

Abstract
Fourteen new withanolides 1-14, named withalongolides A-N, respectively, were isolated from the aerial parts of Physalis longifolia together with eight known compounds (15-22). The structures of compounds 1-14 were elucidated through spectroscopic techniques and chemical methods. In addition, the structures of withanolides 1, 2, 3, and 6 were confirmed by X-ray crystallographic analysis. Using a MTS viability assays, eight withanolides (1, 2, 3, 7, 8, 15, 16, and 19) and four acetylated derivatives (1a, 1b, 2a, and 2b) showed potent cytotoxicity against human head and neck squamous cell carcinoma (JMAR and MDA-1986), melanoma (B16F10 and SKMEL-28), and normal fetal fibroblast (MRC-5) cells with IC50 values in the range between 0.067 and 9.3 μM.
Classically-defined withanolides are a group of C28 ergostane-type steroids with a C-22,26 δ-lactone group, first isolated from the genus Withania.1 They are present primarily in the Solanaceae family, which includes the genera Acnistus, Datura, Dunalia, Jaborosa, Nicandra, Physalis, and Withania.2-9 Withanolides have attracted interest in recent years mainly due to their exhibition of significant biological activities, inclusive of antimicrobial, antitumor, anti-inflammatory, immunomodulatory, and insect-antifeedant activities.2,3, 6 It has been reported that those withanolides displaying the most promising antitumor characteristics contain an α,β-unsaturated ketone in ring A, a 5β,6β-epoxy group in ring B, and a nine-carbon side chain with an α,β-unsaturated δ-lactone group.10 The typical withanolide, withaferin A (16) (Figure 1) contains these three moieties and has been shown in vitro and in vivo to suppress the growth of an array of tumor cells, including breast, pancreatic, prostate, lung, leukemia, and head and neck squamous cell carcinoma (HNSCC), by inducing apoptosis,11 thus possessing potential application as an antiproliferative agent. As part of an ongoing study of withanolides from plant sources,11,12 a library of 224 native plant extracts from the U.S. Great Plains was evaluated for cytotoxic activities against HNSCC and melanoma cell lines using the MTS viability assay. One of the most promising leads, Physalis longifolia Nutt. (Solanaceae), commonly known as “long leaf groundcherry”, was subjected to a phytochemical investigation and the results are presented herein, including the details of the isolation, and structure elucidation of fourteen new withanolides (1-14), four acetylated derivatives (1a, 1b, 2a, and 2b), and eight known compounds (15-22). Their cytotoxicity was determined against HNSCC (JMAR and MDA-1986), melanoma (B16F10 and SKMEL-28), and normal fetal fibroblast (MRC-5) cells. This constitutes the first report of a phytochemical and bioactivity study of P. longifolia.
Figure 1.
Withanolides 1-21 and rutin (22) isolated from Physalis longifolia
RESULTS AND DISCUSSION
The CH2Cl2–MeOH (1:1) extract of the aerial parts of the title plant, and the EtOAc-soluble and n-BuOH soluble fractions, showed cytotoxicity against the above-mentioned cells with IC50 values in the range between 0.7 and 9.8 μg/mL using a MTS assay. All compounds (1-22) were isolated from the EtOAc-soluble or n-BuOH soluble fractions (see Experimental Section).
Compound 1 was isolated as colorless cuboid crystals obtained from a CH2Cl2–CH3CN mixture, a major metabolite in the EtOAc-soluble fraction. Its molecular formula, C28H38O7, was determined by HRESIMS and NMR experiments, equating to ten double-bond equivalents. Its IR absorptions revealed the presence of hydroxy (3431 and 3233 cm-1), keto (1671 cm-1), and ester (1706 cm-1) groups. The 1H NMR spectrum (Table 1) showed the presence of three methyl groups at δ 0.60 (3H, s), 0.90 (3H, d, J = 6.6 Hz), 1.97 (3H, s), seven protons attached to oxygenated carbons at δ 3.18 (1H, brs), 3.52 (1H, d, J = 6.1 Hz), 3.65 (1H, d, J = 9.6 Hz), 4.17 (1H, d, J = 9.6 Hz), 4.23 (1H, d, J = 12.5 Hz), 4.28 (1H, d, J = 12.5 Hz), and 4.33 (1H, dt, J = 13.3, 3.4 Hz), and two olefinic methine groups at δ 6.16 (1H, d, J = 10.0 Hz) and 6.95 (1H, dd, J = 10.0, 6.1 Hz). The 13C NMR (APT) and HSQC spectra for 1 (Table 2) displayed 28 carbon signals differentiated as three CH3, eight CH2 (including two oxygenated at δ 61.0 and 56.7), ten CH (including two olefins at δ 145.7 and 132.9, three oxygenated at δ 78.8, 68.0 and 61.8), and seven C (including one keto carbonyl at δ 200.7, one ester carbonyl at δ 167.4, two olefins at δ 154.3 and 125.6, and one oxygenated at δ 61.5), corresponding to C28H35. The remaining three hydrogen atoms were therefore assigned to three OH groups, indicating that six rings must be present in the structure.
Table 1.
1H NMR Data for Withanolides 1, 1a, 1b, 2, 2a, 2b and 3 (400 or 500 MHz)
| Pos. | 1a | 1b | 1ac | 1bc | 2c | 2ac | 2bc | 3c |
|---|---|---|---|---|---|---|---|---|
| 2 | 6.16 d (10.0) | 6.46 d (10.0) | 6.23 d (10.0) | 6.21 d (10.0) | 6.23 d (10.4) | 6.23 d (9.9) | 6.23 d (9.9) | 6.22 d (10.0) |
| 3 | 6.95 dd (10.0, 6.1) | 7.26 dd (10.0, 6.1) | 7.00 dd (10.0, 6.1) | 7.00 dd (10.0, 6.1) | 7.01 dd (10.4, 6.4) | 7.00 dd (9.9, 6.0) | 7.01 dd (9.9, 6.0) | 6.96 dd (10.0, 6.5) |
| 4 | 3.52 d (6.1) | 4.01 d (6.1) | 4.79 d (6.1) | 4.73 d (6.1) | 3.63 d (6.4) | 4.79 d (6.0) | 4.74 d (6.0) | 3.78 d (6.5) |
| 6 | 3.18 brs | 3.42 brs | 3.12 brs | 3.20 brs | 3.25 brs | 3.16 brs | 3.20 brs | 3.23 brs |
| 7 | 2.14 m, 1.25 m | 2.15 ddd (2.2, 4.0, 14.5) | 2.15 m | 2.01 m, 1.96 m | 2.19 ddd (2.2, 4.2, 15.0) | 2.15 m | 2.17 m | 2.26 ddd (2.4, 4.7, 15.1) |
| 1.37 dd (11.1, 14.5) | 1.25 m | 1.30 m | 1.25 ddd (1.4, 11.3, 15.0) | 1.28 m | 1.29 m | |||
| 8 | 1.44 m | 1.64 dq (4.0, 11.1) | 1.77 dq (4.2, 11.1) | 1.85 m | 1.51 dq (4.2, 11.1) | 1.76 dq (4.2, 11.3) | 1.85 m | 1.87 dq (4.5, 10.8) |
| 9 | 0.85 m | 1.15 m | 1.00 m | 0.91 m | 0.94 m | 1.00 m | 0.92 m | 1.25 dd (3.3, 10.8) |
| 11 | 1.90 m, 1.32 m | 2.31 m, 1.55 m | 1.96 m, 1.62 m | 1.94 m, 1.85 m | 1.94 m, 1.39 m | 1.96 m, 1.62 m | 1.94 m, 1.84 m | 4.15 (brs) |
| 12 | 1.86 m, 0.95 m | 1.82 m, 0.99 m | 1.97 m, 1.08 m | 1.96 m, 1.05 m | 1.93 m, 1.01 m | 1.97 m, 1.08 m | 1.98 m, 1.05 m | 2.17 dd (13.8, 2.8), 1.38 m |
| 14 | 0.81 m | 0.83 m | 0.87 m | 0.83 m | 0.85 m | 0.86 m | 0.85 m | 0.92 m |
| 15 | 1.57 m, 1.05 m | 1.49 m, 0.92 m | 1.62 m, 1.14 m | 1.58 m, 1.13 m | 1.61 m, 1.12 m | 1.61 m, 1.12 m | 1.61 m, 1.12 m | 1.64 m, 1.22 m |
| 16 | 1.59 m, 1.29 m | 1.49 m, 1.12 m | 1.67 m, 1.36 m | 1.65 m, 1.36 m | 1.65 m, 1.35 m | 1.66 m, 1.36 m | 1.65 m, 1.36 m | 1.63 m, 1.37 m |
| 17 | 0.99 m | 0.99 m | 1.07 m | 1.05 m | 1.04 m | 1.07 m | 1.06 m | 1.04 m |
| 18 | 0.60 s | 0.53 s | 0.73 s | 0.73 s | 0.66 s | 0.72 s | 0.73 s | 0.90 s |
| 19 | 4.17 d, 3.65 d (9.6) | 4.79 d, 4.21 d (9.4) | 5.07 d, 4.32 d (11.5) | 4.36 d, 4.07 d (11.5) | 4.32 d, 3.77 d (9.7) | 5.07 d, 4.32 d (11.5) | 4.37 d, 4.09 d (11.6) | 1.63 s |
| 20 | 1.91 m | 1.88 m | 2.00 m | 1.98 m | 1.95 m | 1.96 m | 1.96 m | 1.99 m |
| 21 | 0.90 d (6.6) | 0.95 d (6.7) | 0.98 d (6.7) | 0.97 d (6.6) | 0.95 d (6.6) | 0.97 d (6.6) | 0.97 (6.6) | 1.00 d (6.6) |
| 22 | 4.33 dt (13.3, 3.4) | 4.37 dt (13.3, 3.5) | 4.38 dt (13.3, 3.4) | 4.36 dt (13.3, 3.4) | 4.31 dt (13.3, 3.4) | 4.31 dt (13.3, 3.4) | 4.32 dt (13.3, 3.4) | 4.39 dt (13.2, 3.5) |
| 23 | 2.42 dd (17.0, 13.3) | 2.49 dd (18.0, 13.3) | 2.50 dd (17.9, 13.3) | 2.48 dd (18.2, 13.3) | 2.40 dd (18.0, 13.3) | 2.41 dd (18.0, 13.3) | 2.41 dd (18.0, 13.3) | 2.47 dd (13.2, 17.0) |
| 1.91 m | 2.17 m | 1.99 m | 2.16 m | 1.87 m | 1.88 m | 1.88 m | 1.94 dd (3.5, 17.0) | |
| 27 | 4.28 d, 4.23 d (12.5) | 4.86 d, 4.76 d (11.8) | 4.88 d, 4.84 d (11.8) | 4.88 d, 4.85 d (11.9) | 1.85 s | 1.85 s | 1.85 s | 4.34 br |
| 28 | 1.97 s | 2.11 s | 2.05 s | 2.04 s | 1.90 s | 1.91 s | 1.90 s | 2.01 s |
| 4-OAc | 2.09 s | 2.07 s | 2.09 s | 2.08 s | ||||
| 19-OAc | 2.06 s | 2.06 s | ||||||
| 27-OAc | 2.03 s | 2.02 s |
In CDCl3 with trace amount of CD3OD.
In C5D5N.
In CDCl3.
Table 2.
