide molecular basis in potential explanation for promotion of HCC by alcohol plus HCV infection. Alcohol administration promotes the expression of Nanog, a stem cell/progenitor marker in the liver in an LPS/TLR4-dependent manner (Machida et al., 2009). Furthermore, the highest expression of Nanog was found in alcohol-fed HCV NS5a-transgenic mice after repetitive LPS injections (Machida et al., 2009). These observations raise important aspects in the potential role of gut-derived LPS in alcoholic liver disease in the development of HCC. Interestingly, increased levels of circulating LPS were reported not only in models of alcoholic liver disease but also in patients with chronic HCV infection and this was associated with increased inflammatory cell activation likely as a result of a loss of TLR tolerance (Dolganiuc et al., 2007). Thus, ethanol-induced increase in portal and systemic endotoxin levels likely promote HCV-induced inflammation and HCC development.
Together, the combined effects of alcohol and HCV infection seem to emerge both at molecular and cellular levels suggesting that amplification of the negative effects of alcohol by HCV infection and vice versa deserves further careful investigations.
Acknowledgments
The research reported here was supported by grants from NIH/NIAAA, #AA014372 to GS, #AA-008169 and AA-02666 to JRW, #AA017232 to NAO, and #AA012863 to SAW.
References
- Abdalla MY, Ahmad IM, Spitz DR, Schmidt WN, Britigan BE. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J Med Virol. 2005;76:489–97. doi: 10.1002/jmv.20388. [DOI] [PubMed] [Google Scholar]
- Aloman C, Gehring S, Wintermeyer P, Kuzushita N, Wands JR. Chronic ethanol consumption impairs cellular immune responses against HCV NS5 protein due to dendritic cell dysfunction. Gastroenterology. 2007;132:698–708. doi: 10.1053/j.gastro.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselah T, Benhamou Y, Marcellin P. Protease and polymerase inhibitors for the treatment of hepatitis C. Liver Int. 2009;29(Suppl 1):57–67. doi: 10.1111/j.1478-3231.2008.01928.x. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Barbaro G, Di Lorenzo G, Asti A, et al. Hepatocellular mitochondrial alterations in patients with chronic hepatitis C: ultrastructural and biochemical findings. Am J Gastroenterol. 1999;94:2198–205. doi: 10.1111/j.1572-0241.1999.01294.x. [DOI] [PubMed] [Google Scholar]
- Bardag-Gorce F, Li J, French BA, French SW. The effect of ethanol-induced cyp2e1 on proteasome activity: The role of 4-hydroxynonenal. Exp Mol Pathol. 2005;78:109–15. doi: 10.1016/j.yexmp.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Benali-Furet NL, Chami M, Houel L, et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921–33. doi: 10.1038/sj.onc.1208673. [DOI] [PubMed] [Google Scholar]
- Blonski W, Reddy KR. Hepatitis C virus infection and hepatocellular carcinoma. Clin Liver Dis. 2008;12:661–74. doi: 10.1016/j.cld.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Bureau C, Bernad J, Chaouche N, et al. Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J Biol Chem. 2001;276:23077–83. doi: 10.1074/jbc.M100698200. [DOI] [PubMed] [Google Scholar]
- Burke ZD, Thowfeequ S, Peran M, Tosh D. Stem cells in the adult pancreas and liver. Biochem J. 2007;404:169–178. doi: 10.1042/BJ20070167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin R, Saccoccio G, Masutti F, Bellentani S, Farinati F, Tiribelli C. DNA oxidative damage in leukocytes correlates with the severity of HCV-related liver disease: validation in an open population study. J Hepatol. 2001;34:587–92. doi: 10.1016/s0168-8278(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Chang ML, Chen JC, Chang MY, et al. Acute expression of hepatitis C core protein in adult mouse liver: Mitochondrial stress and apoptosis. Scand J Gastroenterol. 2008;43:747–55. doi: 10.1080/00365520701875987. [DOI] [PubMed] [Google Scholar]
- Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, Iwama A, Nakauchi H, et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006;44:240–251. doi: 10.1002/hep.21227. [DOI] [PubMed] [Google Scholar]
- Choi J, Forman HJ, Ou JH, Lai MM, Seronello S, Nandipati A. Redox modulation of the hepatitis C virus replication complex is calcium dependent. Free Radic Biol Med. 2006;41:1488–98. doi: 10.1016/j.freeradbiomed.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Choi J, Lee KJ, Zheng Y, Yamaga AK, Lai MM, Ou JH. Reactive oxygen species suppress hepatitis C virus RNA replication in human hepatoma cells. Hepatology. 2004;39:81–9. doi: 10.1002/hep.20001. [DOI] [PubMed] [Google Scholar]
- Corrao G, Aricò S. Independent and combined action of hepatitis C virus infection and alcohol consumption on the risk of symptomatic liver cirrhosis. Hepatology. 1998;27:914–9. doi: 10.1002/hep.510270404. [DOI] [PubMed] [Google Scholar]
- Craig CE, Quaglia A, Selden C, Lowdell M, Hodgson H, Dhillon AP. The histopathology of regeneration in massive hepatic necrosis. Semin Liver Dis. 2004;24:49–64. doi: 10.1055/s-2004-823101. [DOI] [PubMed] [Google Scholar]
- Curry-McCoy TV, Osna NA, Donohue TM., Jr Modulation of lysozyme function and degradation after nitration with peroxynitrite. Biochim Biophys Acta. 2009;1790:778–86. doi: 10.1016/j.bbagen.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- Dionisio N, Garcia-Mediavilla MV, Sanchez-Campos S, et al. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J Hepatol. 2009;50:872–82. doi: 10.1016/j.jhep.2008.12.026. [DOI] [PubMed] [Google Scholar]
- Dolganiuc A, Kodys K, Kopasz A, Marshall C, Mandrekar P, Szabo G. Additive inhibition of dendritic cell allostimulatory capacity by alcohol and hepatitis C is not restored by DC maturation and involves abnormal IL-10 and IL-2 induction. Alcohol Clin Exp Res. 2003;27:1023–1031. doi: 10.1097/01.ALC.0000071745.63433.32. [DOI] [PubMed] [Google Scholar]
- Dolganiuc A, Kodys K, Shin S, Finberg R, Golenbock D, Kurt-Jones E, Szabo G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 2004;127:1513–1524. doi: 10.1053/j.gastro.2004.08.067. [DOI] [PubMed] [Google Scholar]
- Dolganiuc A, Chang S, Kodys K, Mandrekar P, Bakis G, Szabo G. Hepatitis C virus (HCV)core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol. 2006;177(10):6758–6768. doi: 10.4049/jimmunol.177.10.6758. [DOI] [PubMed] [Google Scholar]
- Dolganiuc A, Norkina O, Kodys K, Catalano D, Mandrekar P, Bakis G, Marshall C, Szabo G. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology. 2007;133(5):1627–36. doi: 10.1053/j.gastro.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue TM, Jr, Cederbaum AI, French SW, Barve S, Gao B, Osna NA. Role of the proteasome in ethanol-induced liver pathology. Alcohol Clin Exp Res. 2007;31:1446–59. doi: 10.1111/j.1530-0277.2007.00454.x. [DOI] [PubMed] [Google Scholar]
- Donohue TM, Osna NA, Clemens DL. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome p450 2e1 as a model of ethanol-elicited cytotoxicity. Int J Biochem Cell Biol. 2006;38:92–101. doi: 10.1016/j.biocel.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Edsen-Moore MR, Fan J, Ness KJ, Marietta JR, Cook RT, Schlueter AJ. Effects of chronic ethanol feeding on murine dendritic cell numbers, turnover rate, and dendropoiesis. Alcohol Clin Exp Res. 2008;32:1309–1320. doi: 10.1111/j.1530-0277.2008.00699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encke J, Findeklee J, Geib J, Pfaff E, Stremmel W. Prophylactic and therapeutic vaccination with dendritic cells against hepatitis C virus infection. Clin Exp Immunol. 2005;142:362–369. doi: 10.1111/j.1365-2249.2005.02919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encke J, Wands JR. Ethanol inhibition: the humoral and cellular immune response to hepatitis C virus NS5 protein after genetic immunization. Alcohol Clin Exp Res. 2000;24:1063–1069. [PubMed] [Google Scholar]
