

Abstract
The transcriptional activity of RNA polymerase II (Pol II) is a dynamic process and therefore measuring the kinetics of the transcriptional process in vivo is of importance. Pol II kinetics have been measured using biochemical or molecular methods.1-3 In recent years, with the development of new visualization methods, it has become possible to follow transcription as it occurs in real time in single living cells.4 Herein we describe how to perform analysis of Pol II elongation kinetics on a specific gene in living cells.5, 6 Using a cell line in which a specific gene locus (DNA), its mRNA product, and the final protein product can be fluorescently labeled and visualized in vivo, it is possible to detect the actual transcription of mRNAs on the gene of interest.7, 8 The mRNA is fluorescently tagged using the MS2 system for tagging mRNAs in vivo, where the 3'UTR of the mRNA transcripts contain 24 MS2 stem-loop repeats, which provide highly specific binding sites for the YFP-MS2 coat protein that labels the mRNA as it is transcribed.9 To monitor the kinetics of transcription we use the Fluorescence Recovery After Photobleaching (FRAP) method. By photobleaching the YFP-MS2-tagged nascent transcripts at the site of transcription and then following the recovery of this signal over time, we obtain the synthesis rate of the newly made mRNAs.5 In other words, YFP-MS2 fluorescence recovery reflects the generation of new MS2 stem-loops in the nascent transcripts and their binding by fluorescent free YFP-MS2 molecules entering from the surrounding nucleoplasm. The FRAP recovery curves are then analyzed using mathematical mechanistic models formalized by a series of differential equations, in order to retrieve the kinetic time parameters of transcription.
Protocol
The cell system used must contain an integrated gene construct that includes MS2 repeat sequences for tagging the mRNA in living cells. In this protocol we use a human U2OS cell line harboring a stably integrated β-actin gene, that is fluorescently labeled at the DNA, RNA and protein levels8, as follows (Figure 1A): The 5’ of the gene contains lac operator (lacO) repeats – these are used to tag the gene (DNA) and thereby identify the genomic locus of integration. A fluorescently tagged lac repressor protein (LacI) will bind to the lacO repeats and tag the DNA. The mRNAs are tagged with YFP-MS2 proteins that bind to the MS2 repeats (stem-loops). The gene product is CFP-actin and therefore induction of the gene will result in the appearance of CFP-tagged actin cytoskeleton. The gene construct is transfected into a U2OS Tet-On stable cell line and its expression is under Tet transcriptional control.
1. Cell plating and transfection:
48 hours before the measurements, seed ˜2*105 cells on glass bottomed tissue culture dishes (35 mm petri dishes with a 14 mm glass-bottomed microwell (0.16-0.19 mm thickness; Cat. No. P35G-1.5-14-C, MatTek, Ashland, MA)). Add 2ml of medium to the cells, in order to reach 50–80% confluence.
- 24 hours before the experiment, transiently transfect the cells with constructs that express the fluorescent markers for tagging the DNA and the mRNA. Use FuGENE6 transfection reagent (Roche). The constructs used:
- YFP-MS2-NLS will tag the MS2 stem-loops in the mRNA transcripts. The nuclear localization sequence (NLS) provides the nuclear entry sequence for the YFP-MS2 protein.
- optional - CFP-LacI or RFP-LacI that will bind the lacO repeats in the 5’ of the gene and will label the gene locus. Tagging the gene locus assists in tracking the gene during the FRAP experiment but is not necessary.
Incubate the cells for at least 12 hrs, to obtain good expression of the transient transfected proteins (see note).
6 - 12 hours before the experiment add 1.5 μg/ml doxycycline (Sigma) to the cells for activating of the Tet-inducible CFP-actin gene.
Notes: * It is important not to reach very high expression levels of the transfected proteins so as not to mask the signal obtained specifically from the transcription site, relative to the background signal. Levels of expression can be controlled by: a) adjusting the time between transfection and the experiment; b) changing the strength of the promotor (e.g. CMV is stronger than the L30 promoter); c) adding a nuclear export signal (NES) to the YFP-MS2-NLS protein, which will help distribute the YFP signal in the whole cell.
* The fluorophore used for the measurements should preferably be photostable throughout long acquisitions; therefore green/yellow emitting fluorophores are better than red fluorophores. Information on the different fluorophores can be found in ref 10.
