

Abstract
Resolvins are family of lipid mediators derived from omega-3 polyunsaturated fatty acids, which are generated during the resolution phase of acute inflammation. Resolvin E1 is biosynthesized from eicosapentaenoic acid via 18(R)-hydroxyeicosapentaenoic acid (18R-HEPE) in the Cox-2 and lipoxygenase mediated pathway and has proven to exhibit potent anti-inflammatory activity. We report herein the first total chemical synthesis of 18R-HEPE and demonstrate that this compound displays in vivo bioactivity by blocking neutrophil infiltration in a murine model of zymosan-induced peritonitis.
Inflammation plays a central role in the onset and progression of Alzheimer’s disease, atherosclerosis,1 and cancer,2,3 in addition to arthritis and periodontal disease.4,5 Recently, Serhan and co-workers have identified novel oxygenated products that display potent anti-inflammatory activity within resolving inflammatory exudates. Derived from omega-3 polyunsaturated fatty acids, eicosapentaenoic acid (EPA) and docohexaenoic acid (DHA), these compounds have been termed E- and D-series resolvins, respectively.6–11 As an example, resolvin E1 (RvE1) is a metabolite of EPA, which is produced by neutrophils from 18(R)- hydroxy-5(Z),8(Z),11(Z),14(Z),16(E)-eicosapentaenoic acid (18R-HEPE) through a 5-lipoxygenase-mediated pathway.12,13 RvE1 serves as an on-demand negative feedback switch that dramatically reduces inflammatory responses, including neutrophil and dendritic cell migration and interleukin-12 production.14 Efforts to generate synthetic analogues of resolvins,15–21 as well as other lipid mediators that possess significant anti-inflammatory and pro-resolution properties, have been recently reported. Herein, we report the first total chemical synthesis of 18R-HEPE and characterize its anti-inflammatory activity in vivo.
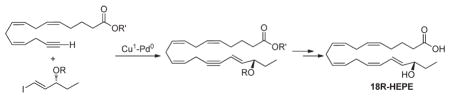
Our retrosynthetic approach for the synthesis of 18R-HEPE is outlined in Scheme 1. According to this strategy, the E,Z-conjugated diene system of 18R-HEPE is constructed using Cu(I)– Pd(0) coupling as the key step. This approach uses the terminal acetylene 7 and the vinyl iodide 13 as requisite building blocks.
Scheme 1.

Retrosynthetic Analysis for 18R-HEPE
The synthesis of 7 began with cross coupling of commercially available 4-chlorobut-2-yn-1-ol and hex-5-ynoic acid methyl ester 2, in the presence of CuI, which afforded 3 in 74% yield.22,23 The alcohol 3 was converted to the corresponding bromide 4 in the presence of CBr4/PPh3.24 Subsequent coupling of 4 with 1(trimethylsilyl)-1,4-pentadiyne in the presence of CuI, NaI, and K2CO3 afforded tetrayne 5 in 71% yield. Selective hydrogenation of 5 with Brown’s P-2 Ni method gave the TMS-protected actetylenic triene 6 in 58% yield with high isomeric purity.25–28 Liberation of the terminal acetylene then led to the key intermediate 7 in 91% yield (Scheme 2).
Scheme 2.

Synthesis of Alkyne Fragment 7
The synthesis of vinyl iodide fragment 13 was achieved from commercially available bis(trimethylsilylacetylene) 8, which was converted to the corresponding ketone 9 via treatment with propionyl chloride in the presence of AlCl3 as an activator.29 The known Noyori’s asymmetric transfer hydrogenation produced the chiral TMS-acetylenic alcohol 10.30–34 In contrast to published work using 5–10% of the Noyori catalyst (R,R)-TSDPEN)Ru(p-cymene) Cl2,32–34 a simple modification of this procedure leads to higher turnover numbers. Thus, freshly prepared catalyst (0.01%) was added to the degassed 2-propanol under argon atmosphere, and this was followed by slow addition of TMS-acetylenic ketone 9 in 2-propanol over 3 h. The reaction mixture was stirred for 12 h, the solvent evaporated, and the product purified by flash chromatography to give the chiral alcohol 10 in 81% yield with very high enantioselectivity.35–39 TIPS protection of the free hydroxyl functionality followed by selective desilylation of the TMS group with potassium carbonate afforded alkyne 12 in 95% yield. Hydrostannylation of 12 was achieved by heating with Bu3SnH in the presence of AIBN as an initiator, followed by exchanging the stannane for iodine. The key vinyl iodide fragment 13 was obtained in 72% overall yield and the Cu(I)– Pd(0) coupling reaction with 7 investigated.40–43 Thus, slow addition of the alkyne 7 (2 equiv) over a 2 h period using a syringe pump to a reaction mixture containing vinyl iodide 13 (1 equiv), tetrakis-triphenylphosphine palladium (0) (0.05 equiv), and copper iodide (0.1 equiv) produced the desired coupled product 14 in 88% yield. Under these reaction conditions, only a minimal amount (<5%) of glacier coupling of alkyne 7 was observed. The triple bond was selectively reduced by zinc–copper couple to provide the protected HEPE 15 in 56% yield.44 All other (Z)-selective methods, such as Lindlar hydrogentation, palladium(0) poisoned with BaSO4, and Brown’s P-2 Ni protocol failed to produce 15, leading only to the recovery of the starting material. Deprotection of the TIPS ether in 15 with excess TBAF followed by alkaline hydrolysis of the methyl ester afforded 18R-HEPE 1 (Scheme 3).