- Encke J, zu Putlitz J, Geissler M, Wands JR. Genetic immunization generates cellular and humoral immune responses against the nonstructural proteins of the hepatitis C virus in a murine model. J Immunol. 1998;161:4917–4923. [PubMed] [Google Scholar]
- Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol. 2002;2:77–84. doi: 10.1038/nri723. [DOI] [PubMed] [Google Scholar]
- Fujita N, Horiike S, Sugimoto R, et al. Hepatic oxidative DNA damage correlates with iron overload in chronic hepatitis C patients. Free Radic Biol Med. 2007;42:353–62. doi: 10.1016/j.freeradbiomed.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Geissler M, Bruss V, Michalak S, Hockenjos B, Ortmann D, Offensperger WB, Wands JR, et al. Intracellular retention of hepatitis B virus surface proteins reduces interleukin-2 augmentation after genetic immunizations. J Virol. 1999;73:4284–4292. doi: 10.1128/jvi.73.5.4284-4292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler M, Gesien A, Tokushige K, Wands JR. Enhancement of cellular and humoral immune responses to hepatitis C virus core protein using DNA-based vaccines augmented with cytokine-expressing plasmids. J Immunol. 1997b;158:1231–1237. [PubMed] [Google Scholar]
- Geissler M, Gesien A, Wands JR. Inhibitory effects of chronic ethanol consumption on cellular immune responses to hepatitis C virus core protein are reversed by genetic immunizations augmented with cytokine-expressing plasmids. J Immunol. 1997a;159:5107–5113. [PubMed] [Google Scholar]
- Geissler M, Gesien A, Wands JR. Chronic ethanol effects on cellular immune responses to hepatitis B virus envelope protein: an immunologic mechanism for induction of persistent viral infection in alcoholics. Hepatology. 1997c;26:764–770. doi: 10.1002/hep.510260332. [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Cascio P, Saric T, Rock KL. The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol Immunol. 2002;39:147–64. doi: 10.1016/s0161-5890(02)00098-6. [DOI] [PubMed] [Google Scholar]
- Gong G, Waris G, Tanveer R, Siddiqui A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci U S A. 2001;98:9599–604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grune T, Blasig IE, Sitte N, Roloff B, Haseloff R, Davies KJ. Peroxynitrite increases the degradation of aconitase and other cellular proteins by proteasome. J Biol Chem. 1998;273:10857–62. doi: 10.1074/jbc.273.18.10857. [DOI] [PubMed] [Google Scholar]
- Harrison-Findik DD, Schafer D, Klein E, et al. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006;281:22974–82. doi: 10.1074/jbc.M602098200. [DOI] [PubMed] [Google Scholar]
- Hassan MM, Hwang LY, Hatten CJ, Swaim M, Li D, Abbruzzese JL, Beasley P, Patt YZ. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206–13. doi: 10.1053/jhep.2002.36780. [DOI] [PubMed] [Google Scholar]
- Hatch HM, Zheng D, Jorgensen ML, Petersen BE. SDF-1alpha/CXCR4: a mechanism for hepatic oval cell activation and bone marrow stem cell recruitment to the injured liver of rats. Cloning Stem Cells. 2002;4:339–351. doi: 10.1089/153623002321025014. [DOI] [PubMed] [Google Scholar]
- Heinz R, Waltenbaugh C. Ethanol consumption modifies dendritic cell antigen presentation in mice. Alcohol Clin Exp Res. 2007;31:1759–1771. doi: 10.1111/j.1530-0277.2007.00479.x. [DOI] [PubMed] [Google Scholar]
- Ho JW, Pang RW, Lau C, Sun CK, Yu WC, Fan ST, Poon RT. Significance of circulating endothelial progenitor cells in hepatocellular carcinoma. Hepatology. 2006;44:836–843. doi: 10.1002/hep.21353. [DOI] [PubMed] [Google Scholar]
