

Abstract
Background
Suppressor of cytokine signaling-3 (SOCS3) is a key negative-feedback regulator of gp130 receptor that provides crucial signaling for cardiac hypertrophy and survival; however, an in vivo role of SOCS3 regulation on cardiac gp130 signaling remains obscure.
Methods and Results
We generated cardiac-specific SOCS3 knockout (SOCS3 cKO) mice. These mice showed increased activation of gp130 downstream signaling targets (STAT3, ERK1/2, AKT and p38) from 15 weeks of age and developed cardiac dysfunction from around 25 weeks of age with signs of heart failure. Surprisingly, SOCS3 cKO failing hearts had minimal histological abnormalities with intact myofibril ultrastructure. In addition, Ca2+ transients were significantly increased in SOCS3 cKO failing hearts compared to wild-type (WT) hearts. We also found that Ser23/24 residues of troponin I were hypophosphorylated in SOCS3 cKO hearts before the manifestation of cardiac dysfunction. These data suggested the presence of abnormalities in myofilament Ca2+ sensitivity in SOCS3 cKO mice. In addition to the contractile dysfunction, we found various ventricular arrhythmias in SOCS3 cKO non-failing hearts accompanied by a sarcoplasmic reticulum Ca2+ overload. To determine the contribution of gp130 signaling to the cardiac phenotype that occurs with SOCS3 deficiency, we generated cardiac-specific gp130 and SOCS3 double knockout mice. Double KO mice lived significantly longer and had different histological abnormalities when compared to SOCS3 cKO mice; thus, demonstrating the importance of gp130 signaling in the SOCS3 cKO cardiac phenotype.
Conclusions
Our results demonstrate an important role of SOCS3 regulation on cardiac gp130 signaling in the pathogenesis of contractile dysfunction and ventricular arrhythmias.
Keywords: SOCS3, gp130, heart failure, ventricular arrhythmia, cardiomyopathy, myofilament Ca2+ sensitivity, SR Ca2+ overload
Introduction
Suppressor of cytokine signaling-3 (SOCS3) is a key negative-feedback regulator of gp130, which is a common receptor component of the interleukin-6 (IL-6) family of cytokines. The IL-6 family of cytokines activates the JAK-STAT signaling pathway through gp130 and induces the expression of the cytokine-responsive genes, including SOCS3. Subsequently, SOCS3 binds to gp130 and inhibits the downstream signaling cascade that includes activation of JAK-STAT, SH2 homology containing tyrosine phosphatase 2 (SHP2)-Ras/ERK and SHP2-AKT.1–3 Accordingly, SOCS3 regulates tightly the duration and intensity of gp130 signaling in a classic negative-feedback manner. While cardiac gp130 signaling has been shown to affect significantly cardiomyocyte survival following acute pressure overload, the role of SOCS3 regulation on cardiac gp130 signaling remains unclear.4
Germ-line knockout (KO) of SOCS3 causes embryonic lethality, via placental deficiency associated with an abnormality of trophoblast giant cell differentiation.5, 6 When placental deficiency is rescued by a tetraploid embryo complementation approach, SOCS3 KO embryos show a perinatal lethality with a markedly enlarged heart and hypertrophy. Furthermore, the placental defect and cardiac abnormalities can be rescued in the embryos with leukemia inhibitory factor (LIF) receptor null background.6 These data demonstrate that SOCS3 is an essential regulator of LIF-gp130 signaling. Moreover, these data suggest that unregulated gp130 activation without SOCS3 regulation leads to cardiac hypertrophy during embryonic development. However, due to embryonic lethality, the ultimate physiological role of SOCS3 regulation on gp130 signaling in the adult heart remains unclear.
In isolated adult cardiomyocytes, gp130 stimulation by IL-6 has been reported to decrease myocyte contractility during acute exposure. In addition, chronic IL-6 stimulation induces cardiac hypertrophy.7–9 These findings indicate a potentially pathological role of gp130 signaling in the cardiomyocyte. In humans, the serum level of IL-6 is known to be elevated in patients with congestive heart failure (CHF) and the elevated levels correspond to worsening functional class, increased hospitalization rates, and poor survival.10–12 In the Framingham heart study, elevated serum IL-6 has been associated with increased risk of CHF in elderly without prior myocardial infarction.13 Furthermore, during heart transplant, myocardial IL-6 expression has been reported to be significantly increased in donor hearts with dysfunction compared to those with normal function.14 Despite the strong association between the activation of gp130 signaling and CHF in humans, the primary role of gp130 activation during the development of cardiac dysfunction has not been thoroughly evaluated using animal models.
It has been reported that SOCS3 protein and mRNA levels are decreased in failing human myocardium.15–17 In many cancer cells, the hypermethylation of CpG islands in the SOCS3 promoter inhibits the transcription of SOCS3 mRNA, resulting in epigenetic silencing of SOCS3 protein.18, 19 These data suggest that abnormalities of SOCS3 regulation on cardiac gp130 signaling could occur in diseased states.
Accordingly, we hypothesized that the loss of gp130-mediated upregulation of SOCS3 induces sustained activation of gp130 signaling in the cardiomyocyte, leading to cardiomyopathy and heart failure. In this study, we found that the absence of SOCS3 regulation in the cardiomyocyte results in early mortality that is inhibited by deletion of gp130 and is accompanied by contractile dysfunction and ventricular arrhythmias. These findings highlight a crucial role of SOCS3 and its apparent effect on gp130 signaling in the homeostasis of adult cardiomyocytes and demonstrate a previously unidentified role of gp130 signaling in the pathogenesis of cardiomyopathy.
Methods
Mice
Floxed SOCS3 (SOCS3F/+, a kind gift from Dr. Kenneth R. Chien, MGH), 20 floxed gp130 (gp130F/+), 21 and alpha myosin heavy chain-Cre (αMHC-Cre) were bred in UCSD vivarium according to institutional guidelines. These mice were backcrossed into Balb/c for at least five generations.
Immunoblot analysis and Antibodies
Protein extraction and western-blot analysis were performed as described previously.21 Primary antibodies used were; rabbit anti-STAT3, p-STAT3, p-STAT1, ERK1/2, p-ERK1/2, AKT, p-AKT, p38, p-p38, JNK, p-JNK, Caspase-3, p-TnI, TnI, p-PKA C, and PKA C (Cell Signaling). Rabbit anti-SCN5A and Cav1.2a (Alamone Labs). Rabbit anti-SOCS3 (AnaSpec Inc.) and anti-CASQ2 (H-60) (Santa Cruz). Mouse anti-RyR, NCX1, KCNQ1 and rabbit anti-KCNH2 (Abcam). Mouse anti-SERCA2a (Thermo Scientific). Rabbit anti-p-PLB (Ser16) (Badrilla).
Echocardiogram
Mice were anesthetized with 1% isoflurane. Recording was performed using a Sonos 5500 (Philips) with a 15MHz probe as described previously.21 Isolation of Adult ventricular myocytes.Cardiomyocytes were isolated as described previously.21 For wheat germ agglutinin (WGA) staining, the cardiomyocytes were plated on laminin precoated plates (10 mg/ml, Sigma) for one hour and fixed with 10% buffered formalin for 5 min followed by washing with PBS.
Ca2+ transients and sarcomere shortening measurement
The measurements were performed as outlined previously.22, 23 The specific conditions were described in supplementary methods.
Telemetry ECG
For unanesthetized ECG studies, an ECG transmitter TA10ETA-F20 or TA10EA-F20 (DSI) was subcutaneously inserted into the backs of the mice. Positive and negative leads were fixed to the right shoulder muscle and the left leg muscle, respectively. To exclude the effect of injury due to the surgery, data were collected for 24 hours after a 1-week recovery period.
Statistical analysis
Data are shown by box plot and statistical significance was evaluated using Mann-Whitney U test unless otherwise noted. The high and low lines on the box plots represent the 90th and 10th percentiles of the data, respectively. Differences in cardiomyocyte size between knockout and littermate control mice were analyzed using paired t test. The changes in echo data before and after TAC in WT and SOCS3 cKO mice were compared using repeated measure ANOVA. For multiple comparisons, 1-way ANOVA using a Tukey-Kramer post hoc test was used. Linear regression analysis was performed using least squares method. For survival rate, the differences between two groups were analyzed by Logrank (Mantel-Cox) test; p values of less than 0.05 were considered statistically significant.