Scheme 3.

Synthesis of 18R-HEPE
The biological activity of 18R-HEPE 1 was examined using a murine model of peritonitis (Figure 1). Eight-week-old male C57BL/6 mice were injected intraperitoneally with zymosan A (1 mg/mL) in sterile saline. 18R-HEPE (2.5 μg) was suspended in 5 μL of ethanol and dissolved in 95 μL of of sterile saline. Test compound or vehicle alone was administered intraperitoneally at the time of zymosan A injection and 1 h later. Mice were sacrificed 4 h after zymosan injection, and peritoneal lavage was performed to characterize the inflammatory cell infiltrate by flow cytometry. 18R-HEPE significantly reduced neutrophil (PMN) infiltration as compared to vehicle alone (6.06 × 106 ± 0.94 × 106 vs 10.52 × 106 ± 2.20 × 106, n = 8/group, *p < 0.01).
Figure 1.

Synthetic 18R-HEPE reduces neutrophil infiltration in a murine model of zymosan-induced peritonitis (*p < 0.01).
In summary, the total synthesis of 18R-HEPE was achieved in a convergent manner using terminal acetylene 7 and vinyl iodide 13 fragments. These fragments were coupled using Cu(I)–Pd-(0) to obtain the E,Z-conjugated diene system of 18R-HEPE. Synthetic 18R-HEPE proved to be biologically active by blocking neutrophil infiltration in a murine peritonitis model.
EXPERIMENTAL SECTION
General Experimental Methods
All reagents were purchased from a commecial supplier and used as received, unless otherwise indicated. 4-Chloro-2-butyn-1-ol, hex-5-ynoic acid methyl ester, and 1-trimethylsilyl-1,4-pentadiyne were purchased from commercial suppliers. All reactions were carried out under nitrogen with anhydrous solvents, unless otherwise stated. All organic extracts were dried over sodium sulfate and concentrated under aspirator vacuum. 1H and 13C were recorded with a Varian 400 MHz spectrometer with CDCl3 solvent. High resolution ESI-mass spectra were recorded by the Emory University Mass Spectrometry Center using a JEOL JMS-SX102 instrument.
Synthesis of 10-Hydroxydeca-5,8-diynoic Acid Methyl Ester (3)
4-Chloro-2-butyn-1-ol (3.0 g, 28.7 mmol) and hex-5-ynoic acid methyl ester (3.62 g, 28.7 mmol) were added to a suspension of CuI (11.0 g, 57.4 mmol), NaI (8.61 g, 57.4 mmol), and K2CO3 (5.94 g, 43 mmol) in 10 mL of anhydrous DMF under Ar atmosphere. The mixture was stirred overnight at room temperature and then quenched with saturated aqueous NH4Cl, and the lipophilic products were extracted with Et2O. The combined organic extracts were washed with water and brine and dried with Na2SO4. After rotary evaporation of solvents, the residue was chromatographed on silica gel to afford alcohol 3 (4.1 g, 74% yield) as colorless oil: Rf = 0.25 (30% EtOAc in hexanes); IR (neat) 3405, 2245, 1730, 1025 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 4.18 (t, J = 2.4 Hz, 2H), 3.61 (s, 3H), 3.11 (p, J = 2.0 Hz, 2H), 2.37 (t, J = 7.2 Hz, 2H), 2.16 (tt, J = 7.0 Hz, J = 2.0 Hz, 2H), 1.74 (p, J = 7.2 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ = 173.9, 80.4, 79.8, 78.8, 74.7, 51.8, 51.2, 33.0, 23.9, 18.3, 9.9; ESI-HRMS calcd for C11H14O3 [M + Na] 217.0835, obsd 217.08346.