13C NMR Data for Withanolides 1-7 and 16-19 (125 MHz)
| Pos. | 1c | 1b | 1aa | 1ba | 2a | 2aa | 2ba | 3a | 4a | 5a | 6a | 7a | 16a | 17a | 18b | 19b | 20b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 200.7 | 201.4 | 198.3 | 198.9 | 200.2 | 198.3 | 198.9 | 204.0 | 208.1 | 213.3 | 211.6 | 208.0 | 202.5 | 210.1 | 210.4 | 209.7 | 210.5 |
| 2 | 132.9 | 133.2 | 133.7 | 134.4 | 133.1 | 133.8 | 134.5 | 131.6 | 40.5 | 39.5 | 129.4 | 42.0 | 132.5 | 39.8 | 44.4 | 42.0 | 32.2 |
| 3 | 145.7 | 147.5 | 141.0 | 140.2 | 145.4 | 140.9 | 140.1 | 143.2 | 77.7 | 76.2 | 155.2 | 73.6 | 142.1 | 77.7 | 67.1 | 73.1 | 27.1 |
| 4 | 68.0 | 69.0 | 71.2 | 71.7 | 68.5 | 71.2 | 71.8 | 69.7 | 72.8 | 74.4 | --- | 75.4 | 70.1 | 75.1 | 78.7 | 76.9 | 72.4 |
| 5 | 61.5 | 61.7 | 59.4 | 61.1 | 61.7 | 59.4 | 61.1 | 64.5 | 62.5 | 66.0 | 147.4 | 63.0 | 64.1 | 65.2 | 65.5 | 65.1 | 65.9 |
| 6 | 61.8 | 62.0 | 59.2 | 59.9 | 62.1 | 59.2 | 59.9 | 63.0 | 57.6 | 59.5 | 123.8 | 57.2 | 62.7 | 60.5 | 57.4 | 57.2 | 58.5 |
| 7 | 31.1 | 31.9 | 31.0 | 30.4 | 31.2 | 30.9 | 30.4 | 31.7 | 31.1 | 32.2 | 31.9 | 31.4 | 31.3 | 31.3 | 32.3 | 31.9 | 29.4 |
| 8 | 30.5 | 30.3 | 30.9 | 31.4 | 30.6 | 31.0 | 31.4 | 27.0 | 30.1 | 27.2 | 32.8 | 30.7 | 29.9 | 29.5 | 30.5 | 30.4 | 31.0 |
| 9 | 43.9 | 44.8 | 44.8 | 44.6 | 44.0 | 44.9 | 44.7 | 48.1 | 42.1 | 47.1 | 42.8 | 42.3 | 44.3 | 42.9 | 43.7 | 43.1 | 42.7 |
| 10 | 54.3 | 55.6 | 51.2 | 52.2 | 54.3 | 51.2 | 52.3 | 49.0 | 56.1 | 51.3 | 47.6 | 55.7 | 47.9 | 50.6 | 49.7 | 49.9 | 50.2 |
| 11 | 22.1 | 23.0 | 22.3 | 21.9 | 22.3 | 22.3 | 21.9 | 69.5 | 21.6 | 69.1 | 22.0 | 22.1 | 22.3 | 21.8 | 21.8 | 21.7 | 21.3 |
| 12 | 39.7 | 40.3 | 39.6 | 39.7 | 39.7 | 39.6 | 39.7 | 47.8 | 39.3 | 47.4 | 39.5 | 39.8 | 39.5 | 39.3 | 39.7 | 39.5 | 38.9 |
| 13 | 42.5 | 42.9 | 42.9 | 43.1 | 42.6 | 42.8 | 43.0 | 42.1 | 42.8 | 42.0 | 43.6 | 43.1 | 42.8 | 42.9 | 43.2 | 43.2 | 42.5 |
| 14 | 56.7 | 57.1 | 56.7 | 57.0 | 56.9 | 56.7 | 57.1 | 58.2 | 56.7 | 58.0 | 56.5 | 56.7 | 56.2 | 56.2 | 56.6 | 56.4 | 56.1 |
| 15 | 24.2 | 24.7 | 24.5 | 24.3 | 24.3 | 24.5 | 24.4 | 24.4 | 24.3 | 24.5 | 24.6 | 24.7 | 24.5 | 24.5 | 24.9 | 24.9 | 24.0 |
| 16 | 27.3 | 27.6 | 27.5 | 27.5 | 27.4 | 27.5 | 27.5 | 27.2 | 27.5 | 27.2 | 27.4 | 27.7 | 27.5 | 27.5 | 27.7 | 27.7 | 27.5 |
| 17 | 51.9 | 52.4 | 52.0 | 52.0 | 52.1 | 52.1 | 52.1 | 52.7 | 52.0 | 52.5 | 52.1 | 52.3 | 52.1 | 52.1 | 52.4 | 52.4 | 51.6 |
| 18 | 11.7 | 12.1 | 11.8 | 12.0 | 11.8 | 11.8 | 11.8 | 14.2 | 11.7 | 13.6 | 12.1 | 12.0 | 11.8 | 11.7 | 11.9 | 11.9 | 11.5 |
| 19 | 61.0 | 61.7 | 64.4 | 65.6 | 62.1 | 64.4 | 65.6 | 21.3 | 60.4 | 19.0 | 19.9 | 59.9 | 17.6 | 15.9 | 16.1 | 15.9 | 15.2 |
| 20 | 38.8 | 39.5 | 39.0 | 39.0 | 39.0 | 39.0 | 39.0 | 39.0 | 38.9 | 39.0 | 39.1 | 39.5 | 39.0 | 38.9 | 39.5 | 39.6 | 38.3 |
| 21 | 13.3 | 13.9 | 13.5 | 13.5 | 13.6 | 13.6 | 13.6 | 13.6 | 13.6 | 13.6 | 13.6 | 14.0 | 13.5 | 13.5 | 14.0 | 14.0 | 13.5 |
| 22 | 78.8 | 78.7 | 78.4 | 78.4 | 78.4 | 78.4 | 78.4 | 78.9 | 78.8 | 78.9 | 79.0 | 78.9 | 78.9 | 78.9 | 78.8 | 78.9 | 78.4 |
| 23 | 29.9 | 30.3 | 30.3 | 30.3 | 29.8 | 29.8 | 29.8 | 30.1 | 30.0 | 30.1 | 30.0 | 30.3 | 30.0 | 30.0 | 30.4 | 30.3 | 29.1 |
| 24 | 154.3 | 154.4 | 157.2 | 157.1 | 149.2 | 149.2 | 149.1 | 153.0 | 153.0 | 152.9 | 153.1 | 154.6 | 153.1 | 153.1 | 154.3 | 154.6 | 152.2 |
| 25 | 125.6 | 127.9 | 122.1 | 122.1 | 122.2 | 122.2 | 122.2 | 125.9 | 125.9 | 125.9 | 125.9 | 127.7 | 125.8 | 125.5 | 127.9 | 127.7 | 125.4 |
| 26 | 167.4 | 166.8 | 165.5 | 165.5 | 167.3 | 167.2 | 167.2 | 167.2 | 167.2 | 167.2 | 167.3 | 167.0 | 167.3 | 167.2 | 166.8 | 167.1 | 167.2 |
| 27 | 56.7 | 56.6 | 58.2 | 58.2 | 12.7 | 12.7 | 12.7 | 57.7 | 57.6 | 57.7 | 57.7 | 56.6 | 57.7 | 57.6 | 56.6 | 56.6 | 57.6 |
| 28 | 20.2 | 20.6 | 20.8 | 20.8 | 20.7 | 20.7 | 20.7 | 20.2 | 20.2 | 20.2 | 20.2 | 20.5 | 20.3 | 20.3 | 20.6 | 20.5 | 20.2 |
| OCOCH3-4 | 170.4 | 170.0 | 170.4 | 170.1 | |||||||||||||
| 21.1 | 21.1 | 21.0 | 21.1 | ||||||||||||||
| OCOCH3-19 | 170.6 | 170.7 | |||||||||||||||
| 21.0 | 21.4 | ||||||||||||||||
| OCOCH3-27 | 171.1 | 171.1 | |||||||||||||||
| 21.4 | 21.0 | ||||||||||||||||
| OCH3-3 | 57.6 | 57.4 | 57.0 |
In CDCl3.
In C5D5N.
In CDCl3 with trace amount of CD3OD.
The NMR data of 1 were very close to those obtained for withaferin A 16,1,13 a six-ring withanolide isolated as another major compound in this study (Tables 1-3 and Figure 1). Compound 1 was found to contain the following moieties also observed in 16: an α,β-unsaturated ketone in ring A ([13C: δ 200.7 (C-1), 132.9 (C-2), 145.7 (C-3); 1H: δ 6.16 (H-2, d, J = 10.0 Hz), 6.95 (H-3, dd, J = 10.0, 6.1 Hz)]; an epoxy group in ring B [13C: δ 61.5 (C-5), 61.8 (C-6); 1H: δ 3.18 (brs, H-6)]; a nine-carbon side chain with an α,β-unsaturated δ-lactone group [13C: δ 78.8 (C-22), 154.3 (C-24), 125.6 (C-25), 167.4 (C-26); 1H: δ 4.33 (H-22, dt, J = 13.3, 3.4 Hz)]), as supported by 1H-1H COSY and HMBC experiments. The obvious differences between 1 and 16 were the presence of an oxygenated methylene [C-19, 13C: δ 61.0; 1H: δ 3.65 (1H, d, J = 9.6 Hz), 4.17 (1H, d, J = 9.6 Hz)] in 1 and a methyl carbon [C-19, 13C: δ 17.6; 1H: 1.38 (3H, s)] in the latter, suggesting that 1 is a 19-hydroxy derivative of 16. This observation was supported by the high-frequency shift of C-10 (δ 54.3 in 1 and δ 47.9 in 16), the low-frequency shifts of C-1 (δ 200.7 in 1 and δ 202.5 in 16), C-5 (δ 61.5 in 1 and δ 64.1 in 16), and C-9 (δ 43.9 in 1 and δ 44.3 in 16) in the 13C NMR spectra, and the HMBC correlations between H2-19 [3.65 (1H, d, J = 9.6 Hz), 4.17 (1H, d, J = 9.6 Hz] and C-1, C-5, C-9, and C-10.
Table 3.
1H NMR Data for Withanolides 4–7 and 16–19 (400 or 500 MHz)
| Pos. | 4a | 5a | 6a | 7b | 16a | 17a | 18b,c | 19b |
|---|---|---|---|---|---|---|---|---|
| 2 | Hβ 2.86 dd (8.3, 16.4) | Hβ 2.86 dd (6.5, 17.5) | 5.93 d (6.4) | Hβ 3.65 dd (8.1, 18.0) | 6.18 d (10.0) | Hβ 2.99 dd (6.3, 15.0) | Hβ 3.62 dd (10.0, 17.1) | Hβ 3.62 dd (9.7, 17.6) |
| Hα 2.56 dd (2.6, 16.4) | Hα 2.56 dd (2.2, 17.5) | Hα 3.22 dd (6.2, 18.0) | Hα 2.56 dd (2.2, 15.0) | Hα 3.02 dd (5.5, 17.1) | Hα 3.30 dd (2.0, 17.6) | |||
| 3 | 3.70 ddd (8.3, 3.4, 2.6) | 3.75 ddd (6.5, 4.0, 2.2) | 7.57 d (6.4) | 5.61 brt (8.1) | 6.91 dd (10.0, 5.9) | 3.68 ddd (6.3, 3.2, 2.2) | 4.67 br | 5.62 ddd (9.7, 6.5, 2.0) |
| 4 | 3.42 d (3.4) | 3.56 d (4.0) | --- | 4.54 br | 3.74 d (5.9) | 3.46 d (3.2) | 4.05 br | 4.51 br |
| 6 | 3.22 brs | 3.15 brs | 5.76 t (3.5) | 3.37 brs | 3.21 s | 3.19 s | 3.30 br | 3.62 br |
| 7 | 2.25 m | 2.31 dd (5.2, 14.8) | 2.37 ddd (3.5, 8.8, 15.0) | 2.35 m | 2.12 ddd (2.2, 3.7, 14.6) | 2.14 ddd (2.2, 3.7, 14.6) | 2.22 ddd (1.7, 4.3, 15.0) | 2.07 m |
| 1.42 m | 1.34 m | 1.70 m | 2.11 m | 1.30 m | 1.31 m | 1.40 ddd (2.0, 10.2, 15.0) | 1.23 m | |
| 8 | 1.41 m | 1.70 dq (4.5, 10.8) | 1.75 m | 1.50 m | 1.39 m | 1.39 m | 1.50 m | 1.41 m |
| 9 | 1.14 m | 1.28 m | 1.17 m | 1.16 m | 1.14 m | 1.16 m | 1.17 m | 1.04 m |
| 11 | 1.27 m, 1.18 m | 3.88 m | 2.24 m, 1.58 m | 1.57 m, 1.34 m | 1.27 m, 1.18 m | 1.35 m | 1.52 m | 1.44 m |
| 12 | 1.90 m, 1.02 m | 2.15 dd (13.9, 2.4), 1.38 m | 1.99 m, 1.18 m | 1.76 m, 1.02 m | 1.88 m, 1.08 m | 1.88 m, 1.08 m | 1.85 m, 1.05 m | 1.80 m, 1.05 m |
| 14 | 0.95 m | 0.94 m | 1.03 m | 0.91 m | 0.95 m | 0.94 m | 0.93 m | 0.92 m |
| 15 | 1.64 m, 1.13 m | 1.66 m, 1.21 m | 1.61 m, 1.19 m | 1.52 m, 0.90 m | 1.64 m, 1.13 m | 1.61 m, 1.12 m | 1.54 m, 0.96 m | 1.54 m, 0.92 m |
| 16 | 1.66 m, 1.35 m | 1.64 m, 1.38 m | 1.64 m, 1.35 m | 1.57 m, 1.13 m | 1.66 m, 1.35 m | 1.64 m, 1.31 m | 1.54 m, 1.15 m | 1.57 m, 1.15 m |
| 17 | 1.07 m | 1.04 m | 1.10 m | 1.13 m | 1.07 m | 1.05 m | 1.07 m | 1.14 m |
| 18 | 0.62 s | 0.85 s | 0.75 s | 0.47 s | 0.67 s | 0.64 s | 0.55 s | 0.52 s |
| 19 | 4.38 d, 3.61 d (10.2) | 1.49 s | 1.15 s | 4.80 d, 4.01 d (9.4) | 1.38 s | 1.28 s | 1.76 s | 1.69 s |
| 20 | 1.96 m | 1.99 m | 1.99 m | 1.86 m | 1.97 m | 1.97 m | 1.90 m | 1.90 m |
| 21 | 0.95 d (6.7) | 1.00 d (6.7) | 1.01 d (6.6) | 0.95 d (6.6) | 0.97 d (6.6) | 0.96 d (6.6) | 0.97 d (6.6) | 0.97 d (6.6) |
| 22 | 4.39 dt (13.2, 3.5) | 4.39 dt (13.2, 3.5) | 4.42 dt (13.3, 3.4) | 4.38 dt (13.2, 3.3) | 4.39 dt (13.2, 3.5) | 4.39 dt (13.2, 3.5) | 4.39 dt (13.2, 3.3) | 4.40 dt (13.2, 3.3) |
| 23 | 2.46 dd (13.2, 17.0) | 2.47 dd (13.7, 17.8) | 2.48 dd (13.6, 17.6) | 2.35 dd (13.9, 18.0) | 2.47 dd (13.6, 17.4) | 2.47 dd (13.6, 17.4) | 2.35 dd (13.4, 17.7) | 2.36 dd (13.7, 17.8) |
| 1.93 dd (3.5, 17.0) | 1.94 dd (3.2, 17.8) | 1.98 dd (3.2, 17.6) | 2.07 m | 1.94 dd (3.2, 17.4) | 1.94 dd (3.2, 17.4) | 2.01 dd (3.3, 17.7) | 2.07 m | |
| 27 | 4.36 d, 4.32 d (12.5) | 4.37 d, 4.31 d (12.6) | 4.37 d, 4.32 d (12.5) | 4.85 d, 4.75 d (11.7) | 4.36 d, 4.31 d (12.6) | 4.36 d, 4.31 d (12.6) | 4.80 d, 4.78 d (11.8) | 4.85 d, 4.75 d (11.7) |
| 28 | 2.02 s | 2.01 s | 2.01 s | 2.09 s | 2.01 s | 2.01 s | 2.10 s | 2.09 s |
| 3-OMe | 3.35 s | 3.34 s | 3.32 s |
In CDCl3.