Results
Generation of cardiac-specific SOCS3 KO mice
Cardiac-specific SOCS3 KO (SOCS3 cKO) mice were generated by breeding floxed SOCS3 mice with α-myosin heavy chain-Cre (αMHC-Cre) transgenic mice.20 These mice were born at expected Mendelian ratios and were fertile. To confirm that there was functional disruption of SOCS3 in the heart, we stimulated isolated adult cardiomyocytes with cardiotrophin-1 (CT-1), a known gp130 activator. After rapid phosphorylation of STAT3, maximal expression of SOCS3 protein in WT cardiomyocytes was observed at four hours post CT-1 stimulation with diminished STAT3 phosphorylation. In contrast, we observed no detectable SOCS3 expression in SOCS3 cKO cardiomyocytes post CT-1 stimulation. Moreover, this resulted in sustained STAT3 activation in the SOCS3 KO cardiomyocytes (Figure 1A). These results demonstrated the functional disruption of SOCS3 in cardiomyocytes from SOCS3 cKO mice.
Figure 1.


Successful disruption of SOCS3 and unregulated activation of gp130 downstream signaling in the heart. A, After 12 hours of serum starvation, isolated adult cardiomyocytes (8 weeks old) were stimulated with CT-1 (1nM) for the indicated time. The expression of SOCS3, phospho-STAT3 and endogenous STAT3 were estimated by western-blot (n=3 separate experiments). B, Known gp130 downstream signaling in the heart was examined at 8 and 15 weeks of age. The level of expression of each phospho-protein was normalized to the endogenous protein expression level. Expression level was calculated relative to the mean value of wild-type (WT) group (n=6 in each group). cKO; SOCS3 cKO * p<0.05 comparing SOCS3 cKO with WT hearts. Representative western-blot from three independent experiments is shown.
Activation of cardiac gp130 signaling in SOCS3 cKO mice
To evaluate the activation of cardiac gp130 signaling in SOCS3 cKO mice, we examined known gp130 downstream signaling molecules. Phosphorylation of STAT3, ERK1/2, AKT and p38 were significantly increased in SOCS3 cKO hearts compared to WT at 15 weeks of age, while no significant differences were observed at 8 weeks of age (Figure 1B). These data suggested the sustained activation of cardiac gp130 signaling in SOCS3 cKO mice by 15 weeks of age. To find potential ligands for the activation of gp130 signaling, we investigated IL-6, LIF, and CT-1 mRNA levels by quantitative PCR and found the significant increase only in CT-1 in both WT and SOCS3 cKO hearts at 15 weeks of age (Supplementary Figure 1). This suggested the physiological activation of cardiac gp130 signaling by CT-1 during postnatal cardiac development.
SOCS3 cKO mice develop dilated cardiomyopathy (DCM)
SOCS3 cKO mice had early mortality and died by 33 weeks of age (Figure 2A). Dying mice showed signs of dyspnea and had significant pleural effusions and ascites with enlarged atria and ventricles (Figure 2B, left panel). Ventricular weight to tibia length ratio (VW/TL) was significantly increased in SOCS3 cKO compared to WT mice at 28 weeks of age (Figure 2B, right panel). Isolated cardiomyocytes from SOCS3 cKO failing hearts were significantly wider and longer than those from gender-matched WT littermates (Figure 2C). Echocardiographic analyses of SOCS3 cKO mice showed significantly decreased cardiac function with dilated left ventricular (LV) cavity compared to WT from approximately 25 weeks of age (Figure 2D). To assess the presence of a potential non-specific effect of Cre overexpression on cardiac function, cardiac-specific heterozygous SOCS3 cKO mice over 28 weeks of age (SOCS3F/+, Cre) were evaluated by echocardiography. We observed no significant cardiac dysfunction in these mice (mean fractional shortening (FS) =40.8 ± 5.9%, n=4, p=0.6). These results demonstrated that SOCS3 cKO mice developed DCM, resulting in heart failure and early mortality. The fact that cardiac gp130 signaling was activated before the manifestation of the cardiac dysfunction suggested a causative association between unregulated gp130 activation and heart failure in SOCS3 cKO mice (Figure 1B and 2D).
Figure 2.

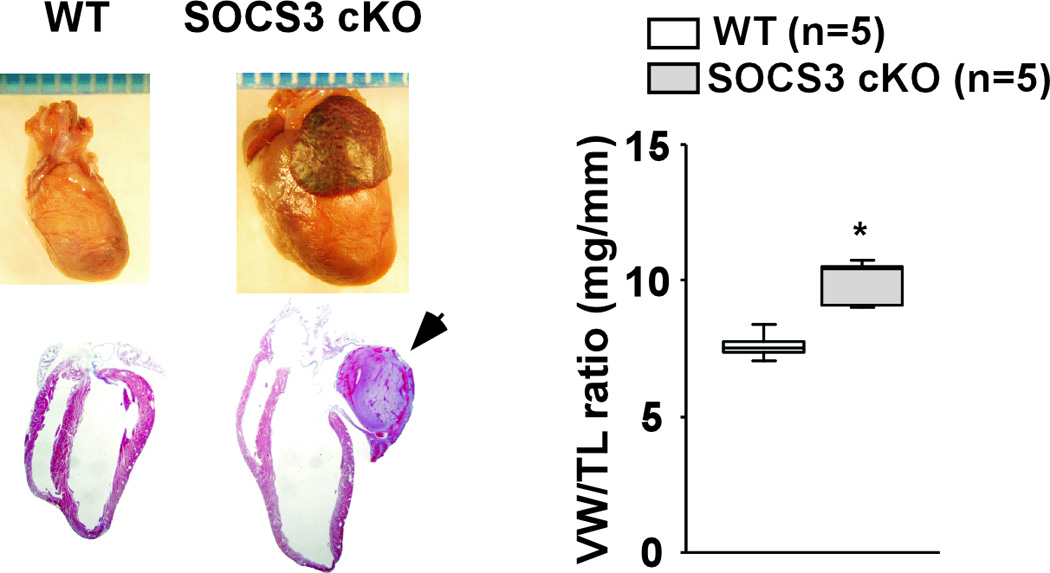
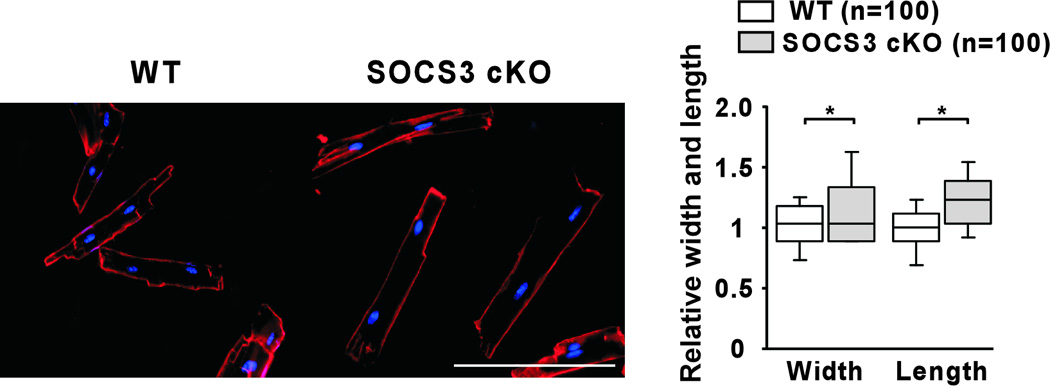


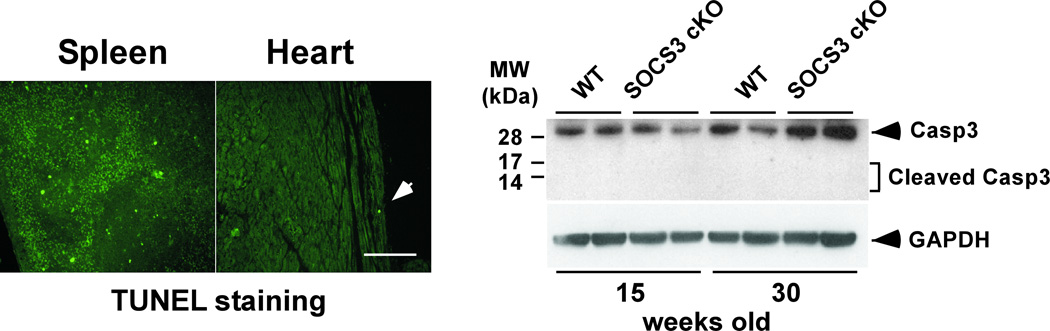
Heart failure and early mortality in SOCS3 cKO mice. A, Kaplan-Meier survival curves of WT and SOCS3 cKO mice. B, Morphology of SOCS3 cKO failing heart (28 weeks old). Mural thrombus in left atrium (arrow) was frequently observed in SOCS3 cKO mice. Scales: 1 mm. Ventricular weight to tibia length ratio (VW/TL) is shown in the panel on the right. C, Isolated cardiomyocytes were stained with WGA conjugated with Texas Red to visualize the sarcolemma membrane clearly (left panels). Width and length of SOCS3 cKO cardiomyocyte were calculated relative to that of WT cardiomyocytes (right panel). Scale bars: 100 µm. D, Echocardiography at 8, 20, 25 and 28 weeks of age (different groups of mice, n=5 in each group, were used at each time point). Representative M-mode images are shown on the top. LVEDD; Left ventricular end-diastolic dimension, LVESD; LV end-systolic dimension, %FS; percent fractional shortening. E, Hematoxylin-eosin (HE) and Masson-Trichrome (MT) staining of WT non-failing and SOCS3 cKO failing heart sections (28 weeks old, left panels). Scale bars: 100 µm (HE) and 200 µm (MT). Ultrastructure of myofibrils in WT non-failing and SOCS3 cKO failing hearts by electron microscopy (right panels). Scale bars: 1.5 µm. F, A representative TUNEL staining of the SOCS3 cKO failing heart using the spleen tissue from the same animal as a positive control (left panels). All the TUNEL positive cells in the heart were blood cells in capillaries (left panel, arrow). Caspase-3 (Casp3) cleavage was examined by western-blot using anti-casp3 antibody that can detect both cleaved and uncleaved Casp3 (right panel). Scale bar: 200 µm. *p<0.05 comparing SOCS3 cKO with WT.