* Determine that the different light channels do not bleed-through to each other. Use slides containing specimens labeled with each of the markers separately, measure in each slide the intensity of both channels, and then calculate the bleed-through.
2. FRAP:
The experiment can be performed on any microscope that is capable of both acquiring images and performing photobleaching with a laser beam. Typically, FRAP experiments are performed with a confocal laser scanning microscope (CLSM). However, since in our case the experiment is relatively long (15-30 min), and we are following one small locus within the nucleus, it is important to be able to keep the gene locus/transcription site in focus throughout the whole experiment. This requires rapid imaging in 3 dimensions over time (4D), and such acquisition rates are hard to obtain using a scanning confocal microscope. Therefore, we use a 3D-FRAP system (Photometrics) built on a wide-field Olympus IX81 microscope (63X Plan-Apo, 1.4 NA) equipped with an EM-CCD (Quant-EM, Roper) that can acquire images at high speed (up to 30 images/sec). The system has 405 nm and 491 nm lasers that can be synchronized with the camera to enable photobleaching on a specific region of interest (ROI) of the sample. The microscope is equipped with a Lambda DG-4 light source (Sutter) and a XY&Z stage (Prior) that allows fast and accurate imaging in the Z-axis. All the equipment is integrated and controlled by MetaMorph (Molecular Devices). The live-cell experiments are performed at 37°C with 5% CO2 using a live-cell chamber system (Tokai).
Procedure: Half an hour before the experiment, turn on the heating device, microscope and laser. During the experiment the temperature needs to remain constant, otherwise it might cause focus changes during the long acquisition times. Laser power must be stable for consistent power output.
- Synchronize the laser with the camera:
- Search for an area in the plate that does not contain cells (using DIC).
- Use the same light channel that will be used for the experiments. Here we use the 491 nm laser for the YFP channel.
- Point the laser to the middle of the image.
- Use the synchronize button (provided macro).
- Try different spots in the field to ensure that the laser and camera are synchronized.
Choose a good candidate cell for the experiment. Although we work with stable cell lines, we find that not all cells have similar expression levels. Good candidates would be cells that: a) exhibit a clear YFP-MS2-tagged transcription site above the diffuse YFP-MS2 background; and b) have enough free YFP-MS2 protein in the cell so as to be able to recover completely after the photobleaching (example in figure 1B - compare the two cells; the left cell is not a good candidate for bleaching).
Choose appropriate measuring conditions. The FRAP procedure requires the acquiring of many images, and therefore it is important to balance between reasonable exposure times and using as low light as possible. High light levels might affect the cell due to phototoxicity, and will cause photobleaching. It is recommended to keep imaging parameters constant between different experiments. Cells are imaged in the YFP channel with 150 msec exposure times, and in the CFP channel for the detection of CFP-lacI (genomic locus). Following the CFP-LacI tagged locus allows the detection of the gene even when the YFP-MS2 mRNA signal is photobleached (optional). For each acquisition, 7 Z-slices are taken every 350 nm. The active transcription site is bleached using the 491 nm laser.
Perform the full experiment but without bleaching. The FRAP experiment measures the time it takes for the transcription site to return to steady state. First, we want to verify that the active transcription site is functional and stable at steady state. Therefore, we perform a complete FRAP experiment but without bleaching to ensure that the measured intensity of the transcription site does not change during the experiment time frame. We use this experiment to also test our imaging conditions by measuring the total photobleaching caused by the full imaging series. As a rule of thumb, the full movie photobleach should not be more than 30% of the initial intensity. This can be measured by using the intensity ratio of the same spot in the nucleus from the last image, divided by first image (ratio should be larger than 0.7).
FRAP: The YFP-MS2 signal at the active transcription site is bleached using the 491 nm laser. 6 pre-bleach images are acquired. Post-bleach images are acquired in a sequence of 3 time frequencies: 15 images every 3 sec, 15 images every 6 sec, and 26 (or 45 for long experiments) images every 30 sec. The changes in frequency of acquiring images allows to acquire more images during the times that show the highest intensity change (initial recovery), and less images towards the end when the changes in signal are less prominent.