- Horner SM, Gale M., Jr Intracellular innate immune cascades and interferon defenses that control hepatitis C virus. J Interferon Cytokine Res. 2009;29:489–98. doi: 10.1089/jir.2009.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Chen Y, Ye J. Inhibition of hepatitis C virus replication by peroxidation of arachidonate and restoration by vitamin E. Proc Natl Acad Sci U S A. 2007;104:18666–70. doi: 10.1073/pnas.0708423104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson SJ, Bird SM, Goldberg DJ. Influence of alcohol on the progression of hepatitis C virus infection: a meta-analysis. Clin Gastroenterol Hepatol. 2005;3:1150–9. doi: 10.1016/s1542-3565(05)00407-6. [DOI] [PubMed] [Google Scholar]
- Jain SK, Pemberton PW, Smith A, et al. Oxidative stress in chronic hepatitis C: not just a feature of late stage disease. J Hepatol. 2002;36:805–11. doi: 10.1016/s0168-8278(02)00060-0. [DOI] [PubMed] [Google Scholar]
- Kessova IG, Cederbaum AI. The effect of cyp2e1-dependent oxidant stress on activity of proteasomes in hepg2 cells. J Pharmacol Exp Ther. 2005;315:304–12. doi: 10.1124/jpet.105.088047. [DOI] [PubMed] [Google Scholar]
- Konishi M, Iwasa M, Araki J, et al. Increased lipid peroxidation in patients with non-alcoholic fatty liver disease and chronic hepatitis C as measured by the plasma level of 8-isoprostane. J Gastroenterol Hepatol. 2006;21:1821–5. doi: 10.1111/j.1440-1746.2006.04420.x. [DOI] [PubMed] [Google Scholar]
- Korenaga M, Okuda M, Otani K, Wang T, Li Y, Weinman SA. Mitochondrial dysfunction in hepatitis c. J Clin Gastroenterol. 2005a;39:S162–6. doi: 10.1097/01.mcg.0000155517.02468.46. [DOI] [PubMed] [Google Scholar]
- Korenaga M, Wang T, Li Y, et al. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem. 2005b;280:37481–8. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- Kuo G, Choo QL, Alter HJ, Gitnick GL, Redeker AG, Purcell RH, Miyamura T, Dienstag JL, Alter MJ, Stevens CE, et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–4. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- Kuroki M, Ariumi Y, Ikeda M, Dansako H, Wakita T, Kato N. Arsenic trioxide inhibits hepatitis C virus RNA replication through modulation of the glutathione redox system and oxidative stress. J Virol. 2009;83:2338–48. doi: 10.1128/JVI.01840-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzushita N, Gregory SH, Monti NA, Carlson R, Gehring S, Wands JR. Vaccination with protein-transduced dendritic cells elicits a sustained response to hepatitis C viral antigens. Gastroenterology. 2006;130:453–464. doi: 10.1053/j.gastro.2005.10.048. [DOI] [PubMed] [Google Scholar]
- Lanford RE, Evans MJ, Lohmann V, Lindenbach B, Gale M, Jr, Rehermann B, Chang KM, Lemon SM. The accelerating pace of HCV research: a summary of the 15th International Symposium on Hepatitis C Virus and Related Viruses. Gastroenterology. 2009;136:9–16. doi: 10.1053/j.gastro.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- Larrea E, Beloqui O, Munoz-Navas MA, Civeira MP, Prieto J. Superoxide dismutase in patients with chronic hepatitis C virus infection. Free Radic Biol Med. 1998;24:1235–41. doi: 10.1016/s0891-5849(97)00437-1. [DOI] [PubMed] [Google Scholar]
- Lemon SM, Walker C, Alter MJ, Yi M. In: Hepatitis C virus in Fields Virology. Fifth edition. Knipe DM, Howley PM, editors. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 1253–1304. [Google Scholar]
- Li Y, Boehning DF, Qian T, Popov VL, Weinman SA. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007;21:2474–85. doi: 10.1096/fj.06-7345com. [DOI] [PubMed] [Google Scholar]
- Libbrecht L, De Vos R, Cassiman D, Desmet V, Aerts R, Roskams T. Hepatic progenitor cells in hepatocellular adenomas. Am J Surg Pathol. 2001;25:1388–1396. doi: 10.1097/00000478-200111000-00006. [DOI] [PubMed] [Google Scholar]
- Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–2556. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–1758. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- Machida K, Cheng KT, Lai CK, Jeng KS, Sung VM, Lai MM. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J Virol. 2006a;80:7199–207. doi: 10.1128/JVI.00321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida K, Cheng KT, Sung VM, Levine AM, Foung S, Lai MM. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol. 2006b;80:866–874. doi: 10.1128/JVI.80.2.866-874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM, Asahina K, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci U S A. 2009;106:1548–1553. doi: 10.1073/pnas.0807390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood S, Kawanaka M, Kamei A, et al. Immunohistochemical evaluation of oxidative stress markers in chronic hepatitis C. Antioxid Redox Signal. 2004;6:19–24. doi: 10.1089/152308604771978318. [DOI] [PubMed] [Google Scholar]
- Maki A, Kono H, Gupta M, et al. Predictive power of biomarkers of oxidative stress and inflammation in patients with hepatitis C virus-associated hepatocellular carcinoma. Ann Surg Oncol. 2007;14:1182–90. doi: 10.1245/s10434-006-9049-1. [DOI] [PubMed] [Google Scholar]
- Mandrekar P, Catalano D, Dolganiuc A, Kodys K, Szabo G. Inhibition of myeloid dendritic cell accessory cell function and induction of T cell anergy by alcohol correlates with decreased IL-12 production. J Immunol. 2004;173:3398–3407. doi: 10.4049/jimmunol.173.5.3398. [DOI] [PubMed] [Google Scholar]
- Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44:1259–72. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RC, Li Y, Liu Q, et al. Manganese Superoxide Dismutase V16A Single-Nucleotide Polymorphism in the Mitochondrial Targeting Sequence Is Associated with Reduced Enzymatic Activity in Cryopreserved Human Hepatocytes. DNA Cell Biol. 2008 Sep 26; doi: 10.1089/dna.2008.0788. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney EM, Semendric L, Helbig KJ, et al. Alcohol metabolism increases the replication of hepatitis C virus and attenuates the antiviral action of interferon. J Infect Dis. 2008;198:1766–75. doi: 10.1086/593216. [DOI] [PubMed] [Google Scholar]
- Mendenhall CL, Seeff L, Diehl AM, Ghosn SJ, French SW, Gartside PS, Rouster SD, Buskell-Bales Z, Grossman CJ, Roselle GA, et al. Antibodies to hepatitis B virus and hepatitis C virus in alcoholic hepatitis and cirrhosis: their prevalence and clinical relevance. The VA Cooperative Study Group (No. 119) Hepatology. 1991;14:581–9. doi: 10.1016/0270-9139(91)90042-t. [DOI] [PubMed] [Google Scholar]
- Mitsuyoshi H, Itoh Y, Sumida Y, et al. Evidence of oxidative stress as a cofactor in the development of insulin resistance in patients with chronic hepatitis C. Hepatol Res. 2008;38:348–53. doi: 10.1111/j.1872-034X.2007.00280.x. [DOI] [PubMed] [Google Scholar]
- Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48:1420–9. doi: 10.1002/hep.22486. [DOI] [PubMed] [Google Scholar]
- Moriya K, Nakagawa K, Santa T, et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001;61:4365–70. [PubMed] [Google Scholar]
- Nguyen GC, Sam J, Thuluvath PJ. Hepatitis C is a predictor of acute liver injury among hospitalizations for acetaminophen overdose in the United States: a nationwide analysis. Hepatology. 2008;48:1336–41. doi: 10.1002/hep.22536. [DOI] [PubMed] [Google Scholar]
- Nishina S, Hino K, Korenaga M, et al. Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology. 2008;134:226–38. doi: 10.1053/j.gastro.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Nomura-Takigawa Y, Nagano-Fujii M, Deng L, et al. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J Gen Virol. 2006;87(Pt 7):1935–45. doi: 10.1099/vir.0.81701-0. [DOI] [PubMed] [Google Scholar]