Synthesis of 10-Bromodeca-5,8-diynoic Acid Methyl Ester (4)
A solution of PPh3 (6.09 g, 23.2 mmol) in dry CH2Cl2 (15 mL) was added dropwise to a stirred solution of alcohol 3 (4.1 g, 21.1 mmol) and CBr4 (7.45 g, 23.2 mmol) in 15 mL of dry CH2Cl2 at 0 °C. Then the mixture was stirred for another 1.5 h at 0 °C. The solvent was evaporated, and the residue was diluted with Et2O and filtered through a short pad of Celite. The filtrate was concentrated and then chromatographed on silica gel to provide bromide 4 (5.33 g, 85% yield) as a yellow oil: Rf = 0.7 (10% EtOAc in hexanes); IR (neat) 2948, 2920, 2845, 2250, 1730, 1460, 1305, 1210 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 3.88 (t, J = 2.4 Hz, 2H), 3.65 (s, 3H), 3.18 (p, J = 2.0 Hz, 2H), 2.40 (t, J = 7.2 Hz, 2H), 2.22 (tt, J = 7.0 Hz, J = 2.0 Hz, 2H), 1.78 (p, J = 7.2 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ = 173.9, 82.0, 80.2, 75.5, 74.0, 51.8, 33.0, 23.9, 18.3, 14.9, 10.2; ESI-HRMS calcd for C11H13O2Br [M + H] 257.01717, obsd 257.01685.
Synthesis of 15-(Trimethylsilyl)pentadeca-5,8,11,14-tetraynoic Acid Methyl Ester (5)
Bromide 4 (1.4 g, 5.4 mmol) and 1-trimethylsilyl-1,4-pentadiyne (0.96 g, 7.1 mmol) were added to a suspension of CuI (1.1 g, 6.0 mmol), NaI (0.89 g, 6.0 mmol), and K2CO3 (0.98 g, 7.1 mmol) in 5 mL of anhydrous DMF under Ar atmosphere. The mixture was stirred overnight at room temperature and then quenched with saturated aqueous NH4Cl, and the lipophilic products were extracted with EtOAc. The combined organic extracts were washed with water and brine and dried with Na2SO4. After rotary evaporation of solvents, the residue was chromatographed on silica gel to afford 5 (1.2 g, 71% yield) as an oil: Rf = 0.4 (12% EtOAc in hexanes); IR (neat) 2960, 2922, 2855, 2190, 1735, 1250 cm−1; 1HNMR(CDCl3, 400 MHz) δ = 3.65 (s, 3H), 3.15 (t, J = 2.4 Hz, 2H), 3.10 (dt, J = 2.4 Hz, J = 1.6 Hz, 2H), 3.07 (p, J = 2.4 Hz, 2H), 2.39 (t, J = 7.6 Hz, 2H), 2.18 (tt, J = 7.0 Hz, J = 2.0 Hz, 2H), 1.74 (p, J = 7.2 Hz, 2H), 0.12 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ = 173.8, 99.6, 85.3, 80.2, 80.1, 79.7, 75.2, 74.8, 74.1, 51.7, 33.0, 23.9, 18.3, 11.1, 10.4, 9.9, 0.1 (3C); ESI-HRMS calcd for C19H24O2Si [M + H] 313.16184, obsd 313.16189.
Synthesis of (5Z,8Z,11Z)-15-(Trimethylsilyl)pentadeca-5,8,11-trien-14-ynoic Acid Methy Ester (6)
Ni(OAc)2 · 4H2O (1.3 g, 5.1 mmol) was dissolved in 20 mL of 95% ethanol and placed under a balloon of H2 atmosphere. NaBH4 (194 mg, 5.1 mmol) was added to this solution, followed after 20 min by ethylenediamine (1.2 g, 20.4 mmol). The diyne 5 (0.8 g, 2.56 mmol) dissolved in 5 mL of absolute ethanol was added, and the reaction was stirred under H2 atmosphere at room temperature for an additional 4 h. After 4 h, the reaction mixture was filtered through a pad of Celite, and the ethanol was removed in vacuo. The reaction mixture was redissolved in EtOAc (50 mL) and washed with satd NH4Cl (30 mL) followed by brine (30 mL). The EtOAc layer was dried, concentrated, and purified by chromatography over silica gel to afford the title compound 6 as colorless oil (475 mg, 58% yield): Rf = 0.75 (12% EtOAc in hexanes); IR (neat) 2990, 2960, 2922, 2855, 2190, 1735, 1250, 890 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 5.44–5.41 (m, 2H), 5.36–5.32 (m, 4H), 3.64 (s, 3H), 2.99 (t, J = 2.4 Hz, 2H), 2.79 (dt, J = 2.4 Hz, J = 1.6 Hz, 2H), 2.76 (p, J = 2.4 Hz, 2H), 2.30 (t, J = 7.6 Hz, 2H), 2.07 (tt, J = 7.0 Hz, J = 2.0 Hz, 2H), 1.68 (p, J = 7.2 Hz, 2H), 0.13 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ = 174.2, 129.9, 129.2, 128.9, 128.8, 127.8, 124.5, 105.2, 84.5, 51.7, 33.8, 27.2, 28.7, 25.8, 24.9, 18.8, 0.3 (3C); ESI-HRMS calcd for C19H30O2Si [M − H] 317.19289, obsd 317.19337.