In C5D5N.
OH-4 signal 7.75 s, OH-3 signal 6.91 brs, OH-27 signal 6.61 brs.
Acetylation of 1 with acetic anhydride in pyridine gave two derivatives: the 4,19,27-triacetate (1a) and the 4,27-diacetate (1b) (Tables 1 and 2), which proved the presence of hydroxy groups at C-4, C-19, and C-27 by a high frequency shift of H-4 (from δ 3.52 in 1 to δ 4.79 in 1a and to δ 4.73 in 1b), of H2-19 (from δ 3.65, 4.17 in 1 to δ 4.32, 5.07 in 1a), and of H2-27 (δ 4.23, 4.28 in 1 to δ 4.84, 4.88 in 1a and to δ 4.85, 4.88 in 1b), and by HMBC correlations between H-4 to the ester carbonyl, between H2-27 and the ester carbonyl, and between H2-19 and the ester carbonyl in 1a and 1b.
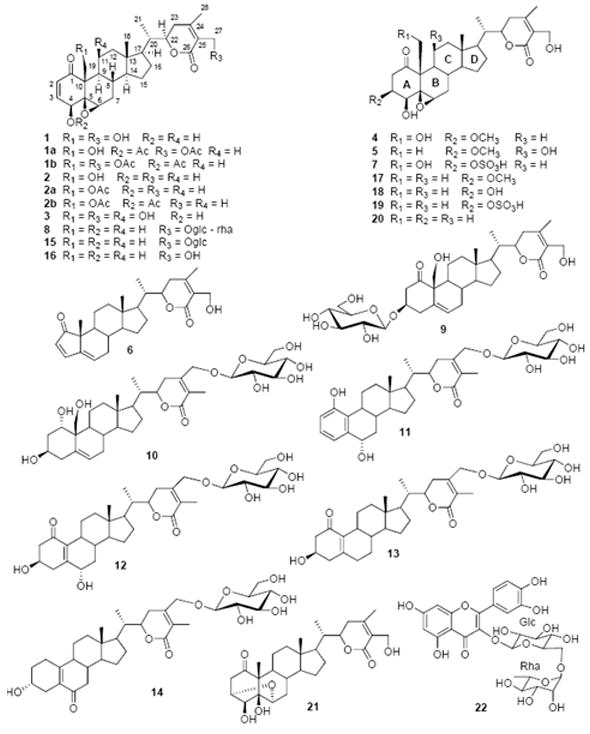
Finally, the structure of 1 was confirmed through a single-crystal X-ray diffraction experiment (Figure 2). Thus, 1 (withalongolide A) was established as 19-hydroxywithaferin A. The full assignments of NMR data of 1, measured in CDCl3 with trace amount of CD3OD and in C5D5N (Tables 1 and 2), were obtained by 2D-NMR methods including 1H-1H COSY, HSQC, HMBC, and ROESY spectra.
Figure 2.
X-ray ORTEP drawing of withalongolide A (1)
Compound 2 was isolated as colorless rod crystals from a CH2Cl2–CH3C6H5 mixture, and was also a major metabolite from the EtOAc-soluble part. Its molecular formula, C28H38O6, was determined by HRESIMS and NMR experiments. The IR and NMR (1H and 13C) data (Tables 1 and 2) were similar to those of 1. Analysis of the 1D- and 2D-NMR data for 2 identified resonances consistent with an α,β-unsaturated ketone in ring A [13C: δ 200.2 (C-1), 133.1 (C-2), 145.4 (C-3); 1H: δ 6.23 (H-2, d, J = 10.4 Hz), 7.01 (H-3, dd, J = 10.4, 6.4 Hz)], an epoxy in ring B [13C: δ 61.7 (C-5), 62.1 (C-6); 1H: δ 3.25 (brs, H-6)], a nine-carbon side chain with a δ-lactone group [13C: δ 78.4 (C-22), 149.2 (C-24), 122.2 (C-25), 167.3 (C-26); 1H: δ 4.31 (H-22, dt, J = 13.3, 3.4 Hz)], and an oxygenated C-19 [13C: δ 62.1; 1H: δ 4.32, 3.77 (d, J = 9.7 Hz)]. The obvious differences between 2 and 1 were the presence of a 27-methyl carbon [13C: δ 12.7; 1H: 1.85 (3H, s)] in 2 and an oxygenated methylene [C-27, 13C: δ 56.7; 1H: δ 4.28 (1H, d, J = 12.5 Hz), 4.23 (1H, d, J = 12.5 Hz)] in 1, suggesting that 2 is a 27-deoxy derivative of 1. This observation was supported by the 13C NMR chemical shift values of the δ-lactone moiety [δ 149.2 (C-24), 122.2 (C-25), 167.3 (C-26) in 2 and δ 154.3 (C-24), 125.6 (C-25), 167.4 (C-26) in 1], by the NMR data comparison of the side chain moiety of 2 to those of 27-deoxywithanolides,14,15 and by HMBC correlations of H3-27/C-24, C-25, and C-26.
Acetylation of 2 with acetic anhydride in pyridine yielded the 4,19-diacetate (2a) and the 4-monoacetate (2b) (Tables 1 and 2), which confirmed the presence of hydroxy groups at C-4 and C-19 by a high frequency shift of H-4 (from δ 3.63 in 2 to δ 4.79 in 2a and to δ 4.74 in 2b) and H2-19 (from δ 3.77, 4.32 in 2 to δ 4.32, 5.07 in 2a).
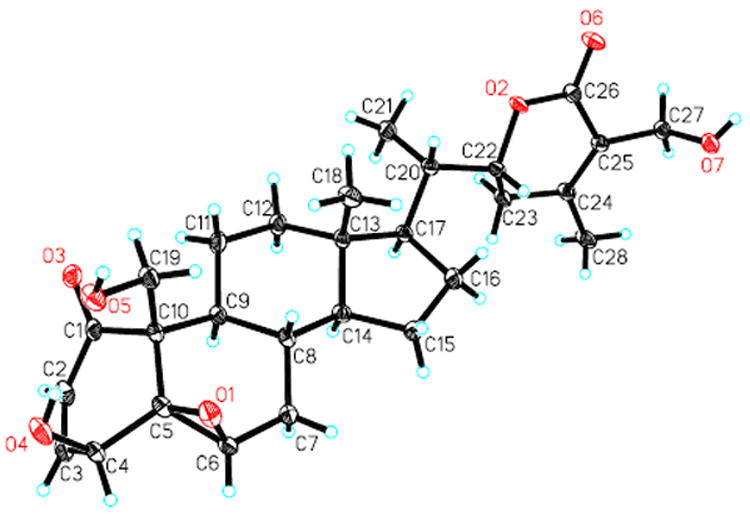
Finally, the structure of 2 was confirmed through a single-crystal X-ray diffraction experiment (Figure 3). Thus, 2 (withalongolide B) was determined as 27-deoxy-19-hydroxywithaferin A.
Figure 3.
X-ray ORTEP drawing of withalongolide B (2)
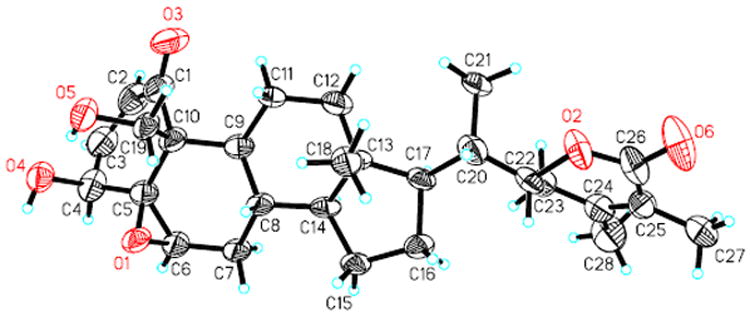
Compound 3 was isolated as colorless cube crystals from a CH2Cl2–CH3CN mixture, a minor component from the EtOAc-soluble fraction. Its molecular formula, C28H38O7, was determined by HRESIMS and NMR experiments. The NMR data of 3 (Tables 1 and 2) were also akin to those of withaferin A 16.1,13 Analysis of the 1D- and 2D-NMR data of 3 identified resonances consistent with an α,β-unsaturated ketone in ring A [13C: δ 204.0 (C-1), 131.6 (C-2), 143.2 (C-3); 1H: δ 6.22 (H-2, d, J = 10.0 Hz), 6.96 (H-3, dd, J = 10.0, 6.5 Hz)], an epoxy in ring B [13C: δ 64.5 (C-5), 63.0 (C-6); 1H: δ 3.23 (brs, H-6)], and a nine-carbon side chain with a δ-lactone group [13C: δ 78.9 (C-22), 153.0 (C-24), 125.9 (C-25), 167.2 (C-26); 1H: δ 4.39 (H-22, dt, J = 13.2, 3.5 Hz)]. The obvious differences between 3 and 16 were the presence of an oxygenated methine [C-11, 13C: δ 69.5; 1H: 4.15 (1H, brs)] in 3 and a low-frequency methylene [C-11, 13C: δ 22.3; 1H: δ 1.27 (1H, m), 1.18 (1H, m)] in 16, implying that 3 is 11-hydroxywithaferin A. This observation was supported by the high-frequency shift of C-9 (δ 48.1 in 3 and δ 44.3 in 16) and C-12 (δ 47.8 in 3 and δ 39.5 in 16), and the low-frequency shift of C-8 (δ 27.0 in 3 and δ 29.9 in 16) in the 13C NMR spectrum; by the presence of a fragment -CH2–CH(OH)–CH–CH(CH2)–CH- (starting with C-12 and ending with C-14) deduced from 1H-1H COSY and HSQC experiments; and by the HMBC correlations between Hα-12 (δ 2.17, dd, J = 13.8, 2.8 Hz) and C-9 (δ 48.1), 11 (δ 69.5), 13 (δ 42.1), 14 (δ 58.2). The orientation of the hydroxy group at C-11 was deduced as β due to the broad single peak pattern of H-11 (δ 4.15, brs), the small coupling constant of 3.3 Hz between H-9 (δ 1.25, dd, J = 10.8, 3.3 Hz) and H-11, and ROESY correlations of H-11/H-9 and H-11/Hα-12. Finally, the structure of 3 was confirmed through a single-crystal X-ray diffraction experiment (Figure 4). Thus, the new withanolide 3 (withalongolide C) was established as 11β-hydroxywithaferin A.
Figure 4.
X-ray ORTEP drawing of withalongolide C (3)
Compounds 4 and 5 were isolated as two presumed artifacts. These two compounds were probably formed from withalongolide A (1) and withalongolide C (3), respectively, by a Michael-type addition due to the use of CH3OH during the extraction procedure. It is possible that these compounds are formed in a similar fashion to 2,3-dihydro-3β-methoxywithaferin A (17)1 (Tables 2 and 3), which was most likely derived from withaferin A (16) during this study. Comparing the NMR data of the methoxy group in 17 [-OCH3 group at C-3: 1H: δ 3.32 (3H, s); 13C: δ 57.0; H-3: δ 3.68 (1H, ddd, J = 6.3, 3.2, 2.2 Hz); C-3: δ 77.7] with both 4 and 5 [-OCH3: 1H: δ 3.35 (3H, s); 13C: δ 57.6; H-3: δ 3.70 (1H, ddd, J = 8.3, 3.4, 2.6 Hz); C-3: δ 77.7 in 4 and -OCH3: 1H: δ 3.34 (3H, s); 13C: δ 57.4; H-3: δ 3.75 (1H, ddd, J = 6.5, 4.0, 2.2 Hz); C-3: δ 76.2 in 5] suggested the presence of a methoxy group at the C-3 positions in 4 and 5. This was confirmed by the presence of 1H-1H COSY fragment of -CH2–CH(OCH3)–CH(OH)- in ring A and HMBC correlation of OCH3/C-3 in both 4 and 5. The structures of 4 (withalongolide D) and 5 (withalongolide E) were determined by spectroscopic methods and complete assignments of their NMR data are listed in Tables 2 and 3.
