

Summary
The Ca2+release-activated Ca2+ (CRAC) channel is a highly Ca2+-selective store-operated channel expressed in T cells, mast cells, and various other tissues. CRAC channels regulate critical cellular processes such as gene expression, motility, and the secretion of inflammatory mediators. The identification of Orai1, a key subunit of the CRAC channel pore, and STIM1, the endoplasmic reticulum (ER) Ca2+ sensor, have provided the tools to illuminate the mechanisms of regulation and the pore properties of CRAC channels. Recent evidence indicates that the activation of CRAC channels by store depletion involves a coordinated series of steps, which include the redistributions of STIM1 and Orai1, direct physical interactions between these proteins, and conformational changes in Orai1, culminating in channel activation. Additional studies have revealed that the high Ca2+ selectivity of CRAC channels arises from the presence of an intrapore Ca2+ binding site, the properties of which are finely honed to occlude the permeation of the much more prevalent Na+. Structure-function studies have led to the identification of the potential pore-binding sites for Ca2+, providing a firm framework for understanding the mechanisms of selectivity and gating of the CRAC channel. This review summarizes recent progress in understanding the mechanisms of CRAC channel activation, pore properties, and modulation.
Keywords: CRAC channel, SOC, Orai1, STIM1, Ca2+ signaling
Introduction
In virtually all animal cells, stimulation of cell surface receptors coupled to the generation of 1,4,5-inositol trisphosphate (IP3) causes the release of Ca2+ from endoplasmic reticulum (ER)-derived Ca2+ stores, followed by Ca2+ influx across the plasma membrane (1). This ubiquitous Ca2+ entry pathway is named store-operated Ca2+ entry to reflect the view that it is regulated by the level of free Ca2+ in the lumen of the ER (2). The store-operated channels (SOCs) of T lymphocytes and mast cells were the first to be studied using patch-clamp techniques and have been extensively characterized in terms of their biophysical properties and downstream functions. This category of SOCs, known as Ca2+ release-activated Ca2+ (CRAC) channels, is distinguished by an extremely high selectivity for Ca2+ and low conductance (2). Opening of CRAC channels enables the refilling of ER Ca2+ stores and sustains long-lasting Ca2+ oscillations and plateau signals, critical for the proliferation of T cells and production of cytokines following antigenic stimulation (3). For nearly two decades following the discovery of CRAC channels, their molecular composition and the mechanisms linking store depletion to channel activation remained obscure. However, recent studies using genetic approaches have identified a gene family that encodes CRAC channels as well as the sensor for ER Ca2+ depletion (4), propelling rapid progress in our understanding of the molecular physiology and modes of regulation of these channels. Here, we review progress in understanding the regulation and molecular physiological properties of CRAC channels.
Historical overview
The earliest descriptions of Ca2+ currents that came to be later identified with CRAC channels were reported two decades ago in mast cells and T cells. Using a combination of patch-clamp and Ca2+ imaging techniques, Penner and coworkers (5, 6) described a small (1–2 pA), low-noise current that was activated by agonists such as substance P and intracellular dialysis with IP3 in rat peritoneal mast cells. This small current developed in parallel with a rise in [Ca2+]i and led the authors to suggest that it is responsible for the rise in [Ca2+]i by agonists and IP3. Likewise, Lewis and Cahalan (7) described a small, highly selective Ca2+-current that developed slowly during whole-cell recordings in lectin-stimulated T cells with low Ca2+-buffered internal pipette solutions. They described several unique aspects of the Ca2+ current, including a lack of voltage-dependent gating, an inwardly rectifying current–voltage relationship, and blockade by Ni2+ and Cd2+. This study (7) also showed a tight temporal correlation between rises in [Ca2+]i and the Ca2+ conductance, supporting a causal role for this conductance in generating [Ca2+]i oscillations in mitogen-activated T cells. Interestingly, at this juncture, these studies did not identify the proximal cause of the activation of the Ca2+ current.