Synthesis of (5Z,8Z,11Z)-Pentadeca-5,8,11-trien-14-ynoic Acid Methyl Ester (7)
Cesium fluoride (154 mg, 1.01 mmol) was added to the alkyne 6 (160 mg, 0.51 mmol) dissolved in DMF (2 mL) and stirred at room temperature for 3 h. After 3 h, the reaction mixture was diluted with EtOAc (10 mL) and NH4Cl (10 mL) followed by brine (10 mL). The aqueous layer was back-extracted with EtOAc (10 mL), and the combined organic layers were dried, concentrated, and purified by chromatography over silica gel to afford the terminal alkyne 7 as a colorless oil (121 mg, 91% yield): Rf = 0.70 (12% EtOAC in hexane); IR 3300, 2990, 2958, 2920, 2840, 2120, 1730, 1640, 1450, 1240, 910 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 5.45–5.44 (m, 2H), 5.37–5.34 (m, 4H), 3.64 (s, 3H), 2.98–2.94 (m, 2H), 2.81–2.77 (m, 4H), 2.32 (t, J = 7.6 Hz, 2H), 2.09 (tt, J = 7.0 Hz, J = 2.0 Hz, 2H), 1.96 (t, J = 2.8 Hz, 1H), 1.70 (p, J = 7.2 Hz, 2H); 13CNMR (CDCl3, 100 MHz) δ = 174.3, 130.3, 129.2, 129.0, 128.8, 127.4, 124.2, 82.7, 68.3, 51.7, 33.6, 29.8, 26.7, 25.7, 24.9, 17.1; ESI-HRMS calcd for C16H22O2 [M + H] 247.16926, obsd 247.16929.
Synthesis of 1-(Trimethylsilyl)-1-pentyn-3-one (9)
Bis-(trimethylsilylacetylene) (3.0 g, 17.6 mmol) and propionyl chloride (1.68 g, 17.6 mmol) were dissolved in dichloromethane (60 mL) and the mixture cooled to 0 °C. To this solution was added aluminum chloride (2.3 g, 17.6 mmol), and the reaction mixture was stirred for 3 h. After 3 h, the mixture was poured in to 3 N HCl (50 mL) and extracted with Et2O (3 × 30 mL). The combined organic layer was dried, concentrated and purified by chromatography over silica gel (1% EtOAC/hexane) to afford the title compound 9 (2.41 g, 89% yield) as a colorless oil: Rf = 0.8 (3% EtOAc in hexanes); IR (neat) 2964, 2904, 2150, 1737, 1680, 1459, 1415, 1353, 1262, 1198 cm−1; 1H NMR (CDCl3, 400 MHz) δ = 2.53 (dd, J = 6.4 Hz, J = 12.0 Hz, 2H), 1.07 (t, J = 7.6 Hz, 3H), 0.18 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ = 188.5, 101.9, 97.7, 38.7, 8.1, −0.6 (3C); ESI-HRMS calcd for C8H14OSi [M + H] 155.08867, observed 155.08865.
Synthesis of 1-Trimethylsilyl-1-pentyn-3(R)-ol (10)
Catalyst ((R,R)-TsDPEN)Ru(p-cymene)Cl2 (22 mg, 0.005 equiv) was dissolved in 50 mL of degassed 2-propanol under Ar atmosphere. To this solution was added a solution of ketone 9 (1.46 g, 9.4 mmol) dissolved in 2-propanol (5 mL) over a period of 3 h using a syringe pump. The reaction was allowed to stir for an additional 12 h, and the solvent was concentrated to provide a crude product which was purified by chromatography over silica gel (5% EtOAc in hexanes) to afford the alcohol 10 which had spectroscopic data consistent with those in the literature39 (1.3 g, 87% yield, > 99% ee) as a colorless oil: Rf = 0.2 (5% EtOAc in hexanes); IR 3350, 2971, 2177, 1737, 1465, 1410 1335, 1255, 1120, 1018, 971, 850 cm−1; [α]19D +6.0 (c 2, CHCl3) (lit.39 [α]24D +6.1, c 2, CHCl3); 1H NMR (CDCl3, 400 MHz) δ = 4.48 (dd, J = 6.4 Hz, J = 12.0 Hz, 1H), 1.96 (br. d, – OH, J = 5.2 Hz, 1H), 1.70–1.66 (m, 2H), 0.97 (t, J = 7.6 Hz, 3H), 0.14 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ = 106.8, 89.6, 64.2, 30.9, 9.5, 0.1; ESI-HRMS calcd for C8H16OSi [M − H2O]+ 139.09376, obsd 139.09372.