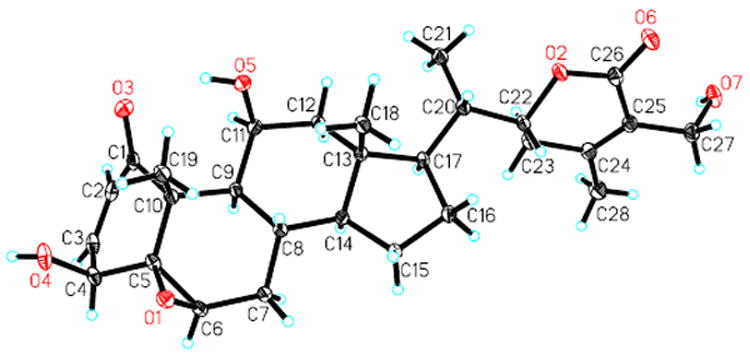
Compound 6 was isolated as colorless cube crystals obtained from a CH2Cl2–CH3CN mixture. Its molecular formula, C27H34O4, was ascertained by HRESIMS and NMR experiments (Tables 2 and 3). Similar to withaferin A (16), it showed signals for four methyl groups [13C: δ 20.2 (C-28), 19.9 (C-19), 13.6 (C-21), 12.1 (C-18); 1H: δ 2.01 (H3-28, s), 1.15 (H3-19, s), 1.01 (H3-21, d, J = 6.6 Hz), 0.75 (H3-18, s)], an α,β-unsaturated ketone in ring A [13C: δ 211.6 (C-1), 129.4 (C-2), 155.2 (C-3); 1H: δ 5.93 (H-2, d, J = 6.4 Hz), 7.57 (H-3, d, J = 6.4 Hz)], and a nine-carbon side chain with an α,β-unsaturated δ-lactone group ([13C: δ 57.7 (C-27), 79.0 (C-22), 153.1 (C-24), 125.9 (C-25), 167.3 (C-26); 1H: δ 4.42 (H-22, dt, J = 13.3, 3.4 Hz), 4.37 (H-27, d, J = 12.5 Hz), 4.32 (H-27, d, J = 12.5)]. A detailed comparison of the NMR data of 6 to those of 16 indicated that both compounds share identical ring C, D and side chain moieties, but are different in their A and B rings. A five-membered ring A for 6 was proposed on the basis of the following evidence: (1) the unusual chemical shift value of the conjugated ketone carbon (C-1, δ 211.6); (2) the coupling pattern of H-3 (δ 7.57, d, J = 6.4 Hz) and the small coupling constant of 6.4 Hz between the olefinic protons H-2 and H-3 when compared to those of H-3 (6.91, dd, J = 10.0, 5.9 Hz) in 16, showing C-3 to be linked with a quaternary carbon in 6. This five-membered ring A and a C-5,6 double bond in ring B were supported by the HMBC correlations of H3-19/C-1, C-5 (quaternary carbon, δ 147.4), C-9 (δ 42.8), and C-10 (δ 47.6); of H-2/C-1, C-3, C-5, and C-10; and of H-3/C-1, C-2, C-5, C-6 (methine, δ 123.8), and C-10. Finally, the observation was confirmed through a single-crystal X-ray diffraction experiment (Figure 5). Thus, 6 (withalongolide F) was deduced to be A-nor-27-hydroxy-1-oxowitha-2,5,24-trienolide. This 4-norwithanolide with a 2,5-dien-1-one system was reported previously as a semi-synthetic product derived from withaferin A by an acid-catalyzed rearrangement.16
Figure 5.
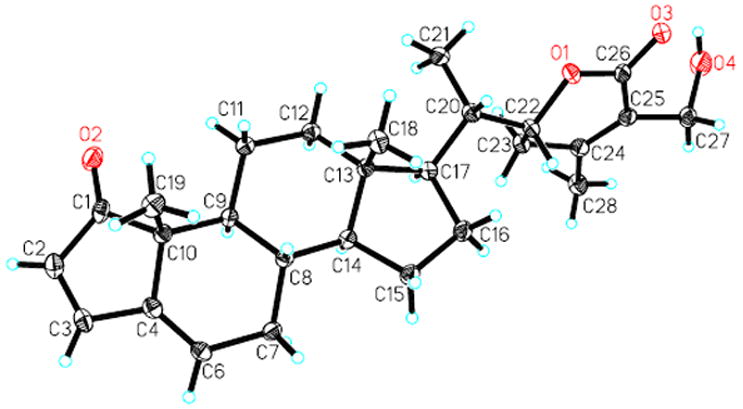
X-ray ORTEP drawing of withalongolide F (6)
Compound 7 was a major compound isolated from the BuOH-soluble fraction. Its molecular formula, C28H40O11S, was determined by HRESIMS and NMR experiments. The NMR data of 7 (Tables 2 and 3) were similar to those of 2,3-dihydro-3β-O-sulfate-withaferin A (19) (Tables 2 and 3),17 another major withanolide isolated from the BuOH-soluble fraction during this study (Figure 1). The obvious differences between 7 and 19 were the presence of an oxygenated methylene [C-19, 13C: δ 59.9; 1H: δ 4.80 (1H, d, J = 9.4 Hz), 4.01 (1H, d, J = 9.4 Hz)] in 7 and a methyl carbon [C-19, 13C: δ 15.9; 1H: δ 1.69 (3H, s)] in the latter, suggesting that 7 is a 19-hydroxy derivative of 19. This observation was supported by the high-frequency shift of C-10 (δ 55.7 in 7 and δ 49.9 in 19), the low-frequency shift of C-1 (δ 208.0 in 7 and δ 209.7 in 19), C-5 (δ 63.0 in 7 and δ 65.1 in 19), and C-9 (δ 42.3 in 7 and δ 43.1 in 19) in the 13C NMR spectrum, and the HMBC correlations of H2-19 [δ 4.01 (1H, d, J = 9.4 Hz), 4.80 (1H, d, J = 9.4 Hz]/C-1, C-5, C-9, and C-10 in 7. Thus, 7 (withalongolide G) was determined as 2,3-dihydro-19-hydroxy-3β-O-sulfate-withaferin A.
Compound 8 was isolated as a major component from the BuOH-soluble fraction. Its molecular formula, C40H58O15, was ascertained by HRESIMS and NMR experiments. The NMR data of 8 (Tables 4 and 5) showed similarities to those of 27-O-β-d-glucopyranosyl-withaferin A (15) (sitoindoside IX)18 (Tables 4 and 5) isolated during this study, suggesting 8 to be a withanolide saponin. The aglycone of 8 was determined to be withaferin A as both 8 and 15, possess the superimposable 1H and 13C NMR signals of the steroid aglycone moieties and both showed the same main LC-MS/MS fragments of m/z 471 and 281 due to the presence of a withaferin A moiety. Differing in the presence of only one glucose residue in 15, two sugar residues were observed in 8 on the basis of the signals of two anomeric carbons [methines, δ 105.3 (C-1′) and 103.1 (C-1″)] and their corresponding anomeric protons [δ 4.97 (H-1′, 1H, d, J = 7.8) and 5.94 (H-1″, 1H, s)]. Furthermore, the data for 8 suggested that the compound had, besides a glucose unit, an additional five oxygenated carbons (five methines) and one low frequency methyl group [13C: δ 19.0; 1H: δ 1.74 (3H, d, J = 6.1 Hz)], corresponding to a rhamnose in the pyranose form. The rhamnose moiety was deduced by the detailed comparison the NMR data of 8 with those of rutin 22 {3-O-[α-l-rhamnopyranosyl-(1→6)]-β-d-glucopyranosyl-quercetin} also isolated in this study and confirmed from the 1H-1H COSY, HSQC and HMBC spectra when starting with the characteristic methyl group [C-6″, 13C: δ 19.0; 1H: δ 1.74 (3H, d, J = 6.1 Hz)]. The α-anomeric configuration of the rhamnose unit was assigned from the small coupling constant between H-1″ (1H, δ 5.94, s) and H-2″ (1H, δ 4.73, s). Furthermore, the rhamnose was confirmed to be attached at C-4′ (δ 78.5) on the basis of HMBC correlations of H-1″/C-4′ and H-4′/C-1″, also supported by the glycosylation shifts of C-4′ (δ 78.5 in 8 and δ 72.1 in 15), C-3′ (δ 77.1 in 8 and δ 79.0 in 15) and C-5′ (δ 77.8 in 8 and δ 79.1 in 15). Thus, the structure of 8 (withalongolide H) was determined as 27-O-[α-l-rhamnopyranosyl(1→4)]-β-d-glucopyranosyl-withaferin A.
Table 4.
1H NMR Data for Withanolides 8-15 in C5D5N (500 MHz)
| Pos. | 8a | 9b | 10c | 11d | 12e | 13f | 14g | 15h |
|---|---|---|---|---|---|---|---|---|
| 1 | --- | --- | 4.61 m | --- | --- | --- | Hα 2.74 m, Hβ 2.10 m | --- |
| 2 | 6.43 d (10.0) | Hβ 3.48 dd (11.0, 12.4) | Hα 2.73 d (18.8) | 7.14 d (7.7) | 3.01 dd (7.1, 15.5) | 2.94 dd (8.1, 15.2) | Hα 1.96 m, Hβ 1.80 m | 6.43 d (10.0) |
| Hα 3.26 ddd (1.5, 5.3, 12.4) | Hβ 2.35 dd (10.2, 18.8) | 2.91 dd (3.7, 15.5) | 2.91 dd (4.1, 15.2) | |||||
| 3 | 7.25 dd (10.0, 6.3) | 4.29 m | 4.84 m | 7.34 t (7.7) | 4.58 m | 4.47 m | 4.33 br | 7.25 dd (10.0, 6.3) |
| 4 | 4.05 dd (6.3, 3.8) | Hβ 3.07 dt (2.0, 12.2) | Hβ 2.95 t (12.5) | 7.87 d (7.7) | 3.63 td (3.2, 18.0) | 2.68 dd (5.9, 16.8) | Hα 2.90 d (15.8) | 4.05 dd (6.3, 3.7) |
| Hα 2.99 ddd (1.5, 5.5, 12.2) | Hα 2.88 dd (12.5, 5.0) | 2.92 m | 2.58 td (2.7, 16.8) | Hβ 2.81 dd (2.2, 15.8) | ||||
| 6 | 3.28 s | 5.75 brd (5.3) | 5.92 brd (4.5) | 5.26 dt (11.0, 6.5) | 4.69 m | Hα 2.26 m, Hβ 2.09 m | --- | 3.28 s |
| 7 | 2.13 dt (13.0, 1.8) | 1.92 m, 1.53 m | Hα 1.66 dd (11.0, 16.0) | 1.63 q (11.0) | 2.23 m | 1.98 m, 1.53 m | 2.47 dd (3.6, 16.0) | 2.13 dt (13.0, 1.8) |
| 1.30 dd (11.5, 13.0) | Hβ 2.02 m | 2.32 dd (6.5, 11.0) | 2.00 m | 1.30 dd (11.5, 13.0) | ||||
| 8 | 1.58 m | 1.74 dt (5.3, 10.7) | 2.20 m | 1.73 q (11.0) | 1.48 m | 1.24 m | 1.62 m | 1.57 m |
| 9 | 1.06 m | 1.94 m | 2.32 m | 2.71 t (11.0) | 2.20 m | 2.04 m | 1.86 m | 1.06 m |
| 11 | 2.02 m, 1.57 m | 2.37 m, 1.96 m | 2.15 m, 1,98 m | 3.68 m, 1.46 m | 2.96 m, 1.17 m | 2.80 m, 2.34 m | 1.84 m, 1.24 m | 2.02 m, 1.57 m |
| 12 | 1.83 m, 1.03 m | 1.95 m, 1.26 m | 1.95 m, 1.02 m | 1.95 m, 1.30 m | 1.83 m, 1.17 m | 1.82 m, 1.13 m | 1.85 m, 1.03 m | 1.85 m, 1.02 m |
| 14 | 0.86 m | 0.97 m | 0.86 m | 1.10 m | 0.95 m | 0.90 m | 0.88 m | 0.86 m |
| 15 | 1.50 m, 0.96 m | 1.50 m, 1.02 m | 1.49 m, 1.06 m | 1.52 m, 1.12 m | 1.45 m, 1.02 m | 1.44 m, 1.02 m | 1.33 m, 0.88 m | 1.50 m, 0.95 m |
| 16 | 1.49 m, 1.11 m | 1.50 m, 1.14 m | 1.91 m, 1.24 m | 1.98 m, 1.27 m | 1.95 m, 1.23 m | 1.95 m, 1.23 m | 1.91 m, 1.21 m | 1.49 m, 1.11 m |
| 17 | 0.99 m | 1.05 m | 1.04 m | 1.12 m | 1.07 m | 1.07 m | 1.06 m | 0.99 m |
| 18 | 0.57 s | 0.67 s | 0.80 s | 0.70 m | 0.59 s | 0.58 s | 0.50 s | 0.57 s |
| 19 | 1.87 s | 4.30 m, 4.23 dd (3.9, 12.3) | 4.32 d, 4.11d (11.0) | --- | --- | --- | --- | 1.86 s |
| 20 | 1.88 m | 1.90 m | 1.98 m | 2.01 m | 1.96 m | 1.96 m | 1.96 m | 1.88 m |
| 21 | 0.96 d (6.6) | 0.97 d (6.6) | 0.99 d (6.6) | 1.01 d (6.6) | 0.97 d (6.6) | 0.96 d (6.6) | 0.99 d (6.6) | 0.95 d (6.6) |
| 22 | 4.34 m | 4.40 dt (13.2, 3.4) | 4.25 m | 4.55 dt (13.2, 3.4) | 4.53 dt (13.2, 3.4) | 4.53 dt (13.2, 3.4) | 4.53 dt (13.2, 3.4) | 4.34 dt (13.2, 3.4) |
| 23 | 2.27 dd (13.3, 18.0) | 2.34 dd (18.0, 14.0) | 2.78 m, 2.33 m | 2.83 dd (18.0, 2.2) | 2.81 dd (18.0, 2.2) | 2.81 dd (18.0, 2.2) | 2.81 dd (18.0, 2.2) | 2.28 dd (13.3, 18.0) |
| 1.98 dd (3.1, 18.0) | 2.03 dd (3.1, 18.0) | 2.34 dd (18.0, 14.0) | 2.35 dd (18.0, 14.0) | 2.35 dd (18.0, 14.0) | 2.37 dd (18.0, 14.0) | 1.98 dd (3.1, 18.0) | ||
| 27 | 5.02 d, 4.80 d (10.8) | 4.88 d, 4.78 d (11.0) | 2.04 s | 2.05 s | 2.05 s | 2.05 s | 2.06 s | 5.07 d, 4.86 d (10.8) |
| 28 | 2.07 s | 2.14 s | 4.87 d, 4.49 d (13.5) | 4.89 d, 4.47 d (13.5) | 4.89 d, 4.49 d (13.5) | 4.89 d, 4.49 d (13.5) | 4.90 d, 4.50 d (13.5) | 2.10 s |
| 1′ | 4.97 d (7.8) | 5.08 d (7.8) | 4.84 d (7.8) | 4.84 d (7.8) | 4.84 d (7.8) | 4.84 d (7.8) | 4.85 d (7.8) | 5.06 d (7.8) |
| 2′ | 4.01 td (7.8, 3.6) | 4.05 t (7.8) | 4.10 m | 4.10 m | 4.10 m | 4.10 m | 4.11 dd (7.8, 7.0) | 4.08 t (7.8) |
| 3′ | 4.23 m | 4.30 m | 4.25 m | 4.25 m | 4.25 m | 4.25 m | 4.25 m | 4.31 m |
| 4′ | 4.51 t (9.0) | 4.31 m | 4.24 m | 4.24 m | 4.24 m | 4.24 m | 4.24 m | 4.30 m |
| 5′ | 3.75 ddd (2.5, 5.0, 9.0) | 3.97 ddd (2.5, 5.0, 9.0) | 3.99 m | 4.00 m | 4.00 m | 4.00 m | 4.00 m | 4.00 ddd (2.5, 5.6, 9.0) |
| 6′ | 4.31 m, 4.16 m | 4.54 ddd (2.5, 5.0, 11.6) | 4.62 m | 4.62 ddd (2.0, 5.6, 11.6) | 4.62 ddd (2.0, 5.6, 11.6) | 4.62 ddd (2.0, 5.6, 11.6) | 4.62 ddd (2.0, 5.6, 11.6) | 4.60 ddd (2.5, 5.6,11.5) |
| 4.42 td (5.0, 11.6) | 4.42 m | 4.42 td (5.6, 11.6) | 4.42 td (5.6, 11.6) | 4.42 td (5.6, 11.6) | 4.42 td (5.6, 11.6) | 4.44 td (5.6, 11.5) |
Note:
H-1″5.94 s, H-2″ 4.73 s, H-3″ 4.60 br, H-4″ 4.38 dd (4.1, 9.9), H-5″ 5.06 m, H-6″ 1.74 d (6.1), OH-4 7.87 d (3.8), OH-2′ 7.36 d (3.5), OH-2″ 6.84 s, OH-4″ 6.83 s, OH-3′ 6.83 s, OH-6″ 6.82 m, OH-3″ 6.55 d (5.5).