- Okuda K. Hepatocellular carcinoma. J Hepatol. 2000;32:225–237. doi: 10.1016/s0168-8278(00)80428-6. [DOI] [PubMed] [Google Scholar]
- Okuda M, Li K, Beard MR, et al. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–75. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- Oshita M, Hayashi N, Kasahara A, Hagiwara H, Mita E, Naito M, Katayama K, et al. Increased serum hepatitis C virus RNA levels among alcoholic patients with chronic hepatitis C. Hepatology. 1994;20:1115–1120. [PubMed] [Google Scholar]
- Osna NA, Clemens DL, Donohue TM., Jr Interferon gamma enhances proteasome activity in recombinant hep g2 cells that express cytochrome p4502e1: Modulation by ethanol. Biochem Pharmacol. 2003;66:697–710. doi: 10.1016/s0006-2952(03)00252-1. [DOI] [PubMed] [Google Scholar]
- Osna NA, Donohue TM., Jr Implication of altered proteasome function in alcoholic liver injury. World J Gastroenterol. 2007;13:4931–7. doi: 10.3748/wjg.v13.i37.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osna NA, Haorah J, Krutik VM, Donohue TM., Jr Peroxynitrite alters the catalytic activity of rodent liver proteasome in vitro and in vivo. Hepatology. 2004;40:574–82. doi: 10.1002/hep.20352. [DOI] [PubMed] [Google Scholar]
- Osna NA, White RL, Krutik VM, Wang T, Weinman SA, Donohue TM., Jr Proteasome activation by hepatitis c core protein is reversed by ethanol-induced oxidative stress. Gastroenterology. 2008;134:2144–52. doi: 10.1053/j.gastro.2008.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osna NA, White RL, Thiele GM, Donohue TM., Jr Ethanol metabolism alters major histocompatibility complex class i-restricted antigen presentation in liver cells. Hepatology. 2009;49:1308–15. doi: 10.1002/hep.22787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osna NA, White RL, Todero S, McVicker BL, Thiele GM, Clemens DL, Tuma DJ, Donohue TM., Jr Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology. 2007;45:53–61. doi: 10.1002/hep.21442. [DOI] [PubMed] [Google Scholar]
- Otani K, Korenaga M, Beard MR, et al. Hepatitis C virus core protein, cytochrome P450 2E1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology. 2005;128:96–107. doi: 10.1053/j.gastro.2004.10.045. [DOI] [PubMed] [Google Scholar]
- Paradis V, Kollinger M, Fabre M, Holstege A, Poynard T, Bedossa P. In situ detection of lipid peroxidation by-products in chronic liver diseases. Hepatology. 1997;26:135–42. doi: 10.1053/jhep.1997.v26.pm0009214462. [DOI] [PubMed] [Google Scholar]
- Perlemuter G, Letteron P, Carnot F, et al. Alcohol and hepatitis C virus core protein additively increase lipid peroxidation and synergistically trigger hepatic cytokine expression in a transgenic mouse model. J Hepatol. 2003;39:1020–7. doi: 10.1016/s0168-8278(03)00414-8. [DOI] [PubMed] [Google Scholar]
- Piasecki BA, Lewis JD, Reddy KR, Bellamy SL, Porter SB, Weinrieb RM, Stieritz DD, Chang KM. Influence of alcohol use, race, and viral coinfections on spontaneous HCV clearance in a US veteran population. Hepatology. 2004;40:892–9. doi: 10.1002/hep.20384. [DOI] [PubMed] [Google Scholar]
- Piccoli C, Quarato G, Ripoli M, et al. HCV infection induces mitochondrial bioenergetic unbalance: causes and effects. Biochim Biophys Acta. 2009;1787:539–46. doi: 10.1016/j.bbabio.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Piccoli C, Scrima R, Quarato G, et al. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology. 2007;46:58–65. doi: 10.1002/hep.21679. [DOI] [PubMed] [Google Scholar]
- Qadri I, Iwahashi M, Capasso JM, et al. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: role of JNK, p38 MAPK and AP-1. Biochem J. 2004;378(Pt 3):919–28. doi: 10.1042/BJ20031587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian SB, Princiotta MF, Bennink JR, Yewdell JW. Characterization of rapidly degraded polypeptides in mammalian cells reveals a novel layer of nascent protein quality control. J Biol Chem. 2006;281:392–400. doi: 10.1074/jbc.M509126200. [DOI] [PubMed] [Google Scholar]