Synthesis of 3(R)-Triisopropylsilyoxy-1-trimethylsilyl-1-pentyne (11)
Alcohol 10 (1.3 g, 8.3 mmol) was dissolved in CH2Cl2 (20 mL) to which TIPS-Cl (2.4 g, 12.5 mmol) and imidazole (0.85 g, 12.5 mmol) were added at 0 °C. The reaction was slowly warmed to room temperature and stirred for 12 h. After 12 h, the reaction mixture was washed with saturated NH4Cl (30 mL) and brine (20 mL). The dichloromethane layer was collected, dried, and concentrated under reduced pressure. Subsequent purification by chromatography over silica gel (5% EtOAc in hexanes) afforded TIPS ether 11 (2.1 g, 82% yield) as a colorless oil: Rf = 0.8 (5% EtOAc in hexanes); IR (neat) 2950, 1450, 1235, 1077 cm−1; [α]19D+38.3 (c 1, CHCl3); 1H NMR (CDCl3, 100 MHz) δ = 4.37 (t, J = 6.0 Hz, 1H), 1.71–1.64 (m, 2H), 1.14–1.10 (m, 21H), 0.97 (t, J = 7.6 Hz, 3H), 0.12 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ = 107.9, 84.5, 64.8, 32.0, 18.2, 12.4, 9.6, 0.01; ESI-HRMS calcd for C17H36OSi2 [M + H] 313.23775, obsd 313.23773.
Synthesis of 3(R)-Triisopropylsilyoxy-1-pentyne (12)
Compound 11 (1.0 g, 3.2 mmol) was dissolved in MeOH (20 mL) to which anhydrous K2CO3 (0.5 g, 3.8 mmol) solid was added and the mixture stirred at room temperature for 12 h. After 12 h, methanol was removed under reduced pressure, and the crude product was diluted with Et2O (30 mL) and saturated NH4Cl (30 mL). The Et2O layer was separated, dried, and concentrated under reduced pressure. Subsequent purification by chromatography over silica gel (5% EtOAc in hexanes) produced the desired alkyne 12 (730 mg, 95% yield) as colorless oil: Rf = 0.7 (5% EtOAc in hexanes); IR (neat) 3030, 2935, 1465, 1250, 1080 cm−1; Optical Rotation [α]19D + 19.5 (c 1, CHCl3); 1H NMR (CDCl3, 400 MHz) δ = 4.41 (ddd, J = 8.1 Hz, J = 6.2 Hz, J = 2.0 Hz, 1H), 2.35 (d, J = 6.0 Hz, 1H), 1.72 – 1.67 (m, 2H), 1.13 – 1.05 (m, 21H), 0.99 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ = 85.7, 72.1, 64.2,32.0, 18.2, 12.4, 9.3; ESI-HRMS calcd for C14H28OSi [M + H] 241.19822, obsd 241.19822.
Synthesis of 1(E)-Iodo-3(R)-triisopropylsilyoxypentene (13)
Alkyne 12 (200 mg, 0.83 mmol) was taken in a round-bottom flask to which AIBN (14 mg, 0.083 mmol) and tributyltin hydride (360 mg, 1.24 mmol) were added and heated to 130 °C for 3 h. After 3 h, the reaction was allowed to cool down to room temperature and cooled to 0 °C. To this was added a solution of I2 (630 mg, 2.49 mmol) dissolved in CH2Cl2 (5 mL). The reaction was slowly warmed to room temperature and stirred for additional 12 h. After 12 h, the reaction mixture was diluted with CH2Cl2 (30 mL) and washed with saturated aq Na2S2O3 (10 mL) and saturated aq NH4Cl (10 mL). The CH2Cl2 layer was then dried, concentrated, and purified by chromatography over silica gel (1% EtOAc in hexane) to afford the vinyl iodide 13 (220 mg, 72% yield) as colorless oil: IR (neat) 1235, 910, 760 cm−1; [α]19D +30.7 (c 1, CHCl3); 1H NMR (CDCl3, 400 MHz) δ = 6.5 (dd, J = 6.4 Hz, J = 15.0 Hz, 1H), 6.19 (d, J = 14.7 Hz, 1H), 4.14 (dd, J = 0.8 Hz, J = 4.8 Hz, 1H), 1.56 – 1.51 (m, 2H), 1.14 – 1.10 (m, 21H), 0.97 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ = 149.3, 76.5, 75.9, 30.8, 18.2, 12.5, 8.8; ESI-HRMS calcd for C14H29IOSi [M + H] 369.11126, obsd 369.11099.