OH-2′ 7.27 s, OH-19 7.27 d (3.9), OH-6′, 6.56 t (5.0), OH-27 6.49 brs.
OH-2′ 7.51, OH-3′ 7.36 s, OH-4′, 7.24 s, OH-6′, 6.52 t (5.0), OH-3, 6.20 d (5.0), OH-1 5.97 d (5.0), OH-19 5.96 brs.
OH-1 11.33 s, OH-2′, 7.45 d (4.5), OH-3′, 7.35 brs, OH-4′, 7.25 brs, OH-6, 6.81 d (6.5), OH-6′, 6.53 t (5.6).
OH-2′ 7.54 d (4.3), OH-3′, 7.34 brs, OH-4′ 7.26 brs, OH-6 6.93 d (6.5), OH-3 6.84 (4.0), OH-6′ 6.53 t (5.6).
OH-2′, 7.54 d (4.2), OH-3′, 7.34 brs, OH-4′ 7.26 brs, OH-3 6.81 d (4.1), OH-6′, 6.53 t (5.5).
OH-2′ 7.49 brs, OH-3′, 7.38 brs, OH-4′ 7.28 brs, OH-6′ 6.53 t (5.6), OH-3 6.28 brs.
OH-4 7.86 d (3.7), OH-3′, 7.22 brs, OH-4′ 7.28 brs, OH-2′ 7.21 brs, OH-6′ 6.47 t (5.6), OH-3 6.28 brs.
Table 5.
13C NMR Data for Withanolides 8-15 in C5D5N (125 MHz)
| Pos. | 8a | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|
| 1 | 203.1 | 208.9 | 69.5 | 157.8 | 199.4 | 198.3 | 25.6 | 203.1 |
| 2 | 132.8 | 49.6 | 41.7 | 115.1 | 49.4 | 49.7 | 30.6 | 132.8 |
| 3 | 145.6 | 75.9 | 66.6 | 127.4 | 66.3 | 66.1 | 64.7 | 145.6 |
| 4 | 70.8 | 39.9 | 44.4 | 119.3 | 36.9 | 41.9 | 32.9 | 70.8 |
| 5 | 65.0 | 132.8 | 137.8 | 146.2 | 156.4 | 153.6 | 130.4 | 65.0 |
| 6 | 60.6 | 129.1 | 127.0 | 70.7 | 71.2 | 33.8 | 198.6 | 60.6 |
| 7 | 32.2 | 31.9 | 32.6 | 38.3 | 37.3 | 26.4 | 44.0 | 32.2 |
| 8 | 30.7 | 33.5 | 33.8 | 39.0 | 37.5 | 39.9 | 40.0 | 30.7 |
| 9 | 45.1 | 43.6 | 42.4 | 46.6 | 44.3 | 43.6 | 46.6 | 45.1 |
| 10 | 49.0 | 60.2 | 48.2 | 127.8 | 136.5 | 136.1 | 158.7 | 49.0 |
| 11 | 22.2 | 24.1 | 22.3 | 27.1 | 26.4 | 26.6 | 25.1 | 22.2 |
| 12 | 39.9 | 40.6 | 40.7 | 41.5 | 41.0 | 41.0 | 40.2 | 39.9 |
| 13 | 43.1 | 43.5 | 43.6 | 44.2 | 43.9 | 44.1 | 43.3 | 43.1 |
| 14 | 56.5 | 57.0 | 57.5 | 54.9 | 54.2 | 54.9 | 54.8 | 56.5 |
| 15 | 24.9 | 25.0 | 25.2 | 24.7 | 24.5 | 24.5 | 24.0 | 24.9 |
| 16 | 27.6 | 27.6 | 27.8 | 27.7 | 27.6 | 27.7 | 27.5 | 27.6 |
| 17 | 52.4 | 52.5 | 52.6 | 52.9 | 52.7 | 52.7 | 52.3 | 52.4 |
| 18 | 12.0 | 12.5 | 12.6 | 12.9 | 12.6 | 12.7 | 12.2 | 12.0 |
| 19 | 17.7 | 63.7 | 63.9 | --- | --- | --- | --- | 17.7 |
| 20 | 39.5 | 39.6 | 39.9 | 40.0 | 39.9 | 39.9 | 39.8 | 39.5 |
| 21 | 13.9 | 13.9 | 14.1 | 13.9 | 13.9 | 13.9 | 14.0 | 13.9 |
| 22 | 78.6 | 78.9 | 79.7 | 79.6 | 79.6 | 79.6 | 79.5 | 78.7 |
| 23 | 30.4 | 30.3 | 25.2 | 25.2 | 25.2 | 25.2 | 25.2 | 30.4 |
| 24 | 157.4 | 154.5 | 148.8 | 148.7 | 148.8 | 148.7 | 148.7 | 157.4 |
| 25 | 124.3 | 127.8 | 125.2 | 125.3 | 125.2 | 125.2 | 125.2 | 124.4 |
| 26 | 166.3 | 166.9 | 167.1 | 167.1 | 167.1 | 167.1 | 167.1 | 166.4 |
| 27 | 64.0 | 56.6 | 13.0 | 12.9 | 12.9 | 12.9 | 12.9 | 63.9 |
| 28 | 21.0 | 20.6 | 67.1 | 66.9 | 67.0 | 67.0 | 67.0 | 21.0 |
| 1′ | 105.3 | 103.5 | 103.5 | 103.4 | 103.5 | 103.5 | 103.5 | 105.4 |
| 2′ | 75.9 | 75.6 | 75.4 | 75.4 | 75.4 | 75.4 | 75.4 | 75.7 |
| 3′ | 77.1 | 79.1 | 79.0 | 79.0 | 79.0 | 79.0 | 79.1 | 79.0 |
| 4′ | 78.5 | 71.9 | 72.1 | 72.1 | 72.1 | 72.1 | 72.1 | 72.1 |
| 5′ | 77.8 | 79.1 | 79.3 | 79.3 | 79.3 | 79.3 | 79.3 | 79.1 |
| 6′ | 61.9 | 63.0 | 63.3 | 63.3 | 63.3 | 63.3 | 63.3 | 63.2 |
1″ (103.1), 2″ (73.1), 3″(73.3), 4″ (74.5), 5″ (70.8), 6″ (19.0)
Compounds 9-14 were isolated as the minor components from the BuOH-soluble fraction. Withanolide 9 was assigned a molecular formula of C34H50O11 by HRESIMS and NMR experiments. Its NMR data (Tables 4 and 5) exhibited a close resemblance to those of withalongolide A (1), possessing the same nine-carbon side chain with an α,β-unsaturated δ-lactone, identical rings C and D, and an oxygenated C-19 methylene group. In addition, the remaining rings A and B showed similarities to those of 3-O-β-d-glucopyranosyl-20,27-dihydroxy-1-oxowitha-5,24-dienolide, a withanolide saponin reported from Physalis peruviana,19 with the following signals: (1) the occurrence of a ketone (δ 208.9, C-1) and a double bond [13C: C-5 quaternary carbon, δ 132.8, and C-6 methine, δ 129.1; 1H: δ 5.75 (1H, brd, J = 5.3 Hz, H-6)]; (2) a glucose moiety attached to C-3 in ring A [13C: δ 75.9 (CH, C-3); characteristic signals for glucose: δ 103.5 (CH, C-1′), 79.1 (CH, C-5′), 79.1 (CH, C-3′), 75.6 (CH, C-2′), 71.9 (CH, C-4′), and 63.0 (CH2, C-6′)]; (3) a fragment of - CH2–CH(O)–CH2- in ring A deduced from the 1H–1H COSY and HSQC spectra; (4) HMBC correlations of H-3/C-1′ and H-1′/C-3; of H2-19/C-1, C-5, C-9 and C-10; of H2-2/C-1, C-3, C-4, and C-10; and of Hβ-4/C-2,C-3, C-5, C-6, and C-10. Thus, the structure of 9 (withalongolide I) was determined as 3-O-β-d-glucopyranosyl-19,27-dihydroxyl-1-oxo-witha-5,24-dienolide.
Compound 10 was assigned a molecular formula of C34H52O11 by HRESIMS and NMR experiments. Similar to those of 9, the NMR data of 10 (Tables 4 and 5) displayed the presence of an oxygenated C-19 methylene group [13C: δ 63.9 CH2; 1H: δ 4.32 (1H, d, J = 11.0 Hz), 4.11 (1H, d, J = 11.0 Hz)] and three methyl groups [13C: δ 12.6, 13.0, 14.1; 1H: δ 0.80 (s), 0.99 (d, J = 6.6 Hz), 2.04 (s)]. The obvious differences between 10 and 9 were the presence of an oxygenated methine (δ 69.5) in 10 instead of the keto carbon (C-1, δ 208.9) in 9, implying that a hydroxy group is attached to C-1. This was supported by the presence of a 1H-1H COSY fragment of -CH(O)–CH2–CH(O)–CH2-assigned as a C-1 to C-4 moiety in ring A and confirmed by HMBC correlations of H2-19/C-1, C-5, C-9, and C-10, of H2-4/C-2, C-3, C-5, C-6, and C-10. The orientation of the hydroxy group at C-1 was assigned as α, based on the small coupling constant of H-1 (δ 4.61, s)/H2-2. Furthermore, it was determined that the glucose was attached to C-28 by the HMBC correlations of H2-28/C-23, 24, 25, 1′, of H-1′/C-28, as well as the chemical shifts of C-23 (δ 25.2), C-24 (δ 148.8), C-25 (δ 125.2), C-26 (δ 167.1), C-27 (δ 13.0), and C-28 (δ 67.1) and detailed comparison to those of withanolides with a 28-O-glucoside moiety.20 Thus, the structure of 10 (withalongolide J) was determined as 28-O-β-d-glucopyranosyl-1α,3β,19-trihydroxywitha-5,24-dienolide.
Similar to withalongolide J (10), compounds 11-14 were shown to possess the same nine-carbon side chain with an α,β-unsaturated δ-lactone and a glucose moiety at C-28, based on their superimposable NMR signals assigned to the side chain (Tables 4 and 5).
Saponin 11 was assigned a molecular formula of C33H46O10 by HRESIMS and NMR experiments. Excluding the six carbons corresponding to the glucose moiety, the 27-carbon-aglycon implied that one carbon in the C28 withanolide scaffold must be lost. The NMR data of its aglycon were similar to those of 1,6,27-trihydroxy-19-norwitha-1,3,5(10),24-tetraenolide (a 19-norwithanolide with an aromatic ring A).21 A trisubstituted aromatic ring A in 11 was observed from the 1H NMR (H-2: δ 7.14, 1H, d, J = 7.7 Hz; H-3: δ 7.34, 1H, t, J = 7.7 Hz; and H-4: δ 7.87, 1H, d, J = 7.7 Hz) and 13C NMR (C-1: δ 157.8 C; C-2: δ 115.1, CH; C-3: δ 127.4 CH; C-4: δ 119.3 CH; C-5: δ 146.2 C; and C-10: δ 127.8, C) experiments. This was confirmed by 1H-1H COSY, HSQC experiments and HMBC correlations of H-2/C-4 and C-10, H-3/C-1 and C-5, and of H-4/C-2 and C-10. Moreover, the HMBC correlations of H-4/C-6 (δ 70.7 CH), of OH-6 (δ 6.81, d, J = 6.5 Hz)/C-5 and C-6, of H-7β/C-5, and the 1H-1H COSY fragment -CH(OH)–CH2–CH–CH–CH2–CH2- (corresponding to -C6–C7–C8–C9–C11–C12-) showed that both the aglycone of 16 and 1,6,27-trihydroxy-19-norwitha-1,3,5(10),24-tetraenolide have the same planar structural moieties in rings A and B. However, the orientation of the hydroxy group at C-6 was assigned as α because of the large coupling constants (11.0 Hz) between H-6 (δ 5.26, dt, J = 11.0, 6.5 Hz) and H-7α (δ 1.63, q, J = 11.0 Hz), and (6.5 Hz) between H-6 and H-7β (δ 2.32, dd, J = 11.0, 6.5 Hz). Thus, the structure of 11 (withalongolide K) was determined as 28-O-β-d-glucopyranosyl-1,6α-dihydroxy-19-norwitha-1,3,5(10),24-tetraenolide.