The identification that ER Ca2+ store depletion activates SOCs arose, in large part following the discovery of thapsigargin, a plant-derived lactone that inhibits the SERCA [sarcoendoplasmic reticulum calcium adenosine triphosphatase (ATPase)] ATPase in the ER without concomitantly generating IP3 (8). With this tool, Putney and coworkers (9) showed in rat parotid cells that Ca2+ influx across the plasma membrane (i.e., store-operated Ca2+ entry) was triggered following the depletion of an intracellular Ca2+ pool. They also showed that IP3 and thapsigargin activated the same Ca2+ influx process (9). Likewise, several groups used fluorescence measurements of [Ca2+]i to demonstrate that ER Ca2+ store depletion by thapsigargin triggers Ca2+ influx across the plasma membrane in various cell types, including T cells and mast cells (10–15). This was followed by the well-known study of Hoth and Penner (16, 17) that described a Ca2+ current activated by various stimuli that depleted Ca2+ stores in mast cells, which was presumably the same current that they had previously studied (5, 6). These authors termed the Ca2+ current ICRAC for Ca2+ release-activated Ca2+ current. Using thapsigargin, Zweifach and Lewis (18) similarly showed that the Ca2+ current identified in the earlier study of Lewis and Cahalan (7) was activated by ER Ca2+ store depletion. Significantly, they showed that same current is also induced by stimulation of endogenous T-cell antigen receptors (TCRs), suggesting an important role for CRAC channels in mediating Ca2+ influx in response to physiological stimulation of T cells (18). In the ensuing decade, extensive investigations of the functions and biophysical characteristics of ICRAC firmly established CRAC channels as a model for Ca2+-selective SOCs.
The CRAC channel is essential for gene expression in activated T cells
In the years following the discovery of CRAC channels, substantial evidence accumulated showing that Ca2+ influx through these channels is essential for activation of Ca2+-dependent gene transcription in T cells. SKF96365, an imidazole compound that inhibits CRAC channels, was also found to block interleukin-2 (IL-2) production in T cells with similar efficacy (19). Likewise, nanomolar concentrations of La3+ were found to block ICRAC, the [Ca2+]i rise, and the induction of T-cell activation markers such as CD25 and CD69 in response to CD3 or thapsigargin stimulation (20). However, in the absence of selective pharmacological inhibitors, genetic studies have provided the most compelling evidence for the involvement of CRAC channels in T-cell activation. In mutant Jurkat T cells that exhibit <10% of the normal ICRAC, T-cell activation as measured by the production of cytokines such as IL-2 was found to be severely attenuated (21). Additionally, several studies of human patients have shown that severe immunodeficiencies arise from mutations in CRAC channels that render them inactive (22–26). The abrogation of CRAC channel function in these cells results in the elimination of Ca2+ elevations necessary to drive nuclear translocation of nuclear factor of activated T cells (NFAT) (22), an important and widely expressed transcription factor involved in cytokine gene expression (27). Collectively, these studies established the critical role of CRAC channels for antigen-mediated T-cell activation and generated strong interest in understanding the regulation and identity of the CRAC channel at a molecular level.
The molecular components of the CRAC channel pathway
The decade following the discovery of ICRAC saw an intense quest to determine its molecular composition and identify the signal linking store depletion to channel activation. Mammalian homologs of the Drosophila TRP channel were proposed as the likely pore-forming subunits of CRAC channels, and several competing hypotheses were proposed for the nature of the activation signal (28). These hypotheses included a diffusible activator released from the ER following store depletion, the insertion of active channels into the plasma membrane through a vesicle fusion mechanism, and direct physical coupling of CRAC channels with proteins in the ER membrane (28). Many of these models were highly controversial, and genuine progress in resolving these issues was stalled for years due to a lack of clearly defined proteins in the pathway. This era of uncertainty ended recently with the successful application of forward and reverse genetic approaches. In 2005, STIM1 (stromal interaction molecule 1) was identified as the mammalian ER Ca2+ sensor, followed closely in 2006 by the identification of Orai1 (also known as CRACM1) as a key subunit of the CRAC channel pore (23, 29–34). The discovery of these proteins has propelled rapid progress in illuminating the molecular mechanisms of this pathway and its functions in various tissues.