Synthesis of 18(R)-(5Z,8Z,11Z,16E)-Triisopropylsilyloxy-5,8,11-trien-14-ynoic Acid Methyl Ester (14)
Piperidine (56 μL, 0.57 mmol) and cuprous iodide (6 mg, 0.028 mmol) were added at room temperature to a solution of vinyl iodide 13 (105 mg, 0.28 mmol) and tetrakis(triphenylphosphine)palladium (17 mg, 0.014 mmol). To this solution was added dienyne 7 (140 mg, 0.57 mmol) in benzene (2 mL) dropwise over a period of 2 h. The mixture was stirred for an additional 6 h, diluted with EtOAc (10 mL), and washed with a saturated solution of NH4Cl (10 mL) and brine (10 mL). The EtOAc layer was collected, dried over Na2SO4, and purified by chromatography over silica gel (eluent: 8% EtOAC in hexane) to provide 14 (243 mg, 88% yield) as a colorless oil: Rf = 0.5 (8% EtOAc in hexanes); IR (neat) 2210, 1735, 730, 710 cm−1; [α]19D +4.6 (c 0.6, CHCl3); 1HNMR (400 MHz, CDCl3) δ = 6.01 (dd, J = 6.0 Hz, J = 16.0 Hz, 1H), 5.63 (dd, J = 5.2 Hz, J = 15.5 Hz, 1H), 5.45 - 5.44 (m, 2H), 5.38 - 5.33 (m, 4H), 4.20 (q, J = 6.0 Hz, 1H), 3.66 (s, 3H), 3.08 (d, J = 6.4 Hz, 2H), 2.81 (dt, J = 2.0 Hz, J = 6.0 Hz, 2H), 2.78 (t, J = 5.2 Hz, 2H), 2.31 (t, J = 7.2 Hz, 2H), 2.08 (dd, J = 7.2 Hz, J = 13.6 Hz, 2H), 1.68 (dt, J = 15.6 Hz, J = 14.8 Hz, 2H), 1.55 (m, 2H), 1.02 (m, 21H), 0.82 (t, J = 7.6 Hz, 3H); 13CNMR (CDCl3, 100 MHz) δ = 175.0, 145.4, 128.9, 128.8, 129.7, 129.2, 127.8, 124.8, 109.3, 88.1, 78.8, 73.9, 51.8, 33.6, 31.0, 29.5, 26.7, 25.8, 25.0, 24.9, 18.2 (6C), 12.5 (3C), 8.7; ESI-HRMS calcd for C30H50O3Si [M + H] 487.36020, obsd 487.36043.
Synthesis of 18(R)-(5Z,8Z,11Z,14Z,16E)-Triisopropylsilyloxyeicosapentaenoic Acid Methyl Ester (15)
To a solution of compound 14 (22 mg, 0.041 mmol) in MeOH/H2O (1:1, 20 mL) was added activated zinc powder (1 g). The reaction mixture was warmed to 40 °C and stirred for 12 h, after which the mixture was filtered on a pad of Celite. Methanol was removed under reduced pressure, and the reaction mixture was diluted with EtOAc (15 mL) and extracted. The EtOAc layer was dried, concentrated, and purified by chromatography over silica gel (8% EtOAc in hexanes) to afford the protected HEPE 15 (12 mg, 56% yield) as a colorless oil: Rf = 0.6 (8% EtOAc in hexanes); IR (neat) 1733, 1670, 1665, 735, 710 cm−1; [α]19D −11.2 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ = 6.43 (dd, J = 4.0 Hz, J = 15.2 Hz, 1H), 6.03–5.96 (m, 1H), 5.62 (dd, J = 6.4 Hz, J = 14.8 Hz, 1H), 5.45 - 5.30 (m, 7H), 4.21 (q, J = 5.6 Hz, 1H), 3.65 (s, 3H), 3.07 (dd, J = 2.0 Hz, J = 6.4 Hz, 1H), 2.91 (t, J = 2.4 Hz, 1H), 2.80 (t, J = 5.6 Hz, 2H), 2.77 (t, J = 5.2 Hz, 2H), 2.28 (t, J = 7.6 Hz, 2H), 2.08 (dd, J = 6.8 Hz, J = 13.6 Hz, 2H), 1.69 (dt, J = 15.6 Hz, J = 14.8 Hz, 2H), 1.57 (m, 2H), 1.02 (m, 21H), 0.87 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ = 174.1, 137.4, 129.2, 129.1, 128.8, 128.7, 126.6, 126.5, 128.2, 128.1, 124.8, 73.9, 51.8, 33.6, 31.4, 31.0, 26.7, 25.9, 24.9, 24.8, 18.3 (6C), 12.5, (3C), 8.7; ESI-HRMS calcd for C30H52O3Si [M + H] 489.37640, obsd 489.37378.