Compound 12 was assigned a molecular formula of C33H48O11 by HRESIMS and NMR experiments. Its NMR data (Tables 4 and 5) were similar to those of withalongolide K (11), containing the same rings B, C, and D because of their superimposable 1H and 13C NMR signals. The differences observed between 12 and 11 were caused by changes in the ring A moiety. Unlike the aromatic ring A in 11, a conjugated 5(10)-ene-1-one system in 12 was revealed by the chemical shifts of quaternary carbons at δ 199.4 (C-1), 156.4 (C-5), 136.5 (C-10). A 1H-1H COSY fragment of -CH2–CH(OH)–CH2- was assigned as -C2–C3–C4- in ring A, and confirmed by the HMBC correlations of H2-2/C-1, C-3, and C-4, and of H2-4/C-2, C-5, and C-10. Thus, the structure of 12 (withalongolide L) was determined as 28-O-β-d-glucopyranosyl-3β,6α-dihydroxy-1-oxo-19-norwitha-5(10),24-dienolide.
Compound 13 was assigned a molecular formula of C33H48O10 by HRESIMS and NMR experiments. Its NMR data were similar to those observed for withalongolide L (12), containing a conjugated 5(10)-ene-1-one system [13C: δ 198.3 (C-1), 153.6 (C-5), 136.1 (C-10)] in ring A. The obvious differences between 13 and 12 were the presence of a methylene (13C: δ 33.8; 1H: δ 2.30, 2H, m) instead of an oxygenated methine (13C: δ 71.2; 1H: δ 4.69), suggesting that 13 is a 6-deoxy derivative of 12. This observation was supported by the 13C NMR high-frequency shift of C-4 (δ 41.9 in 13 and δ 36.9 in 12) and C-8 (δ 39.9 in 13 and δ 37.5 in 12), the low-frequency shift of C-7 (δ 26.4 in 13 and δ 37.3 in 12), and HMBC correlations of OH-3 (δ 6.81, 1H, d, J = 4.1 Hz)/C-2 (δ 49.7), C-3 (δ 66.1), and C-4 (δ 41.9), of H2-4 (δ 2.68, dd, J = 5.9, 16.8 Hz and δ 2.58, dd, J = 2.7, 16.8 Hz)/C-2, C-3, C-5 (δ 153.6), C-6 (δ 33.8), and C-10 (δ 136.1), and of H-6β (δ 2.09, m)/C-4, C-5, C-7 (δ 26.4), C-8 (39.9), and C-10 in 13. Thus, the structure of 13 (withalongolide M) was determined as 28-O-β-d-glucopyranosyl-3β-hydroxy-1-oxo-19-norwitha-5(10),24-dienolide.
Compound 14 was assigned a molecular formula of C33H48O10 by HRESIMS and NMR experiments, and as an isomer of withalongolide M (13). The NMR data of these two compounds (Tables 4 and 5) were similar to each other, having the same functional groups and the same multiplicities for all other carbons present. A conjugated 5(10)-ene-6-one system [13C: δ 198.6 (C-6), 158.7 (C-10), 130.4 (C-5)] in 14 was proposed instead of the 5(10)-ene-1-one one in 13 on the basis of the following observations: (1) a 1H-1H COSY fragment of -CH2–CH2–CH(OH)–CH2- (from C-1 to C-4) in ring A of 14 replaced the ring A fragment -CH2–CH(OH)–CH2- (from C-2 to C-4) in 13; (2) a 1H-1H COSY fragment of -CH2–CH–CH- (from C-7 to C-9) in ring B of 14 replaced the ring B fragment -CH2–CH2–CH–CH- (from C-6 to C-9) in 13; (3) HMBC correlations of H-2/C-1 (δ 25.6), C-3 (δ 64.7), and C-4 (δ 32.9); of H2-7/C-5 (δ 130.4), C-6 (δ 198.6), C-8 (δ 40.0), and C-9 (δ 46.6); H-1/C-3, C-5, and C-10 (δ 158.7). Furthermore, the orientation of the hydroxyl group at C-3 was determined as α due to the small coupling constant (J = 2.2 Hz) between H-3 (δ 4.33, brs)/H-4β (δ 2.81, dd, J = 2.2, 15.8 Hz) and the NOESY correlations of H-3/H-1β, H-2β, H-4β. Thus, the structure of 14 (withalongolide N) was assigned as 28-O-β-d-glucopyranosyl-3α-hydroxy-6-oxo-19-norwitha-5(10),24-dienolide.
Eight known compounds were identified by comparison of their data with those published in the literature, as seven withanolides, sitoindoside IX (15),18 withaferin A (16),1,13 2,3-dihydro-3β-methoxywithaferin A (17),15 viscosalactone B (18),22 2,3-dihydro-3β-O-sulfate withaferin A (19),17 2,3-dihydrowithaferin A (20),15 and 3α,6α-epoxy-4β,5β,27-trihydroxy-1-oxowitha-24-enolide (21),23 and a flavonoid glucoside, rutin (22).24 The full assignments of the NMR data of 15, 16, 17, 18, and 19 are listed in Tables 2-5 as these data were either unavailable or incomplete or in need of revision within the published literature.
The classically defined withanolide-type steroids (1-21) isolated from the title plant showed a diversity of oxygenation patterns that may be summarized as follows: (1) Six withanolides (1, 2, 4, 7, 9 and 10) have an oxygenated C-19 group, which is rare in Nature. A literature investigation showed that from the approximately 520 unmodified withanolides only nine C-19 oxygenated withanolides have been reported so far. They are as follows: jaborosalactones O,25 V, W, X,26 46, 47, 48,27 cinerolide 28 and bracteosin B.29 (2) Compounds 3 and 5 are rare examples of unmodified withanolides having an oxygenated C-11 although withasomniferanolide (with 11β-OH group),30 somniferanolide (with 11β-OH group),30 and witharifeen (with 11α-OH group)31 have been previously reported. (3) Saponins 10-14 have a sugar constituent attached at the C-28. Only two previously published withaholide saponins (physagulins E and G)20 were shown to have a sugar moiety at C-28 thus far. (4) Compounds 11-14 are the first reported examples of C-19 nor withanolide saponins. It should be noted, however, that there are only four C-19 nor withanolides (jaborosalactone Q,21 7,32 45, 12-O-methyl-jaborosalactone 45 27) reported in the literature. (5) Most withanolides have an oxygenated C-1 in ring A, but 15 is the exception by not being oxidized at C-1. In addition, the presence of a 3-O-sulfate group in naturally occurring withanolides is extremely rare. Besides withanolides 7 and 19, there are only five other 3-O-sulfate withanolides previously reported from Datura metel,33 Solanum cilistum,34 and Withania somnifera.35
All the withanolides (1-21) and the four acetylated derivatives (1a, 1b, 2a and 2b) were tested against the HNSCC (JMAR, MDA-1986), melanoma (B16F10 and SKMEL-28), or/and normal fetal fibroblast (MRC-5) cells for their cytotoxicity. As summarized in Table 6, withanolides 1-5, 7, 8, 15, 16, and 19 and the four derivatives (1a, 1b, 2a, and 2b) showed cytotoxic effects against the cells tested with IC50 values in the range 0.067–9.3 μM, while the other withanolides were inactive. Similar to withaferin A 16, withanolides 1-3 containing the functional groups of a 2-en-1-one in ring A, a 5β,6β-epoxy in ring B, and a lactone ring in the side chain, were active, showing the importance of these three groups. The activity of the 3-O-sulfate withanolides 7 and 19 was due to their transformations to 1 and 16, respectively. Withanolide glycosides 8 and 15 displayed less cytotoxicity relative to their aglycone withaferin A (16). However, the esterification of the hydroxy groups at C-4, C-19, and C-27 increased the resultant cytotoxicity, as shown for the acetylated derivatives 1a and 2a with IC50 values less than 1 μM against all the cells tested. These results are in agreement with previous structure-activity relationship reports.10,36,37 In addition, it should be noted that withalongolide A (1), withalongolide B (2), and withaferin A (16) are most likely responsible for the cytotoxic activities of the extract prepared from the title plant due to their relative high abundance levels (0.16% for 1, 0.10% for 16, and 0.03% for 2).
Table 6.
Cytotoxicity IC50 of Isolated Withanolides (μM) against Five Cell Linesa
| Compound | B16F10 | SKMEL-28 | JMAR | MDA-1986 | MRC-5 |
|---|---|---|---|---|---|
| 1 | >10 | 5.1 | 5.3 | 3.3 | >10 |
| 1a | 0.067 | 0.54 | 0.16 | 0.91 | 0.58 |
| 1b | 0.098 | 0.81 | 0.14 | 2.2 | 0.41 |
| 2 | 0.20 | 3.9 | 0.17 | 1.3 | 0.40 |
| 2a | 0.13 | 0.27 | 0.24 | 0.11 | 0.51 |
| 2b | 0.19 | 0.64 | 0.12 | 0.49 | 0.16 |
| 3 | 0.49 | 3.0 | 0.77 | 2.6 | 3.6 |
| 4 | 3.2 | 9.3 | 4.7 | >10 | 6.5 |
| 5 | 5.6 | >10 | >10 | 8.3 | 7.3 |
| 7 | 1.3 | 4.8 | 2.3 | 2.0 | 3.3 |
| 8 | >10 | >10 | 8.2 | 8.1 | 8.7 |
| 15 | 3.7 | 8.3 | 4.2 | >10 | 5.2 |
| 16 | 0.29 | 4.0 | 2.0 | 0.80 | 0.20 |
| 19 | 0.18 | 5.1 | 0.48 | 0.27 | 1.4 |
| cisplatin (positive control) | 1.0 | 1.1 | 1.1 | 1.6 | 8.9 |
For cell lines used, see text. Withanolides 6, 9-14, 17, 18, 20 and 21 were inactive for all cell lines used (IC50 > 10 μM).
EXPERIMENTAL SECTION
General Experimental Procedures
Melting points were obtained using an MPA100 melting point apparatus. Optical rotations were measured with a Rudolph RS Autopol IV automatic polarimeter. IR data were obtained with a Thermo Nicolet Avatar 360 FT-IR spectrometer. NMR spectra were recorded with a Bruker AV-400 or AV-500 instrument with a cryoprobe for 1H, APT, COSY/DQF-COSY, HSQC, HMBC, and NOESY/ROESY. Chemical shift values are given in δ (ppm) using the peak signals of the solvent C5D5N (δH 8.74, 7.58, and 7.22; and δC 150.35, 135.91, and 123.87) or CDCl3 (δH 7.24 and δC 77.23) as references and coupling constants were reported in Hz. ESIMS data were measured with an Agilent 1200 Series LC-MS/MS ion trap 6300 mass spectrometer. HRESIMS data were collected with a LCT Premier time of flight mass spectrometer (Waters Corp., Milford, MA). Column chromatography was performed on silica gel (particle size 12–25 μm) (Sorbent Technologies, Atlanta, GA), or MCI CHP20P (particle size 75–150 μm) (Sigma-Aldrich, Saint Louis, MO), or Sephadex LH-20 (GE Healthcare, Piscataway, NJ), or C18 reversed-phase silica gel (particle size 40–65 μm) (Sigma-Aldrich, Saint Louis, MO). Normal-phase silica gel G TLC plates (w/UV 254) and reversed-phase C18 TLC plates (w/UV 254) (Sorbent Technologies, Atlanta, GA) were used for fraction detection. The spots were visualized using UV light at 254 nm and spraying with 10% EtOH-sulfuric acid reagent. Semi-preparative HPLC was performed on an Agilent 1200 unit equipped with a DAD detector, utilizing a Lichrospher RP-18 column (250 × 10 mm, 5 μm).
Plant Material
Fresh aerial parts of the plant P. longifolia were collected from the Kanopolis wildlife area (latitude: 38.74206°; longitude: 97.98467°), Ellsworth County, Kansas, in August, 2009. It was identified by plant taxonomist Dr. Kelly Kindscher at the Kansas Biological Survey, University of Kansas. A voucher specimen (Hillary Loring 3583) was deposited in the R.L. McGregor Herbarium of the University of Kansas.