STIM1 is a 77 kDa single pass transmembrane protein with several functional domains including a luminal EF-hand motif that senses the ER Ca2+ concentration and multiple protein–protein interaction motifs (4). Knockdown of STIM1 in mammalian cells or in Drosophila S2 cells results in suppressed ICRAC (29, 30). STIM1 is expressed in nearly all mammalian tissues and is conserved from Drosophila to humans, consistent with the widespread prevalence of SOCs in mammalian tissues and other organisms. Homozygous nonsense mutations in the STIM1 gene produce symptoms of severe immunodeficiency, autoimmune disease, and myopathy in human patients (35). These symptoms are remarkably similar to the indications seen in patients with Orai1 mutations (23), underscoring the critical role of CRAC channels in immune function and host defense. STIM1 is distributed throughout the ER network in resting cells with replete ER Ca2+ stores. Remarkably, ER Ca2+ store depletion induces a rapid redistribution of STIM1 from the bulk ER into discrete puncta that accumulate near the plasma membrane (30, 36, 37). EF-hand mutants of STIM1 with impaired Ca2+ binding do not display the wildtype distribution in the ER but instead form puncta near the plasma membrane, mimicking the store-depleted state (30, 36). As if tricked into reporting the emptying of Ca2+ stores, cells expressing these mutants exhibit constitutive opening of CRAC channels without store depletion (30, 36). Moreover, careful measurements of [Ca2+]ER, STIM1 redistribution, and ICRAC activation show that the [Ca2+]ER dependence of ICRAC activation is identical to the [Ca2+]ER dependence of STIM1 translocation to the plasma membrane, indicating that CRAC channels derive their dependence on store depletion from the ER Ca2+ dependence of STIM1 redistribution (38). Collectively, these properties firmly established STIM1’s role as the Ca2+ sensor involved in coupling ER Ca2+ store depletion to CRAC channel activation.
Although STIM1 is essential for CRAC channel function, with one report even speculating on a possible role for STIM1 in forming Ca2+ channels in the plasma membrane (36), it is clearly not sufficient to reconstitute all aspects of store-operated Ca2+ entry. An early investigation of the possible molecular basis of defective CRAC channel function in human patients with the SCID syndrome failed to reveal any defects in STIM1 expression or its function (26). Moreover, overexpression of STIM1 in the mutant cells from the patient failed to rescue the defect, pointing to another component that is responsible for the inactivity of CRAC channels (26). Feske et al. (23) subsequently employed linkage analysis and positional cloning to identify a region in chromosome 12 containing approximately 74 genes as the locus of the defect. Simultaneously, a genome-wide RNA interference (RNAi) screen in Drosophila S2 cells led to the identification of a previously uncharacterized molecule that had a human homologue within the 74-gene region on chromosome 12 (23). Two other screens of the same RNAi library for suppression of SOC function identified the same gene in S2 cells (34, 39). This molecule, termed Orai1, is a widely expressed 33 kDa cell surface protein with four predicted transmembrane domains, intracellular N- and C-termini (Fig. 1), and no significant sequence homology to other previously identified ion channels. The human SCID defect was found to arise from a single point mutation in Orai1 (R91W) that abrogated CRAC channel activity (23).
Fig. 1. Predicted topology of Orai1.
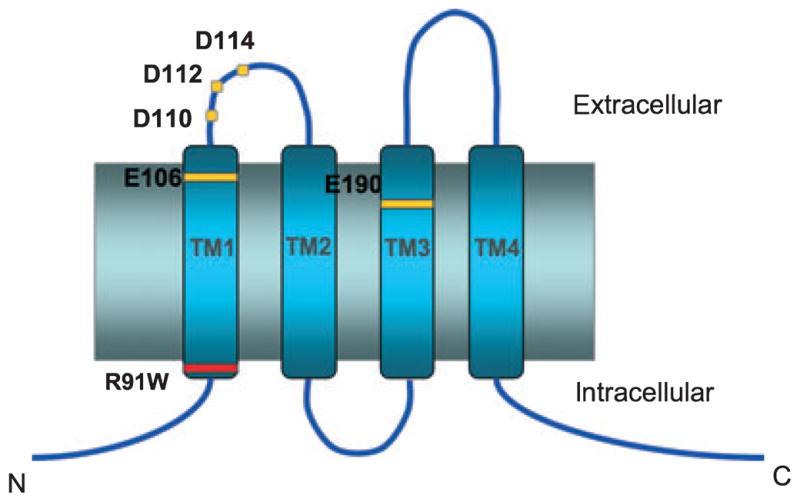
Critical amino acids residues identified from structure-function and human linkage-analysis studies are highlighted. Mutations of E106 in TM1 and E190 in TM3 (yellow) affect ion selectivity and permeation, whereas mutations of the aspartate residues in the TM1-TM2 loop segment (D110/112/114, yellow) affect La3+ block while producing relatively small changes in ion permeation. An inherited mutation (R91W, red) produces non-functional CRAC channels and immunodeficiency in human patients.