- Ratinier M, Boulant S, Crussard S, McLauchlan J, Lavergne JP. Subcellular localizations of the hepatitis C virus alternate reading frame proteins. Virus Res. 2009;139:106–10. doi: 10.1016/j.virusres.2008.09.011. [DOI] [PubMed] [Google Scholar]
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest. 2009;119:1745–54. doi: 10.1172/JCI39133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- Roskams TA, Theise ND, Balabaud C, Bhagat G, Bhathal PS, Bioulac-Sage P, Brunt EM, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739–1745. doi: 10.1002/hep.20130. [DOI] [PubMed] [Google Scholar]
- Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818–3822. doi: 10.1038/sj.onc.1209558. [DOI] [PubMed] [Google Scholar]
- Rountree CB, Senadheera S, Mato JM, Crooks GM, Lu SC. Expansion of liver cancer stem cells during aging in methionine adenosyltransferase 1A-deficient mice. Hepatology. 2008;47:1288–1297. doi: 10.1002/hep.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff ER, Ozden N. Hepatitis C and alcohol. Alcohol Res Health. 2003;27:232–239. [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Ren S, Pietschmann T, et al. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J Virol. 2004;78:7958–68. doi: 10.1128/JVI.78.15.7958-7968.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimovic D, Hassan M. Inhibition of hepatitis C virus (HCV) core protein- induced cell growth by non-structural protein 4A (NS4A) is mediated by mitochondrial dysregulation. Bosn J Basic Med Sci. 2008;8:4–11. doi: 10.17305/bjbms.2008.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serejo F, Emerit I, Filipe PM, et al. Oxidative stress in chronic hepatitis C: the effect of interferon therapy and correlation with pathological features. Can J Gastroenterol. 2003;17:644–50. doi: 10.1155/2003/710693. [DOI] [PubMed] [Google Scholar]
- Shmelkov SV, St Clair R, Lyden D, Rafii S. AC133/CD133/Prominin-1. Int J Biochem Cell Biol. 2005;37:715–719. doi: 10.1016/j.biocel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, Halfon P, Inchauspé G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin-I T, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–73. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- Singal AK, Anand BS. Mechanisms of synergy between alcohol and hepatitis C virus. J Clin Gastroenterol. 2007;41:761–72. doi: 10.1097/MCG.0b013e3180381584. [DOI] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Siu L, Foont J, Wands JR. Hepatitis C virus and alcohol. Semin Liver Dis. 2009;29:188–99. doi: 10.1055/s-0029-1214374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stylianou E, Saklatvala J. Interleukin-1. Int J Biochem Cell Biol. 1998;30:1075–1079. doi: 10.1016/s1357-2725(98)00081-8. [DOI] [PubMed] [Google Scholar]
- Sumida Y, Nakashima T, Yoh T, et al. Serum thioredoxin levels as an indicator of oxidative stress in patients with hepatitis C virus infection. J Hepatol. 2000;33:616–22. doi: 10.1034/j.1600-0641.2000.033004616.x. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Sakamoto S, Tsutsumi T, et al. Molecular determinants for subcellular localization of hepatitis C virus core protein. J Virol. 2005;79:1271–81. doi: 10.1128/JVI.79.2.1271-1281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Dolganiuc A, Mandrekar P, White B. Inhibition of antigen-presenting cell functions by alcohol: implications for hepatitis C virus infection. Alcohol. 2004;33:241–249. doi: 10.1016/j.alcohol.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res. 2009;33:220–32. doi: 10.1111/j.1530-0277.2008.00842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardif KD, Waris G, Siddiqui A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005;13:159–63. doi: 10.1016/j.tim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Thoren F, Romero A, Lindh M, Dahlgren C, Hellstrand K. A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: role of NADPH oxidase-derived oxygen radicals. J Leukoc Biol. 2004;76:1180–6. doi: 10.1189/jlb.0704387. [DOI] [PubMed] [Google Scholar]