Extraction and Isolation
The collected biomass was air dried at room temperature. The dried material was then ground to a coarse powder (930 g), and extracted three times with CH2Cl2–MeOH (50:50, 4.0 L) at room temperature. After removing the solvents under vacuum, the extract (107 g) was suspended in 400 mL H2O, followed by partitions with n-hexane, EtOAc, and n-butanol (3 × 500 mL). The resulting ethyl acetate fraction (22 g) collected was applied to silica gel flash CC (column chromatography), and eluted subsequently with hexane–acetone mixtures of increasing polarities. The fraction obtained on elution with hexane–acetone (80:20) (1.0 g), was again subjected to silica gel CC [eluted with CH2Cl2–CH3COCH3 (90:10)] to afford compound 2 (280 mg). The fraction obtained on elution with hexane–acetone (70:30) (3.0 g), was subjected to silica gel CC [eluted with CH2Cl2–CH3COCH3 (80:20)] to yield compounds 16 (730 mg), 17 (7 mg) and 20 (10 mg). The fraction obtained on elution with hexane–acetone (65:35) (700 mg), was subjected further to silica gel CC [eluted with hexane–acetone (65:35)] to afford compounds 3 (30 mg) and 5 (5 mg). The fraction acquired on elution with hexane–acetone (60:40) (2.2 g), was applied to silica gel CC [eluted with hexane–acetone (60:40)] to afford compounds 1 (1200 mg) and 4 (15 mg). The n-butanol fraction (29 g) obtained was subjected to a MCI CHP20P gel CC (500 g), eluted with a mixture of H2O–MeOH (100:0, 80:20, 60:40, 40:60, 85:15, 0:100), in order of increasing concentrations of methanol. The 85% MeOH fraction (3.5 g) was subjected to silica gel CC, eluted with CH2Cl2-CH3COCH3 with increasing amounts of acetone to afford compounds 1 (240 mg), 6 (40 mg), 16 (200 mg), 18 (250 mg) and 21 (220 mg). The 60% MeOH fraction (4.2 g) was subjected to silica gel CC, eluted with CH2Cl2-MeOH-H2O (7:1:0.1) with increasing amounts of MeOH-H2O (10:1), then the fractions were further subjected to reverse-phase C18 Si gel column chromatography (200 g, particle size 40-63 μm), eluted by MeOH-H2O (40:60, 50:50, 60:40, 65:35). The fractions obtained were subjected to semi-preparative HPLC, with the mobile phase CH3CN-H2O (26:74; 28:72), to afford compounds 7 (40 mg), 8 (35 mg), 9 (9 mg), 10 (12 mg), 11 (6 mg), 12 (5 mg), 13 (6 mg), 14 (7 mg), 15 (22 mg), 19 (35 mg), and 22 (80 mg).
Withalongolide A (1)
colorless cuboid crystals (CH3CN); mp 216-217 °C; [α]25D +14.2 (c 0.16, CHCl3); IR (neat) υmax 3431 (br), 3233 (br), 2922, 1706, 1671, 1400, 1037, 962 cm-1; 1H NMR and 13C NMR, see Tables 1 and 2; ESIMS (positive-ion mode) m/z 487 (M+H, 6), 469 (M–H2O+H, 100), 451 (M–2 H2O+H, 6); HRESIMS m/z 509.2489 [M+Na]+ (calcd for C28H38O7Na, 509.2471), m/z 487.2674 [M+H]+ (calcd for C28H39O7, 487.2696), m/z 469.2585 [M–H2O+H]+ (calcd for C28H37O6, 469.2590).
Single-Crystal X-ray Structure Determination of Withalongolide A (1)
Crystal analysis was performed with a colorless plate crystal (dimensions 0.42 × 0.35 × 0.21 mm3) obtained from CH2Cl2–CH3CN (1:1) using Mo Kα radiation (λ = 0.71073 Å) on a Bruker SMART APEX diffractometer equipped with a sealed-tube x-ray source and a graphite monochromator. Crystal data for 1: C28H38O7 (formula weight 486.58), monoclinic, space group P21, T = 100(2) K, crystal cell parameters a = 8.370(2) Å, b = 10.523(3) Å, c = 14.280(3) Å, β = 104.552(4)°, V = 1217.4 (5) Å3, Dc = 1.327 Mg/m3, Z = 2, F(000) = 524, absorption coefficient μ = 0.094 mm−1. A total of 11335 reflections were collected in the range 2.43 < θ < 29.21°, with 5809 independent reflections [R(int) = 0.050] and 5478 with I > 2σ(I), completeness to θmax was 93.1%. Multi-scan absorption correction applied; full-matrix least-squares refinement on F2, the number of data/restraints/parameters were 5809/1/468; goodness-of-fit on F2 = 1.015; final R indices [I > 2σ (I)], R1 = 0.045, ωR2 = 0.098; R indices (all data), R1 = 0.048, ωR2 = 0.099; largest difference peak and hole, 0.37 and -0.21 e/Å-3.
Acetylation of Withalongolide A (1)
A solution of 1 (50 mg) in pyridine (8 mL) and acetic anhydride (2 mL), was stirred for 24 hrs at room temperature. Then 50 mL water were added to the solution. The precipitate (70 mg) was subjected to semi-preparative HPLC, eluted with the mobile phase CH3CN–H2O (45:55), to afford triacetate 1a (40 mg) and diacetate 1b (10 mg).
Withalongolide A 4, 19,27-triacetae (1a)
IR (neat) υmax 2953 (br), 1731, 1702, 1674, 1366, 1208, 1023, 966 cm-1; 1H NMR and 13C NMR, see Tables 1 and 2; ESIMS (positive-ion mode) m/z 630 (M+H2O, 30), 613 (M+H, 100); HRESIMS m/z 635.2829 [M+Na]+ (calcd for C34H44O10Na, 635.2832).
Withalongolide A 4,27-diacetae (1b)
IR (neat) υmax 3536 (br), 2922, 1736, 1701, 1674, 1376, 1215, 1021, 967 cm-1; 1H NMR and 13C NMR, see Tables 1 and 2; ESIMS (positive-ion mode) m/z 1163 (2 M+Na, 6), 588 (M+H2O, 10), 571 (M+H, 100); HRESIMS m/z 593.2740 [M+Na]+ (calcd for C32H42O7Na, 593.2727).
Withalongolide B (2)
colorless plate crystals (toluene); mp 197-198 °C; [α]25D +12.3 (c 0.15, CHCl3); IR (neat) υmax 3260 (br), 3006, 2946, 2887, 1693, 706, 1675, 1383, 1082, 763 cm-1; 1H NMR and 13C NMR, see Tables 1 and 2; ESIMS (positive-ion mode) m/z 963 (2 M+Na, 40), 941 (2 M+H, 70), 493 (M+Na, 10), 471 (M+H, 4), 453 (M–H2O+H, 100), 435 (M–2 H2O+H, 45%); HRESIMS m/z 493.2554 [M+Na]+ (calcd for C28H38O6Na, 493.2566).
Single-Crystal X-ray Structure Determination of Withalongolide B (2)
Crystal analysis was performed with a colorless plate (dimensions 0.21 × 0.16 × 0.15 mm3) obtained from CH2Cl2–CH3COCH3–toluene (1:1:1) using Cu Kα radiation (λ = 1.54178 Å) on a Bruker APEX2 diffractometer equipped with a Bruker MicroStar microfocus rotating anode x-ray source and Helios multilayer optics. Crystal data for 2: C28H38O6·C7H8 (formula weight 562.72), Orthorhombic, space group P212121, T = 100(2) K, crystal cell parameters a = 7.1844(3) Å, b = 26.0678(10) Å, c = 49.0852(18) Å, V = 9192.8 (6) Å3, Dc = 1.220 Mg/m3, Z = 12, F(000) = 3648, absorption coefficient μ = 0.65 mm−1. A total of 32032 reflections were collected in the range 1.80 < θ < 69.15°, with 13806 independent reflections [R(int) = 0.035] and 12653 with I > 2σ(I), completeness to θmax was 90.8%. Multi-scan absorption correction applied; full-matrix least-squares refinement on F2, the number of data/restraints/parameters were 13806/0/1108; goodness-of-fit on F2 = 1.083; final R indices [I > 2σ (I)], R1 = 0.096, ωR2 = 0.233; R indices (all data), R1 = 0.102, ωR2 = 0.238; largest difference peak and hole, 0.84 and -0.32 e/Å-3.
Acetylation of Withalongolide B (2)
A solution of 2 (50 mg) in pyridine (8 mL) and acetic anhydride (2 mL), was stirred for 24 hrs at room temperature. Then 50 mL water were added to the solution. The precipitate (65 mg) was subjected to semi-preparative HPLC, eluted with the mobile phase CH3CN–H2O (43:57), to afford diacetate 2a (41 mg) and monoacetate 2b (11 mg).
Withalongolide B 4,19-diacetate(2a)
IR (neat) υmax 2943 (br), 1738, 1698, 1368, 1220, 1127, 1043, 1019, 762 cm-1; 1H NMR and 13C NMR, see Tables 1 and 2; ESIMS (positive-ion mode) m/z 1109 (2 M+H, 60), 577 (M+Na, 40), 555 (M+H, 30), 495 (M–HOAC+H, 100), 435 (M–2 HOAC+H, 30), 296 (70); HRESIMS m/z 577.2764 [M+Na]+ (calcd for C32H42O8Na, 577.2727).
Withalongolide B 4-acetate (2b)
IR (neat) νmax 3536 (br), 2922, 1736, 1701, 1674, 1376, 1215, 1021, 967 cm-1; 1H NMR and 13C NMR, see Tables 1 and 2; ESIMS (positive-ion mode) m/z 1047 (2 M+Na, 30), 1025 (2 M+H, 75), 513 (M+H, 100), 453 (M–HOAc+H, 6); HRESIMS m/z 535.2670 [M+Na]+ (calcd for C30H40O7Na, 535.2672).
Withalongolide C (3)
colorless cuboid crystals, mp 197-198 °C; [α]25D +10.3 (c 0.12, CHCl3); IR (neat) νmax 3550 (br), 3419 (br), 2952, 2879, 1686, 1663, 1394, 1227, 1026, 957 cm-1; 1H NMR and 13C NMR, see Tables 1 and 2; ESIMS (positive-ion mode) m/z 469 (M–H2O+H, 100); HRESIMS m/z 509.2481 [M+Na]+ (calcd for C28H38O7Na, 509.2471).
Single-Crystal X-ray Structure Determination of Withalongolide C (3)
Crystal analysis was performed with a colorless irregular chunk (dimensions 0.45 × 0.32 × 0.25 mm3) obtained from CH2Cl2–CH3CN (1:1) and measured using Mo Kα radiation (λ = 0.71073 Å) on a Bruker APEX diffractometer equipped with a sealed-tube x-ray source and a graphite monochromator. Crystal data for 3: C28H38O7 (formula weight 486.58), Orthorhombic, space group P212121, T = 100(2) K, crystal cell parameters a = 10.679(4) Å, b = 12.245(5) Å, c = 18.674(7) Å, V = 2442 (2) Å3, Dc = 1.324 Mg/m3, Z = 4, F(000) = 1048, absorption coefficient μ = 0.094 mm−1. A total of 22352 reflections were collected in the range 2.53 < θ < 29.12°, with 6145 independent reflections [R(int) = 0.055] and 5925 with I > 2σ(I), completeness to νmax was 95.6%. Multi-scan absorption correction applied; full-matrix least-squares refinement on F2, the number of data/restraints/parameters were 6145/0/468; goodness-of-fit on F2 = 1.080; final R indices [I > 2σ (I)], R1 = 0.045, ωR2 = 0.109; R indices (all data), R1 = 0.046, ωR2 = 0.113; largest difference peak and hole, 0.63 and -0.21 e/Å-3.
Withalongolide D (4)
[α]25D +2.7 (c 0.08, CHCl3); IR (neat) νmax 3411 (br), 2944 (br), 1693, 1393, 1211, 1023 cm-1; 1H NMR and 13C NMR, see Tables 2 and 3; ESIMS (positive-ion mode) m/z 1059 (2 M+Na, 6), 519 (M+H, 25), 501 (M–H2O+H, 100), 483 (M–2 H2O+H, 4); HRESIMS m/z 541.2798 [M+Na]+ (calcd for C28H38O7Na, 541.2777).
Withalongolide E (5)
[α]25D +2.2 (c 0.12, CHCl3); IR (neat) νmax 3550 (br), 2940 (br), 1690, 1390, 1200, 1020 cm-1; 1H NMR and 13C NMR, see Tables 2 and 3; ESIMS (positive-ion mode) m/z 1059 (2 M+Na, 20), 541 (M+Na, 18), 501 (M–H2O+H, 100); HRESIMS m/z 541.2777 [M+Na]+ (calcd for C28H38O7Na, 541.2777).
Withalongolide F (6)
colorless cuboid crystals, mp 190-191 °C; [α]25D +8.9 (c 0.16, CHCl3); IR (neat) νmax 2887, 1683, 1659, 1393, 1002, 851 cm-1; 1H NMR and 13C NMR, see Tables 2 and 3; ESIMS (positive-ion mode) m/z 871 (2 M+Na, 25), 425 (M+H, 100); HRESIMS m/z 447.2503 [M+Na]+ (calcd for C27H38O4Na, 447.2511).
Single-Crystal X-ray Structure Determination of Withalongolide F (6)
Crystal analysis was performed with a colorless rectangular parallelepiped (dimensions 0.39 × 0.37 × 0.20 mm3) obtained from CH2Cl2–CH3CN (1:1) and measured using Mo Kα radiation (λ = 0.71073 Å) on a Bruker APEX diffractometer equipped with a sealed-tube x-ray source and graphite monochromator. Crystal data for 6: C27H36O4 (formula weight 424.56), monoclinic, space group P21, T = 100(2) K, crystal cell parameters a = 10.873(5) Å, b = 9.233(4) Å, c = 12.271(6) Å, β = 113.273°(7), V = 1132 (9) Å3, Dc = 1.246 Mg/m3, Z = 2, F(000) = 460, absorption coefficient μ = 0.082 mm−1. A total of 10347 reflections were collected in the range 2.85 < θ < 29.06°, with 5283 independent reflections [R(int) = 0.041] and 5133 with I > 2σ(I), completeness to θmax was 93.8%. Multi-scan absorption correction applied; full-matrix least-squares refinement on F2, the number of data/restraints/parameters were 5283/1/424; goodness-of-fit on F2 = 1.055; final R indices [I > 2σ (I)], R1 = 0.045, ωR2 = 0.107; R indices (all data), R1 = 0.046, ωR2 = 0.108; largest difference peak and hole, 0.43 and -0.21 e/Å-3.