Minimal histological abnormalities in SOCS3 cKO failing hearts
Despite the severe cardiac dysfunction, there was no obvious cardiomyocyte disarray, necrosis, inflammation, or interstitial fibrosis in SOCS3 cKO failing hearts (Figure 2E, left panel). Consistent with the minimal interstitial fibrosis, levels of matrix metalloproteinase (MMP)-9 and -2 in SOCS3 cKO failing hearts were equivalent to those in WT hearts (Supplementary Figure 2). The ultrastructure of the myofibrils was also intact with clear A, I, Z- and M-bands (Figure 2E, right panel). We also examined the activation of apoptosis in the SOCS3 cKO failing hearts; however, there was no significant increase in TUNEL positive cardiomyocytes (Figure 2F, left panel). Furthermore, caspase-3 cleavage was not detectable in the failing hearts by western-blot (Figure 2F, right panel). These data strongly suggested the presence of non-structural abnormalities in SOCS3 cKO failing hearts.
SOCS3 cKO mice showed cardiac dysfunction under acute-pressure overload
It has been reported that gp130 signaling is activated in the heart under acute-pressure overload induced by transverse aortic constriction (TAC).4, 24 Therefore, we examined the effect of TAC on cardiac function by echocardiography using 8-week-old SOCS3 cKO mice. Cardiac function was significantly decreased with LV chamber dilatation in SOCS3 cKO compared to WT mice at 10 days post TAC surgery. LV weight and tibial length ratio (LV/TL) was also significantly increased in SOCS3 cKO mice (Figure 3A). Consistently, p-STAT3, p-ERK1/2 and p-AKT was significantly increased in SOCS3 cKO hearts at 10 days post TAC (Figure 3B) while there werw no significant differences in the signaling between sham-operated WT and SOCS3 cKO mice at this age (Supplementary Figure 3). These results indicated the association between unregulated gp130 activation and cardiac dysfunction in SOCS3 cKO mice under acute-pressure overload.
Figure 3.


SOCS3 cKO mice developed cardiac dysfunction under acute pressure overload. A, Echocardiography before (Pre TAC) and 10 days after TAC (Post TAC) (8 weeks old, female, n=6 and 7, respectively). The changes in echo data before and after TAC were compared between the two groups using repeated measure ANOVA (*p<0.05). As a control, sham operated mice (n=5 in each group) were also examined at 10 days. PG; pressure gradient between right and left carotid arteries measured 10 days post TAC. LV/TL; LV weight and tibia length ratio. B, Activation of STAT3, ERK1/2 and AKT signaling in the heart 10 days after TAC. **p<0.05 comparing SOCS3 cKO with WT.
Increase in Ca2+ transients and abnormal myofilament Ca2+ sensitivity in SOCS3 cKO failing cardiomyocytes
Given that there were no obvious structural abnormalities in the SOCS3 cKO failing heart, we examined Ca2+ transients and twitch contraction using isolated cardiomyocytes from SOCS3 cKO failing hearts (28 weeks old). Unexpectedly, SOCS3 cKO cardiomyocytes had significantly higher Ca2+ transients with a longer time constant of [Ca2+]i decline compared to age-matched, WT cardiomyocytes (Figure. 4A, upper panels). The time constant of relaxation was significantly prolonged in SOCS3 cKO cardiomyocytes while there was no significant difference in max shortening between the two groups (Figure. 4A, lower panels). We next analyzed the relationship between peak [Ca2+]i and %sarcomere shortening in the scatter-plot graph. The %sarcomere shortening was increased in proportion to the increase in peak [Ca2+]i in WT cardiomyocytes (R2=0.896). In contrast, the proportional relationship was absent in SOCS3 cKO cardiomyocytes (Figure 4B). These results demonstrated the presence of abnormalities in relaxation and myofilament Ca2+ sensitivity in SOCS3 cKO cardiomyocytes. Given the increase in Ca2+ transients, we subsequently examined the expression level of sarcoplasmic reticulum (SR) proteins. While there were no abnormalities in SR Ca2+-ATPase (SERCA2a), phosphorylated phospholamban (p-PLB, Ser16) and endogenous PLB, we found a significant increase in cardiac calsequestrin (CASQ2) in SOCS3 cKO failing hearts (Figure. 4C). Given cardiac calsequestrin is the major Ca2+ binding protein in SR, these data implied an increase in SR Ca2+ contents in SOCS3 cKO cardiomyocytes. Furthermore, we frequently observed triggered activities in SOCS3 cKO failing cardiomyocytes during the Ca2+ transient recording when stimulated with 1.0 Hz (Figure 4D, upper panel). Interestingly, short coupling of the triggered activity sometimes induced abnormal automaticity followed by fibrillation (Figure 4D, lower panel). These results indicated the presence of an electrical instability in SOCS3 cKO failing hearts.
Figure 4.