- Tirnitz-Parker JE, Tonkin JN, Knight B, Olynyk JK, Yeoh GC. Isolation, culture and immortalisation of hepatic oval cells from adult mice fed a choline-deficient, ethionine-supplemented diet. Int J Biochem Cell Biol. 2007;39:2226–2239. doi: 10.1016/j.biocel.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Tokushige K, Wakita T, Pachuk C, Moradpour D, Weiner DB, Zurawski VR, Jr, Wands JR. Expression and immune response to hepatitis C virus core DNA-based vaccine constructs. Hepatology. 1996;24:14–20. doi: 10.1002/hep.510240104. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, Machida K, Dynnyk A, Mkrtchyan H. “Second hit” models of alcoholic liver disease. Semin Liver Dis. 2009;29:178–87. doi: 10.1055/s-0029-1214373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi T, Suzuki T, Moriya K, et al. Hepatitis C virus core protein activates ERK and p38 MAPK in cooperation with ethanol in transgenic mice. Hepatology. 2003;38:820–8. doi: 10.1053/jhep.2003.50399. [DOI] [PubMed] [Google Scholar]
- Tumurbaatar B, Hao J, Weinman SA. Hepatitis C protein expression in conjunction with reduced SOD2 causes marked sensitivity to alcohol-induced liver injury. Hepatology. 2008;48:495A. [Google Scholar]
- Valgimigli M, Valgimigli L, Trere D, et al. Oxidative stress EPR measurement in human liver by radical-probe technique. Correlation with etiology, histology and cell proliferation. Free Radic Res. 2002;36:939–48. doi: 10.1080/107156021000006653. [DOI] [PubMed] [Google Scholar]
- Vendemiale G, Grattagliano I, Portincasa P, Serviddio G, Palasciamo G, Altomare E. Oxidative stress in symptom-free HCV carriers: relation with ALT flare-up. Eur J Clin Invest. 2001;31:54–63. doi: 10.1046/j.1365-2362.2001.00747.x. [DOI] [PubMed] [Google Scholar]
- Vidali M, Occhino G, Ivaldi A, Rigamonti C, Sartori M, Albano E. Combination of oxidative stress and steatosis is a risk factor for fibrosis in alcohol-drinking patients with chronic hepatitis C. Am J Gastroenterol. 2008a;103:147–53. doi: 10.1111/j.1572-0241.2007.01596.x. [DOI] [PubMed] [Google Scholar]
- Vidali M, Tripodi MF, Ivaldi A, et al. Interplay between oxidative stress and hepatic steatosis in the progression of chronic hepatitis C. J Hepatol. 2008b;48:399–406. doi: 10.1016/j.jhep.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Wang T, Weinman SA. Causes and consequences of mitochondrial reactive oxygen species generation in hepatitis C. J Gastroenterol Hepatol. 2006;21(Suppl 3):S34–7. doi: 10.1111/j.1440-1746.2006.04591.x. [DOI] [PubMed] [Google Scholar]
- Williamson CD, Colberg-Poley AM. Access of viral proteins to mitochondria via mitochondria-associated membranes. Rev Med Virol. 2009;19:147–64. doi: 10.1002/rmv.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav D, Hertan HI, Schweitzer P, Norkus EP, Pitchumoni CS. Serum and liver micronutrient antioxidants and serum oxidative stress in patients with chronic hepatitis C. Am J Gastroenterol. 2002;97:2634–9. doi: 10.1111/j.1572-0241.2002.06041.x. [DOI] [PubMed] [Google Scholar]
- Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Yano M, Ikeda M, Abe K, et al. Oxidative stress induces anti-hepatitis C virus status via the activation of extracellular signal-regulated kinase. Hepatology. 2009;50:678–88. doi: 10.1002/hep.23026. [DOI] [PubMed] [Google Scholar]
- Yuan JM, Govindarajan S, Arakawa K, Yu MC. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer. 2004;101:1009–17. doi: 10.1002/cncr.20427. [DOI] [PubMed] [Google Scholar]