Withalongolide G (7)
[α]25D -2.3 (c 0.11, MeOH); IR (neat) νmax 3385 (br), 2950 (br), 1686, 1399, 1234, 992 cm-1; 1H NMR and 13C NMR, see Tables 2 and 3; ESIMS (positive-ion mode) m/z 1031 [2 (M–SO3)+Na, 20], 585 (M+H, 50), 505 (M–SO3+H, 100); (negative-ion mode) m/z 583 (M–H, 100); HRESIMS m/z 607.2169 [M+Na]+ (calcd for C28H40O11SNa, 607.2189).
Withalongolide H (8)
[α]25D -0.9 (c 0.12, MeOH); IR (neat) νmax 3369 (br), 2929 (br), 1686, 1398, 1018, 916 cm-1; 1H NMR and 13C NMR, see Tables 4 and 5; ESIMS (positive-ion mode) m/z 796 [M+H2O, 40], 471 (M–rha–glc+H, 100); LC-MS/MS fragments of m/z 796 peak: m/z 471 (M–rha–glc+H, 100), 281 (10); LC–MS/MS fragments of m/z 471 peak: m/z 281 (100); HRESIMS m/z 801.3683 [M+Na]+ (calcd for C40H58O15Na, 801.3673).
Withalongolide I (9)
[α]25D -7.1 (c 0.08, MeOH); IR (neat) νmax 3381 (br), 2921 (br), 1686, 1398, 1007 cm-1; 1H NMR and 13C NMR, see Tables 4 and 5; ESIMS (positive-ion mode) m/z 652 (M+H2O, 6), 635 (M+H, 5), 473 (M–glc+H, 100); HRESIMS m/z 657.3267 [M+Na]+ (calcd for C34H50O11Na, 657.3251).
Withalongolide J (10)
[α]25D -1.0 (c 0.13, MeOH); IR (neat) νmax 3370 (br), 2931 (br), 1687, 1396, 1017 (br) cm-1; 1H NMR and 13C NMR, see Tables 4 and 5; ESIMS (positive-ion mode) m/z 1273 (2 M+H, 100), 619 (M–H2O+H, 20), 601 (M–2 H2O+H, 15), 583 (M–3 H2O+H, 10), 475 (M–glc+H, 6); HRESIMS m/z 659.3411 [M+Na]+ (calcd for C34H52O11Na, 657.3407).
Withalongolide K (11)
[α]25D +3.1 (c 0.17, MeOH); IR (neat) νmax 3381 (br), 2921 (br), 1686, 1398, 1007 cm-1; 1H NMR and 13C NMR, see Tables 4 and 5; ESIMS (positive-ion mode) m/z 460 (M–glc+H, 40) 603 (M+H, 35), 1205 (2 M+H, 100); HRESIMS m/z 625.33007 [M+Na]+ (calcd for C33H46O10Na, 625.2989).
Withalongolide L (12)
[α]25D -3.1 (c 0.09, MeOH); IR (neat) νmax 3350 (br), 2931 (br), 1687, 1396, 1018 cm-1; 1H NMR and 13C NMR, see Tables 4 and 5; ESIMS (positive-ion mode) m/z 1241 (2 M+H, 15), 1079 (2 M–glc+H, 30), 638 (M+H2O, 80). 459 (M–glc+H, 100); LC-MS/MS fragments of the m/z 638 peak: m/z 621 (M+H, 80), 459 (M–glc+H, 100); HRESIMS m/z 643.3101 [M+Na]+ (calcd for C33H48O11Na, 643.3094).
Withalongolide M (13)
[α]25D -4.4 (c 0.08, MeOH); IR (neat) νmax 3380 (br), 2932 (br), 1687, 1650, 1388, 1070 cm-1; 1H NMR and 13C NMR, see Tables 4 and 5; ESIMS (positive-ion mode) m/z 1209 (2 M+H, 20), 1048 (2 M–glc+H, 30), 622 (M+H2O, 30), 605 (M+H, 10), 443 (M–glc+H, 100); HRESIMS m/z 627.3156 [M+Na]+ (calcd for C33H48O10Na, 627.3145).
Withalongolide N (14)
[α]25D -2.0 (c 0.14, MeOH); IR (neat) νmax 3368 (br), 2931 (br), 1688, 1650, 1384, 1072, 1017 cm-1; 1H NMR and 13C NMR, see Tables 4 and 5; ESIMS (positive-ion mode) m/z 1209 (2 M+H, 35), 605 (M+H, 100); LC-MS/MS fragments of m/z 1209 peak: m/z 605 (M+H, 100%), 443 (M–glc+H, 100); LC-MS/MS fragments of m/z 605 peak: m/z 443 (M–glc+H, 100); HRESIMS m/z 627.3146 [M+Na]+ (calcd for C33H48O10Na, 627.3145).
Cytotoxicity Bioassay
The cytotoxicity assays were performed as previously described.11 In general, five concentrations ranging from 0.1 to 100 μg/mL were tested for the extracts, and ten concentrations ranging from 50 nM to 20 μM were tested for pure compounds. Statistical analysis was carried out by one-way ANOVA on ranks test using GraphPad Prism 5 (GraphPad Software, San Diego, CA). IC50 values were obtained from cell viability plots fitted with a sigmoidal dose-response function with variable slope using GraphPad Prism 5 software.
Supplementary Material
Acknowledgments
This study was supported, in part, by grant IND 0061464 (awarded to B.N.T. and K.K.) from the Kansas Bioscience Authority (KBA) and Center for Heartland Plant Innovations (HPI). The authors also acknowledge partial financial assistance from grant NFP0066367 from the Institute for Advancing Medical Innovation (IAMI) (awarded to M.S.C. and to B.N.T.). Partial support of the in vitro experiments was provided by the University of Kansas Center for Cancer Experimental Therapeutics NIH-COBRE P20 RR015563 (PI: B.N.T., project award PI: M.S.C.). The authors are grateful to NSF grant CHE-0923449 that was used to purchase the new Bruker APEX2 X-ray diffractometer. The authors thank Q. Long, H. Loring and M. Ferreira, botanists at the University of Kansas or at the Kansas Biological Survey at the University of Kansas for assistance in plant collections and identifications.
Footnotes
Supporting Information. 1H and 13C NMR spectra of withanolides 1-21 and the bioassay data of the samples (CH2Cl2-MeOH crude extract, hexane soluble fraction, EtOAc soluble fraction, and n-BuOH soluble fraction) are available free of charge via the Internet at http://pubs.acs.org. Crystallographic data for the structures of 1, 2, 3, and 6 reported in this paper have been deposited with the Cambridge Crystallographic Data Centre, under reference numbers CCDC 840311, CCDC 840312, CCDC 840313 and CCDC 840314, respectively. Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-(0)1223-336033 or deposit@ccdc.cam.ac.uk).
References
- 1.Lavie D, Glotter E, Shvo Y. J Org Chem. 1965;30:1774–1778. [Google Scholar]
- 2.Misico RI, Nicotra VE, Oberti JC, Barboza G, Gil RR, Burton G. Prog Chem Org Nat Prod. 2011;94:127–229. doi: 10.1007/978-3-7091-0748-5_3. [DOI] [PubMed] [Google Scholar]
- 3.Chen LX, Hao H, Qiu F. Nat Prod Rep. 2011;28:705–740. doi: 10.1039/c0np00045k. [DOI] [PubMed] [Google Scholar]
- 4.Eich E. Solanaceae and Convolvulaceae: Secondary Metabolites. Spring-Verlag; Berlin: 2008. pp. 466–483. [Google Scholar]
- 5.Veleiro AS, Oberti JC, Burton G. In: Studies in Natural Products Chemistry; Atta-ur- Rahman. Elsevier Science BV, editor. Vol. 32. Amsterdam: 2005. pp. 1019–1051. [Google Scholar]
- 6.Budhiraja RD, Krishan P, Sudhir S. J Sci Ind Res. 2000;59:904–911. [Google Scholar]
- 7.Anjaneyulu ASR, Rao DS, Lequesne PW. In: Studies in Natural Products Chemistry; Atta-ur-Rahman. Elsevier Science BV, editor. Vol. 20. Amsterdam: 1998. pp. 135–261. [Google Scholar]
- 8.Ray AB, Gupta M. Prog Chem Org Nat Prod. 1994;63:1–106. [Google Scholar]
- 9.Glotter E. Nat Prod Rep. 1991:415–440. doi: 10.1039/np9910800415. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida M, Hoshi A, Kuretani K, Ishiguro M, Ikekawa N. J Pharm Dyn. 1979;2:92–97. [Google Scholar]
- 11.Samadi AK, Tong XQ, Mukerji R, Zhang HP, Timmermann BN, Cohen MS. J Nat Prod. 2010;73:1476–1481. doi: 10.1021/np100112p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong XQ, Zhang HP, Timmermann BN. Phytochemistry Lett. 2011 doi: 10.1016/j.phytol.2011.04.016. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuska J, Prousek J, Rosazza J, Budesinsky M. Steroids. 1982;40:157–170. doi: 10.1016/0039-128x(82)90030-7. [DOI] [PubMed] [Google Scholar]
- 14.Pramanick S, Roy A, Ghosh S, Majumder HK, Mukhopadhyay S. Planta Med. 2008;74:1745–1748. doi: 10.1055/s-2008-1081357. [DOI] [PubMed] [Google Scholar]
- 15.Pelletier SW, Mody NV, Nowacki J, Bhattacharyya J. J Nat Prod. 1979;42:512–521. [Google Scholar]
- 16.Lavie D, Kashman Y, Glotter E, Danieli N. J Chem Soc (C) 1966:1757–1764. [Google Scholar]
- 17.Xu YM, Marron MT, Seddon E, McLaughlin SP, Ray DT, Whitesell L, Gunatilaka AAL. Bioorg Med Chem. 2009;17:2210–2214. doi: 10.1016/j.bmc.2008.10.091. [DOI] [PubMed] [Google Scholar]
- 18.Ghosal S, Kaur R, Srivastava RS. Indian J Nat Prod. 1988;4:12–13. [Google Scholar]
- 19.Ahmad S, Yasmmin R, Malik A. Chem Pharm Bull. 1999;47:477–480. [Google Scholar]
- 20.Shingu K, Yahara S, Okabe H, Nohara T. Chem Pharm Bull. 1992;40:2448–2451. [Google Scholar]
- 21.Veleiro AS, Trocca CE, Burton G, Oberti JC. Phytochemistry. 1992;31:2550–2551. [Google Scholar]
- 22.Pelletier SW, Gebeyehu G, Nowacki J, Mody NV. Heterocycles. 1981;15:317–320. [Google Scholar]
- 23.Zhao J, Nakamura N, Hattori M, Kuboyama T, Tohda C, Komatsu K. Chem Pharm Bull. 2002;50:760–765. doi: 10.1248/cpb.50.760. [DOI] [PubMed] [Google Scholar]
- 24.Li L, Henry GE, Seeram NP. J Agric Food Chem. 2009;57:7282–7287. doi: 10.1021/jf901716j. [DOI] [PubMed] [Google Scholar]
- 25.Monteagudo ES, Burton G, Gros EG, Gonzalez CM, Oberti JC. Phytochemistry. 1989;28:2514–2515. [Google Scholar]
- 26.Misico RI, Oberti JC, Veleiro AS, Burton G. J Nat Prod. 1996;59:66–68. doi: 10.1021/np000022z. [DOI] [PubMed] [Google Scholar]
- 27.Cirigliano AM, Veleiro AS, Misico RI, Tettamanzi MG, Oberti JC, Burton G. J Nat Prod. 2007;70:1644–1646. doi: 10.1021/np070197+. [DOI] [PubMed] [Google Scholar]
- 28.Maldonado E, Alvarado VE, Torres FR, Martínez M, Pérez-Castorena AL. Planta Med. 2005;71:548–553. doi: 10.1055/s-2005-864157. [DOI] [PubMed] [Google Scholar]
- 29.Riaz N, Malik A, Azia-ur-Rehman, Nawaz SA, Muhammad P, Choudhary MI. Chem Biodivers. 2004;1:1289–1295. doi: 10.1002/cbdv.200490091. [DOI] [PubMed] [Google Scholar]
- 30.Ali M, Shuaib M, Ansari SH. Phytochemistry. 1997;44:1163–1168. [Google Scholar]
- 31.Siddiqui BS, Arfeen S, Afshan F, Begum S. Heterocycles. 2005;65:857–863. [Google Scholar]
- 32.Misico R, Veleiro AS, Burton G, Oberti JC. Phytochemistry. 1997;45:1045–1048. [Google Scholar]
- 33.Shingu K, Furusawa Y, Nohara T. Chem Pharm Bull. 1989;37:2132–2135. [Google Scholar]
- 34.Zhu XH, Ando J, Takagi M, Ikeda T, Nohara T. Chem Pharm Bull. 2001;49:161–164. doi: 10.1248/cpb.49.161. [DOI] [PubMed] [Google Scholar]
- 35.Misra L, Lal P, Sangwan RS, Sangwan NS, Uniyal GC, Tuli R. Phytochemistry. 2005;66:2702–2707. doi: 10.1016/j.phytochem.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Llanos GG, Araujo LM, Jiménez IA, Moujir LM, Vázquez JT, Bazzocchi IL. Steroids. 2010;75:974–8. doi: 10.1016/j.steroids.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Machin RP, Veleiro AS, Nicotra VE, Oberti JC, Padròn JM. J Nat Prod. 2010;73:966–968. doi: 10.1021/np9006734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.