Notes: This experiment differs from "regular" FRAP in that: * FRAP of mRNA at the transcription site measures the binding of YFP-MS2 proteins to the nascent mRNA, whereas typical FRAP experiments measure the mobility of different proteins in the cell. Therefore, there is no interest in measuring the diffusive pool of YFP-MS2 proteins. When the time between bleaching and the first acquired image is relatively long it is possible to disregard this diffusing pool of free YFP-MS2 proteins.
* Since the recovery of the YFP-MS2 signal is relatively long (15-30 min), it is important to keep the transcription site in focus. That is why a series of Z-slices is acquired at every time point.
3. Normalization of the FRAP data:
The 4D FRAP data is analyzed using the free Image analysis software ImageJ (NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/). Following is analysis performed with macros available through the ImageJ software, but of course self-written macros can be generated.
Z-stacks are maximum projected at every time-point, resulting in a 2D movie over time for further analysis.
The transcription site is tracked in each frame and the mean intensity is measured using the Spot Tracker tool.11 The data is saved in an Excel sheet for further analysis.
For every time-point, the mean intensities at other regions of interest (ROI) are measured as follows (using the Time Series Analyzer plug-in of ImageJ): a) The background is taken from an ROI outside of the cell = B(t). b) An ROI in the nucleoplasm, but as far as possible from the bleach site = C(t). c) The transcription site = S(t) (as measured by the spot tracker).
Correct the data for photobleaching caused during imaging: Subtract the background from all other measurements and then correct the bleach. Sc(t) is the corrected intensity of the transcription site at time t.
eq. 1:
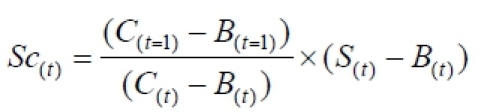
Normalize the data: first calculate the average of the pre-bleach images, which shows the intensity of the transcription site at steady-state ( = <Sc(pre-bleach)>).
Then normalize the data:
eq. 2
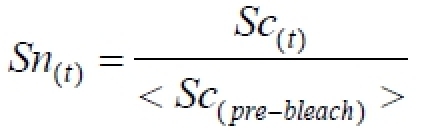
The data are normalized for at least 10 experiments. Calculate the average of the experiments and the standard deviation (STD) for the error-bars at every time-point (Figure 1C).
Notes: The diffusion of the YFP-MS2 protein during the FRAP analysis was disregarded since the diffusion rate of free nucleoplasmic YFP-MS2 is very rapid, while the bound YFP-MS2 is associated with high affinity to the mRNA, and does not reattach to the transcription site. This is why the first image after bleaching should be removed before analysis.
4. Analyzing the data using a mathematical model:
In order to retrieve the kinetic parameters from this kind of data set, we used the simplest described model that can fit this kind of data. The full explanation of this specific model can be read here5. The model contains the following equations that describe the scheme in figure 2A:
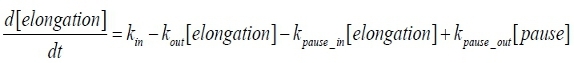
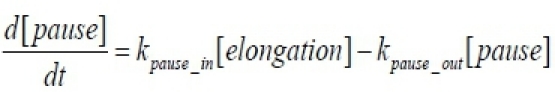
Having the model parameters, it is possible to calculate the elongation rate:
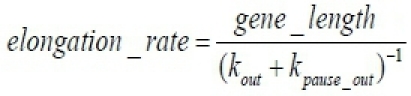
We analyzed the curve using Berkeley Madonna (http://www.berkeleymadonna.com), as previously modeled5 but other options are available (MatLab, Virtual Cell, COPASI etc.). The data was fit to the model and rate constants were extracted. We attach the code in Berkeley Madonna. In order to best fit the data we calculated the steady state and then fit the normalized experimental data to the normalized model.