Synthesis of 18(R)-Hydroxy-(5Z,8Z,11Z,14Z,16E)-eicosapentaenoic Acid (1)
Compound 15 (10 mg, 0.02 mmol) was dissolved in THF (1 mL) to which a 1 M TBAF in THF solution (0.1 mL, 0.1 mmol) was added under Ar atmosphere and stirred at room temperature for 10 h. The reaction was monitored by TLC, and after 10 h, the reaction was diluted with EtOAC (5 mL) and washed with saturated aq NH4Cl (2 × 5 mL). The organic phase was extracted and concentrated under reduced pressure. The resulting crude was subsequently dissolved in THF/H2O (1:1, 2 mL), and the reaction was allowed to stir at room temperature for 5 h. After 5 h, the organic phase was removed under reduced pressure and carefully acidifed to pH 5 using 1 N HCl. The aqueous layer was extracted with EtOAc (3 × 5 mL), dried, concentrated, and purifed by chromatography over silica gel (40% EtOAc in hexanes) to afford 1 (4.6 mg, 71% yield) as colorless oil: Rf = 0.3 (40% EtOAc in hexanes); IR (neat) 3510, 3475, 1730, 1665, 735, 710 cm−1; [α]19D −17.9 (c 0.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ = 6.51 (dd, J = 4.0 Hz, J = 15.2 Hz, 1H), 5.99 (t, J = 10.8 Hz, 1H), 5.63 (dd, J = 6.4 Hz, J = 14.8 Hz, 1H), 5.39–5.33 (m, 7H), 4.09 (q, J = 5.6 Hz, 1H), 2.95 (t, J = 2.4 Hz, 2H), 2.83 (t, J = 5.6 Hz, 2H), 2.78 (t, J = 5.2 Hz, 2H), 2.30 (t, J = 7.6 Hz, 2H), 2.08 (dd, J = 6.8 Hz, J = 13.6 Hz, 2H), 1.68 (dt, J = 15.6 Hz, J = 14.8 Hz, 2H), 1.57 (m, 2H), 0.87 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ = 177.9, 136.0, 130.4, 129.2, 129.0, 128.8, 128.4, 128.2, 128.1, 127.8, 125.7, 74.2, 33.2, 30.3, 30.0, 26.6, 26.3, 25.9, 24.7, 9.9; ESI-HRMS calcd for C20H30O3 [M − H]−, expected 317.21222, obsd 317.21244.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health HL106018. We thank Dr. Fred Strobel, Emory University Mass Spectrometry Center, for valuable suggestions.
Footnotes
Supporting Information. 1H and 13C NMR spectra for new compounds 1, 3–7, and 9–15. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Helgadottir A, Manolescu A, Thorleifsson G, Gretarsdottir S, Jonsdottir H, Thorsteinsdottir U, Samani NJ, Godmundsson G, Grant SFA, Thorgeirsson G, Sveinbjornsdottir S, Valdimarsson EM, Matthiasson SE, Johannsson H, Gudmundsdottir O, Gurney ME, Sainz J, Thorhallsdottir M, Andresdottir A, Frigge ML, Topol EJ, Kong A, Gudnason V, Hakonarson H, Gulcher JR, Stefansson K. Nat Genet. 2004;36:233–239. doi: 10.1038/ng1311. [DOI] [PubMed] [Google Scholar]
- 2.Erlinger TP, Platz EA, Rifai N, Helzlsouer KJ. J Am Med Assoc. 2004;291:585–590. doi: 10.1001/jama.291.5.585. [DOI] [PubMed] [Google Scholar]
- 3.Pasche B, Serhan CN. J Am Med Assoc. 2004;291:623–624. doi: 10.1001/jama.291.5.623. [DOI] [PubMed] [Google Scholar]
- 4.Gallin JI, Snyderman R, Fearon DT, Haynes BF, Nathan C. flammation: Basic Principles and Clinical Correlates. Lippincott Williams & Wilkins; Philadelphia: 1999. p. 1360. [Google Scholar]
- 5.Van Dyke TE, Serhan CN. J Dent Res. 2003;82:82–90. doi: 10.1177/154405910308200202. [DOI] [PubMed] [Google Scholar]
- 6.Serhan CN, Chiang N, Van Dyke TE. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serhan CN, Chiang N. Br J Pharmacol. 2008;153:S200–S215. doi: 10.1038/sj.bjp.0707489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwab JM, Chiang N, Arita M, Serhan CN. Nature. 2007;447:869–875. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. J Exp Med. 2000;192:1197–1204. 6. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. J Exp Med. 2002;196:1025–103. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong S, Gronert K, Devchand P, Moussignac RL, Serhan CN. J Biol Chem. 2003;278:14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- 12.Arita M, Clish CB, Serhan CN. Biochem Biophys Res Commun. 2005;338:149–157. doi: 10.1016/j.bbrc.2005.07.181. [DOI] [PubMed] [Google Scholar]