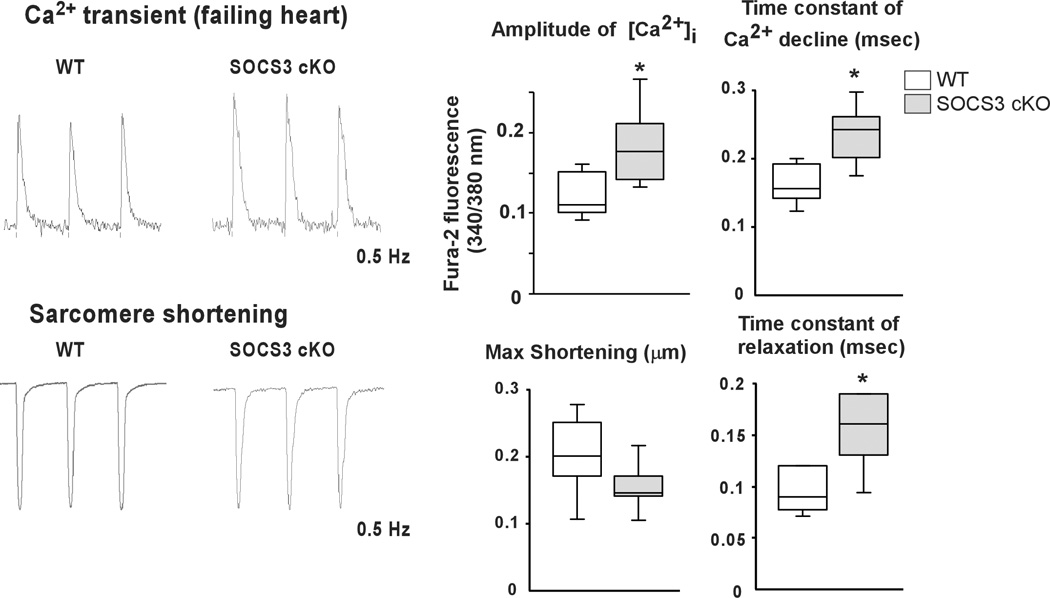
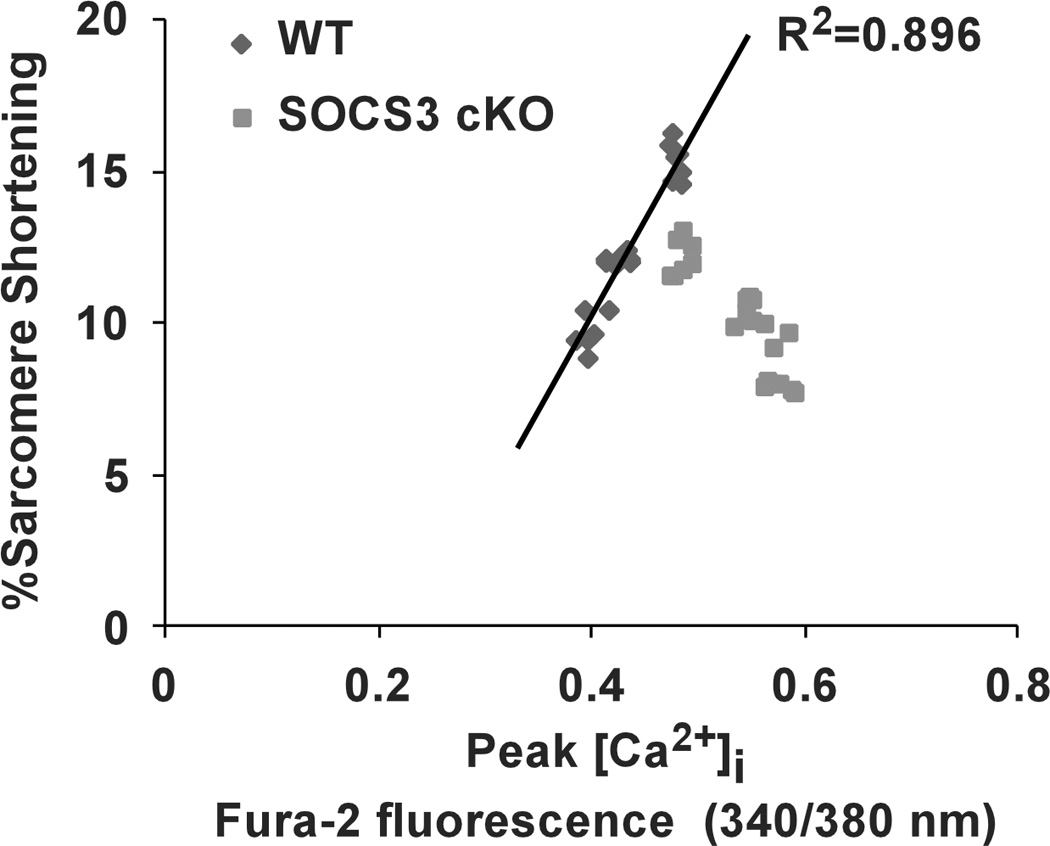

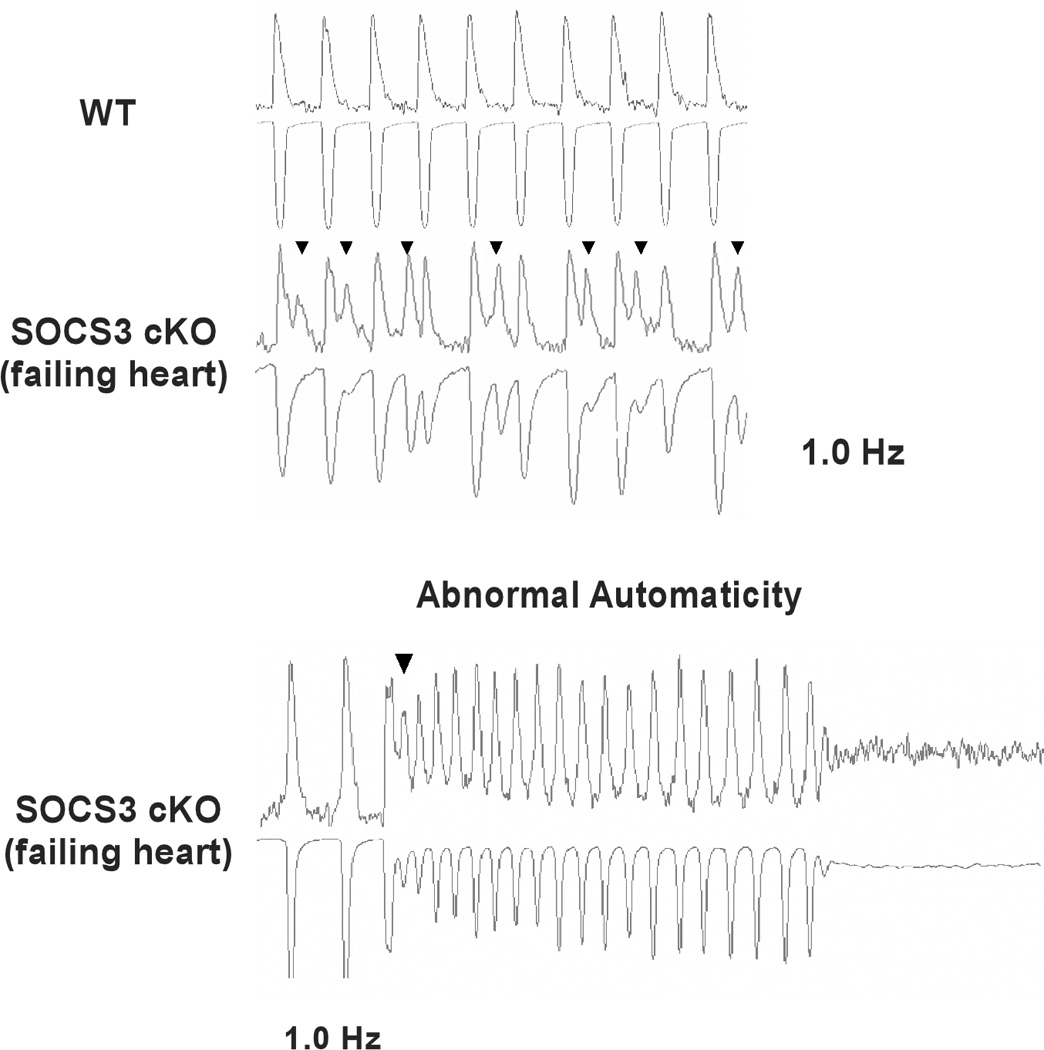
Increased Ca2+ transients and abnormal myofilament Ca2+ sensitivity in SOCS3 cKO mice. A, Ca2+ transient and sarcomere shortening measurements in SOCS3 cKO failing cardiomyocytes (28 week old). Cardiomyocytes were labeled with Fura-2 and voltage paced at 0.5 Hz. Representative recoding images for WT and SOCS3 cKO were shown on the left. Results for amplitude of [Ca2+]i, time constant of [Ca2+]i decline, max shortening, and time constant of relaxation are shown on the right. Representative recordings of 15 cardiomyocytes from three mice are shown. * p<0.05 comparing SOCS3 cKO with WT cardiomyocytes. B, The relationship between peak [Ca2+]i and %sarcomere shortening. Each dot represents each cardiomyocyte recorded. C, The expression level of SR proteins in SOCS3 cKO failing hearts. A representative western-blot from three independent experiments is shown. * p<0.05 comparing SOCS3 cKO with WT (n=5 in each group). D, Increased triggered activities in SOCS3 cKO failing cardiomyocytes. Cardiomyocytes were voltage paced at 1 Hz. Simultaneous recoding images of Ca2+ transient (top) and sarcomere shortening (bottom) are shown. Arrowheads show triggered activity. Short coupling of the triggered activity induced subsequent abnormal automaticity followed by fibrillation (bottom panel).
SR Ca2+ overload and ventricular arrhythmias in SOCS3 cKO non-failing heart
To exclude the secondary effects due to the cardiomyopathy, we investigated Ca2+ transients and SR Ca2+ contents in the SOCS3 cKO non-failing cardiomyocytes (20 weeks old). We found significantly higher Ca2+ transients and SR Ca2+ contents in SOCS3 cKO compared to age-matched WT cardiomyocytes, indicating the presence of SR Ca2+ overload in SOCS3 cKO, non-failing hearts (Figure. 5A). Moreover, we found a variety of ventricular arrhythmias such as premature ventricular contraction (PVC) and aberrancy in ventricular conduction including the possibility of ventricular tachycardia in SOCS3 cKO mice with normal cardiac function (Figure 5B and 5C). These results indicated the increase in arrhythmogenicity caused by SR Ca2+ overload in SOCS3 cKO mice.
Figure 5.