5. Representative Results:
We photobleached the labeled mRNA signal at the active transcription site using high laser power, and followed the recovery of the YFP-MS2- signal over time. The fluorescent signal at the transcription site consists of the steady state kinetics of mRNA synthesis as well as mRNA released from the site. Measuring transcription site intensity over time without bleaching gives a 'constant' signal (figure 1C, the red dots) which implies that this gene is currently in a relative steady state. This means that the signal that accumulates on the newly transcribed mRNA is equal to the signal that leaves when an mRNA is released from the transcription site. When photobleached, this system continues in its steady-state, but is now possible to measure the time it takes for the signal to recover. Therefore, the recovery of fluorescence mostly depends on the elongation properties of the polymerase. The 3D FRAP system allowed the acquisition of the full 3D nuclear volume during FRAP recovery, and thereby no signal was lost during the experiments. For calculating the kinetic parameters from the data, the normalized curve was fitted to the kinetic model describing elongation, that is based on differential equations.5 The advantage of using a simple ODE model is that it can be fit to only a small number of parameters. The initial step of the model (entry point) is elongation, namely the process responsible for synthesizing new MS2-binding sites. The end step (exit point) is mRNA release into the nucleoplasm. The model is based on the recovery curves, which consist of two kinetically resolved components, one fast and one slower. The fast component refers to elongation, whereas the slower component was modeled as polymerase pausing during elongation. The two polymerase states were modeled as elongation with a stochastic transition to pausing. We optimized the differential equations from this model, which yielded an elongation speed of 3.3 kb/minute with a stochastic transition to a slower synthesis rate (pausing for a cumulative time of 2.5 minutes). Modeling showed that this transition from elongation to pausing affected only 11% of the polymerases that entered elongation.
| Kout | 0.0112 | |
| K pause_in | 0.0015 | |
| K pause_out | 0.0067 | |
| Elongation time | (Kout + k pause_out)-1 | 56 sec |
| Pause time | (k pause_out)-1 | 2.5 minutes |
| Pause probability | (K pause_in) / (K pause_in + kout) | 11 % |
Table 1.The model results in a parameter table.
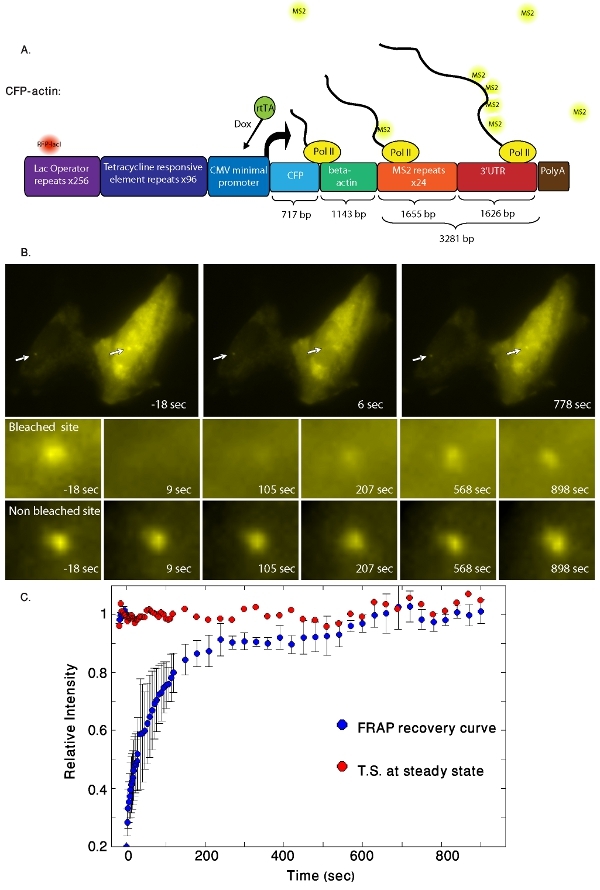 Figure 1. (A) The experimental cell system used for following gene expression in vivo. Schematic representation of the CFP-actin gene
construct. The 5'-end contains a series of 256 lacO repeats that are bound by RFP-LacI (red) and mark the site of gene integration. Transcriptional
induction from the minimal CMV promoter is achieved by the binding of reverse tetracycline transcriptional activator (rtTA or Tet-On) to Tet-responsive elements
(TREs) in the presence of dox. The transcribed mRNA contains the coding sequence for CFP-β-actin, and 24 MS2 repeats that are bound by the YFP-MS2 fusion
protein. At the 3'-end, a portion of a rabbit β-globin exon-intron-exon module is followed by a cleavage-polyA signal. The scheme also describes the elongation
assay showing fluorescent MS2 proteins (yellow) attached to the newly transcribed mRNAs transcribed by RNA Pol II. (B) An active transcription
site (YFP-MS2, right hand cell) was photobleached and the fluorescence recovery was tracked over time (frames on the bottom). We measured the fluorescence
intensity of both bleach (middle line) and non-bleached (bottom line) active transcription sites. Note the left-hand cell is not suitable for a photobleaching
experiment. (C) The experimental measurements performed on the CFP-actin gene transcription sites. In blue the recovery curves of the YFP-MS2
FRAP measurements. In red, a non-bleached cell from the same experiment showing the YFP-MS2 signal at the transcription site at steady state.