- 13.Serhan CN, Gotlinger K, Hong S, Arita M. Prostaglandins Other Lipid Mediators. 2004;73:155–172. doi: 10.1016/j.prostaglandins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. J Exp Med. 2000;192:1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, Yang R, Petasis NA, Serhan CN. J Exp Med. 2005;201:713–722. doi: 10.1084/jem.20042031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kosaki Y, Ogawa N, Kobayashi Y. Tetrahedron Lett. 2010;51:1856–1859. [Google Scholar]
- 17.Seiji O, Urabe D, Yokokura Y, Arai H, Arita M, Inoue M. Org Lett. 2009;11:3602–3605. doi: 10.1021/ol901350g. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez AR, Spur BW. Tetrahedron Lett. 2005;46:3623–3627. [Google Scholar]
- 19.Rodriguez AR, Spur BW. Tetrahedron Lett. 2004;45:8717–8720. [Google Scholar]
- 20.Nicolaou KC, Ramphal JY, Petasis NA, Serhan CN. Angew Chem, Int Ed. 1991;30:1100–1116. [Google Scholar]
- 21.Kato T, Nakai T, Ishikawa R, Iio Y. Heterocycles. 2002;56:119–122. [Google Scholar]
- 22.Lapitskaya MA, Vasiljeva LL, Pivnitsky KKA. Synthesis. 1993:65–66. [Google Scholar]
- 23.Yao FM, Palmer SL, Khanolkar AD, Tian X, Guo J, Makriyannis A. J Labelled Compd Radiopharm. 2003;46:115–129. [Google Scholar]
- 24.Li C, Wei XW, Vadivel SK, Fan P, Makriyannis A. J Med Chem. 2005;48:6423–6429. doi: 10.1021/jm050272i. [DOI] [PubMed] [Google Scholar]
- 25.Brown CA, Ahuja VK. J Chem Soc, Chem Commun. 1973:553–554. [Google Scholar]
- 26.Brown CA, Ahuja VK. J Org Chem. 1973;38:2226–2230. [Google Scholar]
- 27.Nicolaou KC, Ladduwahetty T, Elisseou EM. J Chem Soc, Chem Commun. 1985:1580–1581. [Google Scholar]
- 28.Suh YG, Min KH, Lee YS, Seo SY, Kim SH, Park HJ. Tetrahedron Lett. 2002;43:3825–3828. [Google Scholar]
- 29.Stang PJ, Vandana D, Melvyn DS, Paul D. J Am Chem Soc. 1987;109:1150–1156. [Google Scholar]
- 30.Haack KJ, Hashiguchi S, Fujii A, Ikariya T, Noyori R. Angew Chem, Int Ed. 1997;36:285–288. [Google Scholar]
- 31.Matsumura K, Hashiguchi S, Ikariya T, Noyori R. J Am Chem Soc. 1997;119:8738–8739. The air-stable, purple Ru complex was easily prepared from (1R,2R)-1,2-diphenyl-N-(4-toluenesulfonyl)ethylenediamine with [RuCl2(η6-p-cymene)]2 in 87% yield. [Google Scholar]
- 32.Noyori R, Hashiguchi S. Acc Chem Res. 1997;30:97–102. [Google Scholar]
- 33.Ohkuma T, Utsumi N, Tsutsumi Murata K, Sandoval C, Noyori R. J Am Chem Soc. 2006;128:8724–8725. doi: 10.1021/ja0620989. [DOI] [PubMed] [Google Scholar]
- 34.Yamakawa M, Yamada I, Noyori R. Angew Chem, Int Ed. 2001;40:2818–2821. [PubMed] [Google Scholar]
- 35.Allevi P, Ciuffreda P, Anastasia M. Tetrahedron: Asymmetry. 1997;8:93. [Google Scholar]
- 36.Niwa S, Soai K. J Chem Soc, Perkin Trans. 1990;1:937. [Google Scholar]
- 37.Mukaiyama T, Suzuki K. Chem Lett. 1980:255. [Google Scholar]
- 38.Mori M, Nakai T. Tetrahedron Lett. 1997;38:6233. [Google Scholar]
- 39.Denmark SE, Yang SM. J Am Chem Soc. 2004;126:12432–12440. doi: 10.1021/ja0466863. [DOI] [PubMed] [Google Scholar]
- 40.Nicolaou KC, Veale CA, Webber SE, Katerinopoulos HJ. Am Chem Soc. 1985;105:7515–7518. [Google Scholar]
- 41.Alami M, Ferri F, Linstrumelle G. Tetrahedron Lett. 1993;34:6403–6406. [Google Scholar]
- 42.Crousse B, Alami M, Linstrumelle G. Tetrahedron Lett. 1995;36:4245–4248. [Google Scholar]
- 43.Alami M, Crousse B, Linstrumelle G. Tetrahedron Lett. 1994;35:3543–3544. [Google Scholar]
- 44.Boland W, Schroer N, Sieler C, Feigel M. Helv Chim Acta. 1987;70:1025–1040. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.