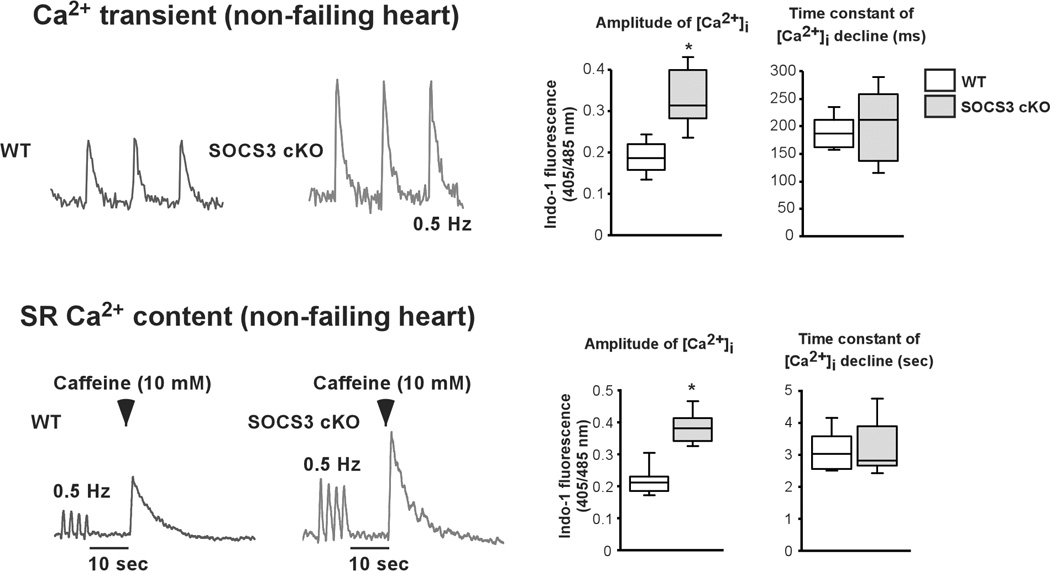



SR Ca2+ overload and ventricular arrhythmias in SOCS3 cKO non-failing hearts. A, Increased Ca2+ transients and SR Ca2+ content in SOCS3 cKO non-failing cardiomyocytes (20 weeks old). Cardiomyocytes were labeled with Indo-1 and voltage paced at 0.5 Hz. For SR Ca2+ content evaluation, cardiomyocytes were treated with 10 mM caffeine after 10 second of resting period following the 0.5 Hz pacing. Representative recordings were shown on the left. Results for amplitude of [Ca2+]i, and time constant of [Ca2+]i decline are shown on the right. * p<0.05 comparing SOCS3 cKO with WT cardiomyocytes (n=15 in each group). B and C, PVCs and ventricular conduction aberrancy that may represent ventricular tachycardia recorded in SOCS3 cKO mice (20 weeks old). In C, arrowheads show occasional PVCs. The beginning and ending of ventricular conduction aberrancy are shown in the box, respectively on the top panel. The ECG in the boxes are enlarged and shown on the bottom panel. D, The increase in SCN5A and p-PKA C, and the decrease in p-TnI found in SOCS3 cKO non-failing hearts. Representative western-blot from three independent experiments is shown with quantification from all samples tested (n=5 in each group). *p<0.05 comparing SOCS3 cKO with WT.
Unregulated gp130-STAT3 activation led to an increase in cardiac voltage-gated sodium channel (SCN5A) in SOCS3 cKO cardiomyocyte
Since we detected SR Ca2+ overload in SOCS3 cKO hearts, we subsequently examined the expression level of proteins that can affect Ca2+ transients and SR Ca2+ content; however, there were no significant abnormalities in the expression of PLB, SERCA2a, Ryanodine receptor 2 (RyR2), Na+-Ca2+ exchanger (NCX1), L-type Ca2+ channel (Cav1.2a) and potassium channels (KCNH2 and KCNQ1) (Supplementary Figure 4). In contrast, we found a significant increase in SCN5A protein and mRNA in SOCS3 cKO non-failing hearts (Figure 5D and Supplementary Figure 5A). To examine whether the gp130 activation directly increases SCN5A mRNA, we stimulated isolated WT and SOCS3 cKO cardiomyocytes with CT-1. We found a significant increase in SCN5A mRNA in SOCS3 cKO cardiomyocytes at 4 hours post CT-1 stimulation while there were no such increases in WT cardiomyocytes (Figure 6A). To identify which signaling pathway may be responsible for the effect of gp130 signaling on the increased SCN5A mRNA, we inhibited STAT3 activation in SOCS3 cKO cardiomyocytes using adenoviral vector expressing dominant-negative STAT3 (dnSTAT3) (Figure 6B, left panel).21 The inhibition of STAT3 activation significantly decreased SCN5A mRNA level both at baseline and 4 hours post CT-1 stimulation (Figure 6B, right panel). These data indicated that unregulated gp130-STAT3 activation is required for the increase in SCN5A mRNA in SOCS3 cKO cardiomyocytes.
Figure 6.


Unregulated gp130-STAT3 is required for the increase in SCN5A mRNA in SOCS3 cKO cardiomyocytes. A, SCN5A mRNA expression levels in isolated WT or SOCS3 cKO cardiomyocytes were examined by quantitative PCR at indicated hours post CT-1 (1 nM) stimulation (n=5 in each group). *p<0.05 B, Inhibition of STAT3 activation significantly decreased SCN5A mRNA level in SOCS3 cKO cardiomyocytes. Dominant-negative STAT3 (dnSTAT3) was expressed in SOCS3 cKO cardiomyocytes with adenovirus vector (Ad-dnSTAT3) infection for 12 hours. Adenovirus expressing lacZ (Ad-lacZ) was used as a negative control. Then, the cardiomyocytes were stimulated with CT-1, and the activation of STAT3 (left panel) and SCN5A mRNA (right panel) levels were examined at 4 hours post CT-1 stimulation by western-blot and quantitative PCR, respectively (n=5 in each group). *, **, ***, **** p<0.05 (1-way ANOVA using a Tukey-Kramer post hoc test).
Hypo-phosphorylation of TnI (Ser23/24) in SOCS3 cKO non-failing hearts
The absence of enhanced cardiac function under the increased Ca2+ transients in SOCS3 cKO hearts (Figure 5A) suggested the presence of abnormalities in myofilament Ca2+ sensitivity that preceded the manifestation of cardiac dysfunction. Since ultrastructure of myofibrils was intact even in the failing hearts (Figure 2E), we next focused on the phosphorylation of contractile proteins. We found hypo-phosphorylation of troponin-I (TnI) (Ser23/24) in SOCS3 cKO non-failing and failing hearts (Figure 5D and Supplementary Figure 2, respectively). This suggested a role for abnormal myofilament Ca2+ sensitivity in the cardiac dysfunction found in SOCS3 cKO mice. Unexpectedly, phospho-protein kinase A catalytic domain (p-PKA C) that phosphorylates TnI was not decreased in SOCS3 cKO non-failing and failing hearts (Figure 5D and Supplementary Figure 2, respectively).
The importance of gp130 in the development of cardiomyopathy in SOCS3 cKO mice
To determine whether unregulated gp130 activation is a primary cause of cardiomyopathy in SOCS3 cKO mice, we generated cardiac-specific gp130 and SOCS3 double KO mice (gp130&SOCS3 cDKO). We found that the increase in p-STAT3, p-ERK1/2, p-AKT and p-p38 in SOCS3 cKO hearts was abolished in gp130&SOCS3 cDKO hearts at 15 weeks of age (Figure 7A). We next examined the survival curve of gp130&SOCS3 cDKO mice. The double KO mice survived significantly longer than SOCS3 cKO mice, but died at time points that were very similar to cardiac-specific gp130 KO mice (gp130 cKO) (Figure 7B). The survival curve of gp130 cKO mice was comparable to a previous report.25 At 33 weeks of age, gp130&SOCS3 cDKO mice showed significantly decreased cardiac function by echocardiography (Supplementary Figure 6). Unlike SOCS3 cKO, histological analysis of both gp130&SOCS3 cDKO and gp130 cKO hearts revealed typical cardiomyopathic findings such as myocyte necrosis and diffuse fibrosis. The heart weight to tibia length ratio (HW/TL) was also significantly increased in both gp130&SOCS3 cDKO and gp130 cKO mice compared to SOCS3 cKO mice (Figure 7C). The similarity of cardiac phenotype between gp130 cKO and gp130&SOCS3 cDKO mice, and the clearly distinct cardiac phenotype between SOCS3 cKO and gp130&SOCS3 cDKO mice demonstrated the importance of gp130 in the development of the cardiac dysfunction in SOCS3 cKO mice.