Figure 1. (A) The experimental cell system used for following gene expression in vivo. Schematic representation of the CFP-actin gene
construct. The 5'-end contains a series of 256 lacO repeats that are bound by RFP-LacI (red) and mark the site of gene integration. Transcriptional
induction from the minimal CMV promoter is achieved by the binding of reverse tetracycline transcriptional activator (rtTA or Tet-On) to Tet-responsive elements
(TREs) in the presence of dox. The transcribed mRNA contains the coding sequence for CFP-β-actin, and 24 MS2 repeats that are bound by the YFP-MS2 fusion
protein. At the 3'-end, a portion of a rabbit β-globin exon-intron-exon module is followed by a cleavage-polyA signal. The scheme also describes the elongation
assay showing fluorescent MS2 proteins (yellow) attached to the newly transcribed mRNAs transcribed by RNA Pol II. (B) An active transcription
site (YFP-MS2, right hand cell) was photobleached and the fluorescence recovery was tracked over time (frames on the bottom). We measured the fluorescence
intensity of both bleach (middle line) and non-bleached (bottom line) active transcription sites. Note the left-hand cell is not suitable for a photobleaching
experiment. (C) The experimental measurements performed on the CFP-actin gene transcription sites. In blue the recovery curves of the YFP-MS2
FRAP measurements. In red, a non-bleached cell from the same experiment showing the YFP-MS2 signal at the transcription site at steady state.
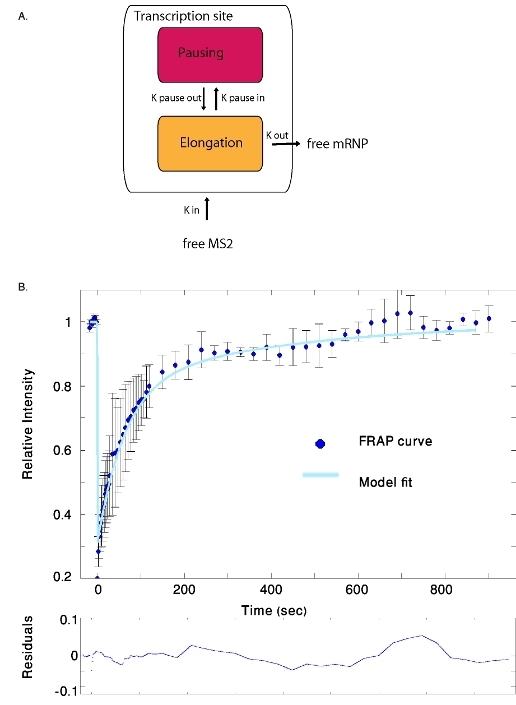 Figure 2. (A) Scheme of the model describing the entrance and exit kinetics of mRNAs marked with YFP-MS2 molecules at the transcription
site. (B) YFP-MS2 FRAP data and the fitted simulation (the average of ten experiments was fitted to the model). The calculated residuals of the fit
are presented at the bottom.
Figure 2. (A) Scheme of the model describing the entrance and exit kinetics of mRNAs marked with YFP-MS2 molecules at the transcription
site. (B) YFP-MS2 FRAP data and the fitted simulation (the average of ten experiments was fitted to the model). The calculated residuals of the fit
are presented at the bottom.
Discussion
The method for measuring polymerase kinetic activity in living cells can be divided into two parts. The first part describes the 'wet' procedure which enables the measuring and retrieving of the kinetic data of mRNA transcription, whereas in the second part, the data is analyzed using an ODE model.5 A number of studies have now utilized this approach for extracting transcription kinetics in living cells5, 6, 8, 12-14. The major difference between these studies was the way the FRAP recovery curve was converted into kinetic parameters. In many cases a mathematical model is used. A model should describe the biological process in a simple manner and it is then compared to the FRAP data. Once there is a good correlation between the data and the model, one should verify the model by additional biological experiments such as measurements of drug effects or comparing between different genes.