Figure 7.


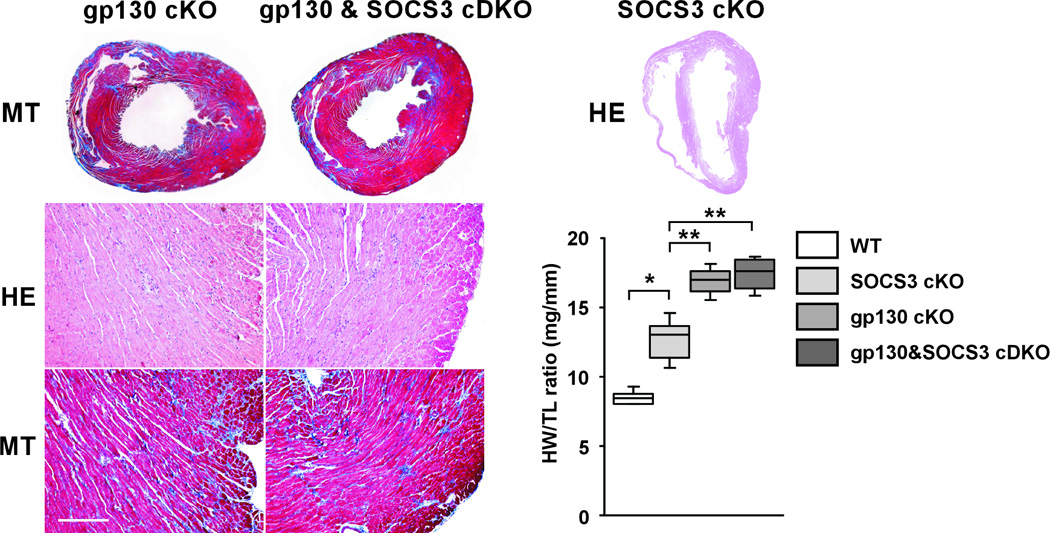
Role of gp130 activation in the development of mortality and cardiomyopathy in SOCS3 cKO mice. A, gp130 signaling activation was examined in gp130&SOCS3 cDKO (cDKO) hearts at 8 and 15 weeks of age. Representative western-blot from three independent experiments is shown (left panel). The activation of STAT3, ERK1/2, AKT and p38 signaling in SOCS3 cKO mice was abolished in the double KO mice (right panels). Expression level was calculated relative to the mean value of wild-type (WT) group (n=6 in each group). B, gp130&SOCS3 cDKO survived significantly longer than SOCS3 cKO (Logrank test p<0.0001). There was no significant difference in the survival rate between gp130&SOCS3 cDKO and gp130 cKO. C, Histological differences between SOCS3 cKO and gp130&SOCS3 cDKO failing hearts. The transverse section of gp130 cKO (35 weeks old), gp130&SOCS3 cDKO (35 weeks old) and SOCS3 cKO (28 weeks old) failing hearts are shown on the top panels. HE and MT staining of the heart tissue from gp130 cKO and gp130&SOCS3 cDKO are shown on the left lower panel. Unlike SOCS3 cKO, significant fibrosis can be observed in both groups. Scale bar: 200 µm. Heart weight and tibia length ratio (HW/TL) is shown on the right lower panel. Total five hearts in each group were examined. Error bars are ±SEM. *,** p<0.05
Discussion
In this study, we sought to determine the pathophysiological role of SOCS3 regulation in the cardiomyocyte using a cardiac-specific SOCS3 KO animal model. We demonstrated that the absence of SOCS3 allows for unregulated activation of gp130 signaling. This led to contractile dysfunction and ventricular arrhythmias accompanied by abnormal myofilament Ca2+ sensitivity and SR Ca2+ overload. We further determined that acute pressure overload in the absence of SOCS 3 results in cardiac dysfunction that is associated with increased activation of downstream markers of gp130 signaling (Figure 3). These results demonstrate the apparently important role for homeostatic regulation of gp130 signaling via SOCS3 in the cardiomyocyte (Figure 8). It is possible that there are other potential mechanisms that might account for the improvement in the phenotype that occurs with SOCS3 deficiency when gp130 is also absent in the heart. This could include the possibility that complete absence of the gp130 receptor intracellular domain could make available other gp130 interacting proteins such as JAKs and SHP-2 to be more available to interact with other receptor systems.
Figure 8.
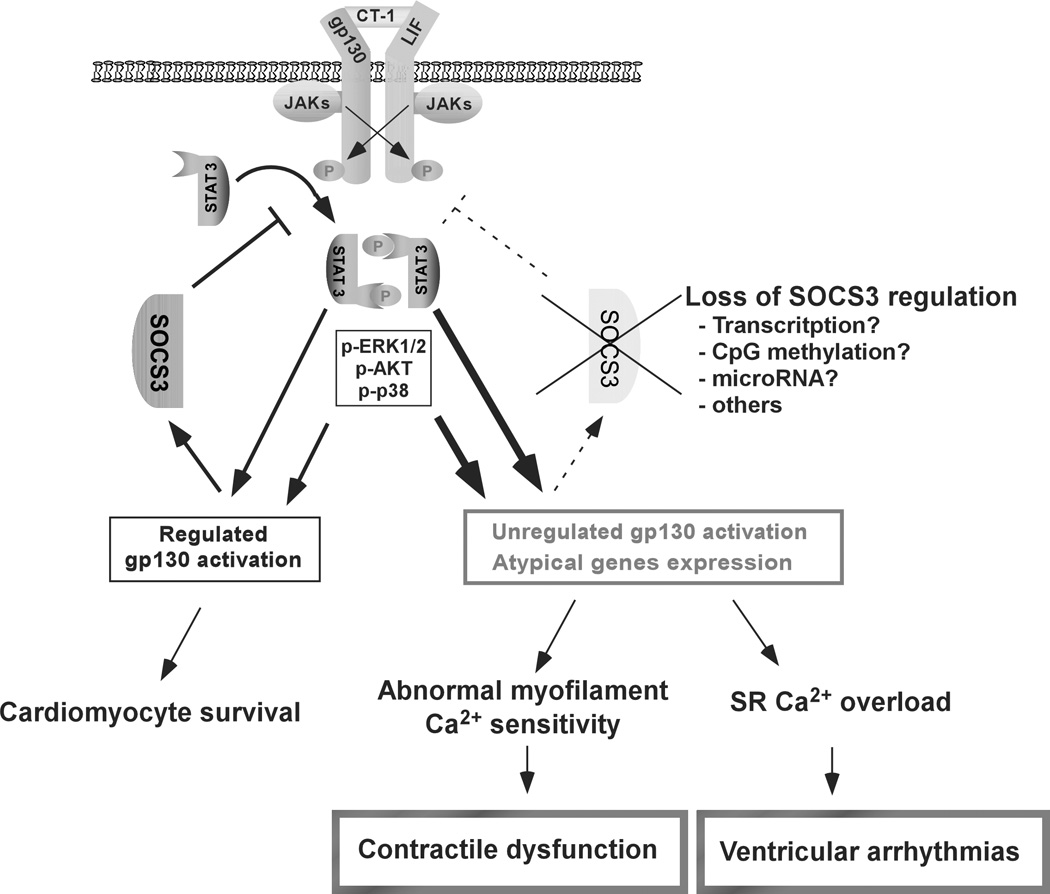
SOCS3 regulation on gp130 signaling is crucial for the maintenance of cardiomyocyte homeostasis. Under SOCS3 regulation, gp130 signaling stimulates a cardiomyocyte survival signaling cascade. Absence of SOCS3 regulation induces unregulated gp130 activation and atypical gene expression in the cardiomyocyte, which leads to contractile dysfunction and ventricular arrhythmias accompanied by abnormal myofilament Ca2+ sensitivity and SR Ca2+ overload. There are various factors that can inhibit SOCS3 protein expression such as alterations in transcription, promoter silencing through CpG methylation and microRNA expression that could potentially modulate the plasticity of gp130 signaling.
Factors that can affect SOCS3 expression level in the cardiomyocyte are poorly understood. We are not aware of any association between SOCS3 gene mutation and cardiomyopathy that has been identified in humans to date. However, decreases in SOCS3 protein and mRNA in failing human hearts have been reported.16, 17 Furthermore, epigenetic SOCS3 silencing by the hypermethylation of CpG islands in SOCS3 promoter has been reported in many cancer cells.18, 19 These data indicate that abnormalities in SOCS3 protein expression can occur in the heart.
It has been reported that IL-6 stimulation decreases cardiomyocyte contractility through a cardiac nitric oxide synthase (NOS)-dependent mechanism.7, 26, 27 However, we did not observed a significant increase in NOS activity in SOCS3 cKO failing hearts (data not shown). Furthermore, while chronic IL-6 exposure decreases Ca2+ transients via inducible NOS,28 we found an increase in Ca2+ transients in SOCS3 cKO cardiomyocytes (Figure 4A and 5A). These differences suggest that while activation of gp130 signaling can induce contractile abnormalities, underlying mechanisms may be different in the absence of SOCS3 regulation. It has been reported that SOCS3 determines the plasticity of gp130 signaling. Typically, gp130 activation by IL-6 promotes proinflammatory responses; however, in macrophage-specific SOCS3 knockout mice, IL-6 conversely attenuates the inflammatory responses. Furthermore, gp130 stimulation in SOCS3-null macrophages and liver activates transcription of genes not typical of gp130 signaling such as IFN-γ–responsive genes.20, 29, 30 Similarly, we found an atypical increase in SCN5A mRNA and protein in SOCS3 cKO hearts (Figure 5D and Supplementary Figure 5A). The increases in SCN5A mRNA and protein were abolished in gp130 and SOCS3 cDKO hearts (Supplementary Figure 5B). Moreover, the activation of gp130 by CT-1 increased SCN5A mRNA only in SOCS3 cKO cardiomyocytes (Figure 6A). These data demonstrate that unregulated gp130 activation is required for the augmented expression of SCN5A mRNA and protein.
The SCN5A gene encodes the α-subunit of cardiac voltage-gated Na+ channel. Evidence is accumulating that SCN5A mutations are associated with not only arrhythmias but also DCM; however, the underlying mechanisms are still obscure.31 In our preliminary results, the increase in SCN5A protein expression was associated with the enhanced voltage-gated Na+ current density and [Na+]i in SOCS3 cKO cardiomyocytes (data not shown). It is well-known that an increase in [Na+]i can negatively affect forward mode NCX activity, leading to an increase in [Ca2+]i.32 Although additional studies with more comprehensive electrophysiological assessments are necessary, but given the minimal histological abnormalities and the normal expression of SR proteins, L-type Ca2+ and NCX1 in SOCS3 cKO cardiomyocytes (Figure 2E and Supplementary Figure 4), we speculate that the increase in SCN5A may, at least in part, be involved in the mechanisms underlying the increase in Ca2+ transients and SR Ca2+ contents in SOCS3 cKO cardiomyocytes. The increase in SR Ca2+ content is known to enhance the open probability of ryanodine receptors.33 Thus, SR Ca2+ leak due to the Ca2+ overload is thought to be an important factor that mediates triggered activity and arrhythmogenicity in SOCS3 cKO cardiomyocytes (Figure 4D, 5B and 5C).
The relationship between peak [Ca2+]i and %sarcomere shortening indicated that there were abnormalities in myofilament Ca2+ sensitivity in SOCS3 cKO cardiomyocytes (Figure 4B). We found the hypo-phosphorylation of TnI (Ser23/24) in SOCS3 cKO hearts before the mice developed significant cardiac dysfunction (Figure 5D). Of the known TnI phosphorylation sites, Ser23/24 are the most well characterized phosphorylation sites affecting myofilament contractility.34, 35 Indeed, the hypo-phosphorylation of TnI (Ser23/24) is frequently observed in patients with heart failure.36, 37 While the down-regulation and/or desensitization of β-adrenergic receptor can induce the hypo-phosphorylation of TnI (Ser23/24),38 the increase in p-PKA C in SOCS3 cKO hearts implies that the inactivation of β-adrenergic receptor-PKA signaling pathway may not be involved in the hypo-phosphorylation of TnI (Ser23/24) (Figure 5D). However, given a possibility that whole cell PKA catalytic activity may not reflect local and targeted activity for TnI phosphorylation, further studies are necessary to determine whether unregulated gp130 activation is sufficient to change phosphorylation status of TnI in a PKA independent manner.
Since it has been shown that expression of Cre might induce a toxic effect on the myocardium in certain circumstances,39–42 we controlled for such an effect by examining the survival curve of Cre only mice (+/+, Cre) and cardiac function of cardiac-specific heterozygous SOCS3 KO mice (SOCS3F/−, Cre). Another potential problem is the timing of SOCS3 gene ablation. Given that Cre expression starts during cardiac development in our system, we could not completely exclude the possibility of myofilament remodeling as an additional factor accelerating cardiac dysfunction in SOCS3 cKO mice. Postnatal SOCS3 ablation in the cardiomyocyte would give us insights into this point.
In conclusion, disruption of SOCS3 regulation in the cardiomyocyte leads to contractile dysfunction and ventricular arrhythmias associated with increases in downstream markers of gp130 activation. Furthermore, the improvement in mortality and cardiac function in the gp130 and SOCS3 double knockout when compared with the SOCS3-deficient hearts provides strong evidence that at least part of the phenotype in the SOCS3-deficient hearts is secondary to unregulated activation of gp130 signaling. Our results are the first to support the possibility that abnormalities in SOCS3 regulation of cardiac gp130 signaling could be an important disease mechanism underlying fatal arrhythmias and DCM in humans.
Clinical Summary.
The serum level of interleukin-6 (IL-6), a ligand for gp130 receptor, is elevated in patients with congestive heart failure (CHF) and the elevated levels correspond to worsening functional class, increased hospitalization rates, and poor survival. This suggests that chronic activation of cardiac gp130 signaling may be detrimental to cardiac function. The IL-6 family of cytokines is known to induce the expression of SOCS3 through gp130 signaling activation. The expressed SOCS3 binds to gp130 and inhibits the receptor’s downstream signaling. Accordingly, SOCS3 tightly regulates the duration and intensity of gp130 signaling in a classic negative-feedback manner. It has recently been reported that there is a significant decrease in SOCS3 protein or mRNA in human failing hearts. This also implies that chronic gp130 activation and alterations in SOCS3 levels may be involved in the pathogenesis of CHF. In this study, we sought to determine the role of SOCS3 regulation on gp130 signaling in the heart using cardiac-specific SOCS3 KO mice. We demonstrated that the absence of SOCS3 allows for unregulated, chronic activation of gp130 signaling, leading to contractile dysfunction and ventricular arrhythmias accompanied by abnormal myofilament Ca2+ sensitivity and SR Ca2+ overload. These results demonstrated the importance of homeostatic regulation of cardiac gp130 signaling via SOCS3, and are the first to support the possibility that abnormalities in SOCS3 regulation on cardiac gp130 signaling could be a novel disease mechanism underlying CHF and fatal arrhythmias in humans as well.
Supplementary Material
Acknowledgments
We thank Dr. Indroneal Banerjee for critical reading of this manuscript.