It is possible to take the above analysis further to obtain additional kinetic results. For instance, by using a photoactivatable form of the MS2 fluorescent protein it is possible to photoactivate already generated mRNA transcripts and follow their release ( = decrease in intensity vs the recovery of signal measured by FRAP) from the transcription site.5 Additionally, while FRAP of MS2 tagged mRNAs measures elongation kinetics only, FRAP of GFP-RNA Pol II at the transcription site yields the kinetics of the whole transcriptional process, namely, pre-initiation, initiation, elongation and termination5, 13, 15, 16.
Disclosures
No conflicts of interest declared.
Acknowledgments
YS-T is supported by the European Research Council (ERC), the Israel Science Foundation (ISF) (250/06), ISF-Bikura, Israel Cancer Research Fund (ICRF), German-Israeli Foundation for Scientific Research and Development (GIF), USA-Israel Binational Science Foundation (BSF), German-Israeli Project Cooperation (DIP), and the Israeli Ministries of Science and Health, and is the Jane Stern Lebell Family Fellow in Life Sciences at Bar-Ilan University. YB is grateful to the Azrieli Foundation for the award of an Azrieli fellowship.
References
- Wada Y. A wave of nascent transcription on activated human genes. Proc. Natl. Acad. Sci. U. S. A. 2009;106:18357–18361. doi: 10.1073/pnas.0902573106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol. 2009;16:1128–1133. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody Y, Shav-Tal Y. Visualizing transcription in real-time. Cent Eur J Biol. 2008;3:11–18. [Google Scholar]
- Lamond AI, Swedlow JR. RNA polymerase II transcription in living color. Nat Struct Mol Biol. 2007;14:788–790. doi: 10.1038/nsmb0907-788. [DOI] [PubMed] [Google Scholar]
- Darzacq X. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boireau S. The transcriptional cycle of HIV-1 in real-time and live cells. J Cell Biol. 2007;179:291–304. doi: 10.1083/jcb.200706018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki SM. From silencing to gene expression; real-time analysis in single cells. Cell. 2004;116:683–698. doi: 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. The life of an mRNA in space and time. J Cell Sci. 2010;123:1761–1774. doi: 10.1242/jcs.062638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shav-Tal Y. Dynamics of single mRNPs in nuclei of living cells. Science. 2004;304:1797–1800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers GJ, Gilbert SG, Cranfill PJ, Davidson MW, Piston DW. Fluorescent proteins at a glance. J. Cell Sci. 2011;124:157–160. doi: 10.1242/jcs.072744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage D, Neumann FR, Hediger F, Gasser SM, Unser M. Automatic tracking of individual fluorescence particles: application to the study of chromosome dynamics. IEEE Trans Image Process. 2005;14:1372–1383. doi: 10.1109/tip.2005.852787. [DOI] [PubMed] [Google Scholar]
- Yunger S, Rosenfeld L, Garini Y, Shav-Tal Y. Single-allele analysis of transcription kinetics in living mammalian cells. Nat Methods. 2010;7:631–633. doi: 10.1038/nmeth.1482. [DOI] [PubMed] [Google Scholar]
- Brody Y. The in vivo kinetics of RNA polymerase II elongation during co-transcriptional splicing. PLoS Biol. 2011;9:e1000573–e1000573. doi: 10.1371/journal.pbio.1000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz MJ. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell. 2009;137:708–720. doi: 10.1016/j.cell.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Hieda M, Winstanley H, Maini P, Iborra FJ, Cook PR. Different populations of RNA polymerase II in living mammalian cells. Chromosome Res. 2005;13:135–144. doi: 10.1007/s10577-005-7720-1. [DOI] [PubMed] [Google Scholar]
- Kimura H, Sugaya K, Cook PR. The transcription cycle of RNA polymerase II in living cells. J Cell Biol. 2002;159:777–782. doi: 10.1083/jcb.200206019. [DOI] [PMC free article] [PubMed] [Google Scholar]