Funding Sources
This work was supported by grants from the National Heart Lung and Blood Institute of the NIH (5R01HL092116 to T. Yajima, and 5P01HL046345 to K.U. Knowlton), and American Heart Association Scientist Development grant (0730333N to T. Yajima).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
References
- 1.Ernst M. Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. 2004;20:23–32. doi: 10.1016/j.tig.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Hilfiker-Kleiner D, Shukla P, Klein G, Schaefer A, Stapel B, Hoch M, Muller W, Scherr M, Theilmeier G, Ernst M, Hilfiker A, Drexler H. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122:145–155. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- 3.Yajima T, Knowlton KU. Viral myocarditis: From the perspective of the virus. Circulation. 2009;119:2615–2624. doi: 10.1161/CIRCULATIONAHA.108.766022. [DOI] [PubMed] [Google Scholar]
- 4.Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J, Jr, Muller W, Chien KR. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–198. doi: 10.1016/s0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- 5.Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, Metcalf D, Hilton DJ, Alexander WS. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9324–9329. doi: 10.1073/pnas.161271798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi Y, Carpino N, Cross JC, Torres M, Parganas E, Ihle JN. Socs3: An essential regulator of lif receptor signaling in trophoblast giant cell differentiation. EMBO J. 2003;22:372–384. doi: 10.1093/emboj/cdg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–389. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- 8.Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N. Proinflammatory cytokines in heart failure: Double-edged swords. Heart Failure Reviews. 2010;15:543–562. doi: 10.1007/s10741-010-9168-4. [DOI] [PubMed] [Google Scholar]
- 9.Szabo-Fresnais N, Lefebvre F, Germain A, Fischmeister R, Pomérance M. A new regulation of il-6 production in adult cardiomyocytes by β-adrenergic and il-1β receptors and induction of cellular hypertrophy by il-6 trans-signalling. Cell. Signal. 2010;22:1143–1152. doi: 10.1016/j.cellsig.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Hirota H, Izumi M, Hamaguchi T, Sugiyama S, Murakami E, Kunisada K, Fujio Y, Oshima Y, Nakaoka Y, Yamauchi-Takihara K. Circulating interleukin-6 family cytokines and their receptors in patients with congestive heart failure. Heart Vessels. 2004;19 doi: 10.1007/s00380-004-0770-z. [DOI] [PubMed] [Google Scholar]
- 11.MacGowan GA, Mann DL, Kormos RL, Feldman AM, Murali S. Circulating interleukin-6 in severe heart failure. Am. J. Cardiol. 1997;79:1128–1131. doi: 10.1016/s0002-9149(96)00063-x. [DOI] [PubMed] [Google Scholar]
- 12.Maeda K, Tsutamoto T, Wada A, Mabuchi N, Hayashi M, Tsutsui T, Ohnishi M, Sawaki M, Fujii M, Matsumoto T, Kinoshita M. High levels of plasma brain natriuretic peptide and interleukin-6 after optimized treatment for heart failure are independent risk factors for morbidity and mortality in patients with congestive heart failure. J. Am. Coll. Cardiol. 2000;36:1587–1593. doi: 10.1016/s0735-1097(00)00912-8. [DOI] [PubMed] [Google Scholar]
- 13.Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, Sawyer DB, Levy D, Wilson PW, D'Agostino RB. Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: The framingham heart study. Circulation. 2003;107:1486–1491. doi: 10.1161/01.cir.0000057810.48709.f6. [DOI] [PubMed] [Google Scholar]
- 14.Bulcao CF, D'Souza KM, Malhotra R, Staron M, Duffy JY, Pandalai PK, Jeevanandam V, Akhter SA. Activation of jak-stat and nitric oxide signaling as a mechanism for donor heart dysfunction. The Journal of Heart and Lung Transplantation. 2010;29:346–351. doi: 10.1016/j.healun.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mann DL, Topkara VK, Evans S, Barger PM. Innate immunity in the adult mammalian heart: For whom the cell tolls. Trans. Am. Clin. Climatol. Assoc. 2010;121:34. [PMC free article] [PubMed] [Google Scholar]
- 16.Margulies KB, Matiwala S, Cornejo C, Olsen H, Craven WA, Bednarik D. Mixed messages: Transcription patterns in failing and recovering human myocardium. Circ. Res. 2005;96:592–599. doi: 10.1161/01.RES.0000159390.03503.c3. [DOI] [PubMed] [Google Scholar]
- 17.Podewski EK. Alterations in janus kinase (jak)-signal transducers and activators of transcription (stat) signaling in patients with end-stage dilated cardiomyopathy. Circulation. 2003;107:798–802. doi: 10.1161/01.cir.0000057545.82749.ff. [DOI] [PubMed] [Google Scholar]
- 18.Fourouclas N, Li J, Gilby DC, Campbell PJ, Beer PA, Boyd EM, Goodeve AC, Bareford D, Harrison CN, Reilly JT, Green AR, Bench AJ. Methylation of the suppressor of cytokine signaling 3 gene (socs3) in myeloproliferative disorders. Haematologica. 2008;93:1635–1644. doi: 10.3324/haematol.13043. [DOI] [PubMed] [Google Scholar]
- 19.Isomoto H. Epigenetic alterations in cholangiocarcinoma-sustained il-6/stat3 signaling in cholangio- carcinoma due to socs3 epigenetic silencing. Digestion. 2009;79 Suppl 1:2–8. doi: 10.1159/000167859. [DOI] [PubMed] [Google Scholar]
- 20.Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A. Il-6 induces an anti-inflammatory response in the absence of socs3 in macrophages. Nat Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- 21.Yajima T, Yasukawa H, Jeon ES, Xiong D, Dorner A, Iwatate M, Nara M, Zhou H, Summers-Torres D, Hoshijima M, Chien KR, Yoshimura A, Knowlton KU. Innate defense mechanism against virus infection within the cardiac myocyte requiring gp130-stat3 signaling. Circulation. 2006;114:2364–2373. doi: 10.1161/CIRCULATIONAHA.106.642454. [DOI] [PubMed] [Google Scholar]
- 22.Kondo RP. Comparison of contraction and calcium handling between right and left ventricular myocytes from adult mouse heart: A role for repolarization waveform. The Journal of Physiology. 2005;571:131–146. doi: 10.1113/jphysiol.2005.101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonough PM, Stella SL, Glembotski CC. Involvement of cytoplasmic calcium and protein kinases in the regulation of atrial natriuretic factor secretion by contraction rate and endothelin. J. Biol. Chem. 1994;269:9466–9472. [PubMed] [Google Scholar]
- 24.Yasukawa H, Hoshijima M, Gu YS, Nakamura T, Pradervand S, Hanada T, Hanakawa Y, Yoshimura A, Ross J, Chien KR. Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J. Clin. Invest. 2001;108:1459–1467. doi: 10.1172/JCI13939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: The gp130-stat3 axis. Basic Res. Cardiol. 2007;102:279–297. doi: 10.1007/s00395-007-0658-z. [DOI] [PubMed] [Google Scholar]
- 26.Kinugawa K, Takahashi T, Kohmoto O, Yao A, Aoyagi T, Momomura S, Hirata Y, Serizawa T. Nitric oxide-mediated effects of interleukin-6 on ca2+ (i) and cell contraction in cultured chick ventricular myocytes. Circ. Res. 1994;75:285–295. doi: 10.1161/01.res.75.2.285. [DOI] [PubMed] [Google Scholar]
- 27.Yu X. Jak2/stat3, not erk1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes. J. Biol. Chem. 2003;278:16304–16309. doi: 10.1074/jbc.M212321200. [DOI] [PubMed] [Google Scholar]
- 28.Yu XW. Inhibition of sarcoplasmic reticular function by chronic interleukin-6 exposure via inos in adult ventricular myocytes. The Journal of Physiology. 2005;566:327–340. doi: 10.1113/jphysiol.2005.086686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. Socs3 negatively regulates il-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 30.Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. Socs3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 31.Bezzina CR, Remme CA. Dilated cardiomyopathy due to sodium channel dysfunction: What is the connection? Circ Arrhythm Electrophysiol. 2008;1:80–82. doi: 10.1161/CIRCEP.108.791434. [DOI] [PubMed] [Google Scholar]
- 32.Bers DM, Barry WH, Despa S. Intracellular na+ regulation in cardiac myocytes. Cardiovasc. Res. 2003;57:897–912. doi: 10.1016/s0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- 33.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 34.Solaro RJ, van der Velden J. Why does troponin i have so many phosphorylation sites? Fact and fancy. J. Mol. Cell. Cardiol. 2010;48:810–816. doi: 10.1016/j.yjmcc.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yasuda S, Coutu P, Sadayappan S, Robbins J, Metzger JM. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin i in regulating relaxation in cardiac myocytes. Circ. Res. 2007;101:377–386. doi: 10.1161/CIRCRESAHA.106.145557. [DOI] [PubMed] [Google Scholar]
- 36.Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PA. Troponin i phosphorylation in the normal and failing adult human heart. Circulation. 1997;96:1495–1500. doi: 10.1161/01.cir.96.5.1495. [DOI] [PubMed] [Google Scholar]
- 37.Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: Dephosphorylation of ser23/24 on troponin i could account for the contractile defect in end-stage heart failure. J. Mol. Cell. Cardiol. 2007;42:247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 38.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 39.Buerger A, Rozhitskaya O, Sherwood MC, Dorfman AL, Bisping E, Abel ED, Pu WT, Izumo S, Jay PY. Dilated cardiomyopathy resulting from high-level myocardial expression of cre-recombinase. J. Card. Fail. 2006;12:392–398. doi: 10.1016/j.cardfail.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Loonstra A, Vooijs M, Beverloo HB, Al Allak B, van Drunen E, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by cre recombinase in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9209–9214. doi: 10.1073/pnas.161269798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt EE, Taylor DS, Prigge JR, Barnett S, Capecchi MR. Illegitimate cre-dependent chromosome rearrangements in transgenic mouse spermatids. Proc. Natl. Acad. Sci. U. S. A. 2000;97:13702–13707. doi: 10.1073/pnas.240471297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silver DP, Livingston DM. Self-excising retroviral vectors encoding the cre recombinase overcome cre-mediated cellular toxicity. Mol. Cell. 2001;8:233–243. doi: 10.1016/s1097-2765(01)00295-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.