Two important lines of evidence indicated that Orai1 is an essential pore subunit of the CRAC channel. First, overexpression of Orai1 together with STIM1 in HEK293 cells produced enormous CRAC currents similar to native ICRAC in its biophysical and pharmacological characteristics (40–43). These characteristics included high Ca2+ selectivity and low permeability to Cs+, a narrow pore diameter of approximately 3.8 Å, kinetic properties of Ca2+ block of Na+ currents, and pharmacological responses to 2-APB and La3+ (40, 41, 43). More definitive evidence came from the finding that mutations of highly conserved acidic residues in Orai1 significantly alter the ion selectivity of the CRAC channels (31–33). Together these studies definitively identified Orai1 as a key component of the CRAC channel pore and provided the first glimpse into the molecular mechanisms of ion selectivity in the CRAC channel. Mammalian cells express two other closely related homologues, Orai2 and Orai3. All three isoforms appear to function similarly in producing store-operated Ca2+ entry when co-expressed with STIM1 in HEK293 cells (44–46) and are widely expressed in most tissues (44, 46, 47).
Biophysical characteristics of the CRAC channel pore
Ion selectivity and permeation
A defining feature of the CRAC channel is its extremely high selectivity for Ca2+ over monovalent ions. Using the fura-2 overload method to estimate the fractional Ca2+ current (48, 49), Hoth and Penner estimated that CRAC channels conduct Ca2+ > 1000 times better than Na+ under physiological conditions, placing CRAC channels among the most Ca2+ selective channels known (17). Biophysical studies indicated that the pore of the CRAC channel does not intrinsically exclude monovalent ions; instead, the high Ca2+ selectivity seen under physiological conditions arises via ion-pore and ion–ion interactions. This is readily seen in the absence of extracellular divalents: CRAC channels readily conduct a variety of small monovalent ions, such as Na+, Li+, and K+ in divalent-free solutions, but the addition of micromolar concentrations of Ca2+ blocks the permeation of the more prevalent monovalent ions by high-affinity Ca2+ binding in the channel (Ki approximately 20 μM at −100 mV) (17, 50–54). Occupancy by a single Ca2+ ion appears sufficient to block the large monovalent conductance, and, as expected for a binding site within the pore, Ca2+ block is voltage-dependent (43, 52). These characteristics are qualitatively reminiscent of the properties of voltage-gated (Cav) L-type channels, which have historically served as prototypes for studies of Ca2+ selectivity (55). In these highly Ca2+-selective channels, Ca2+ ions bind tightly to a high-affinity binding site within the selectivity filter, a narrow region of the pore that controls ion selectivity, to occlude Na+ flux. However, at millimolar concentrations of extracellular Ca2+, the Ca2+ binding site is thought to be sufficiently flexible to enable interactions with multiple Ca2+ ions, permitting rapid Ca2+ flux (55–57).
The identification of Orai1 provided the impetus and tools to investigate the molecular underpinnings of the CRAC channel’s high Ca2+ selectivity. Guided by past studies on L-type Cav channels, wherein four glutamate residues, one in each of the four pore-loops converge to form a single high-affinity Ca2+ binding site (the EEEE locus) (57, 58), it was reasoned that the Ca2+ binding site(s) in the CRAC channel pore may similarly arise from the carboxylate side-chains of pore-lining acidic residues. Indeed, sequence comparisons of various Orai proteins revealed the presence of several highly conserved acidic residues in the first and third predicted transmembrane (TM) segments and in the loop connecting TM1 and TM2 (Fig. 1), setting the stage for mutagenesis studies. A Glu → Asp substitution at position 106 in the predicted TM1 segment of human Orai1 strongly diminished the high Ca2+ selectivity of CRAC channels (31–33) and altered the affinity and voltage-dependence of Ca2+ block of the monovalent current (43). A Glu → Gln mutation at position 190 in the TM3 segment similarly compromised the Ca2+ selectivity of mutant channels (31, 33, 43). In addition, mutations of three Asp residues D110A/D112A/D114A in the predicted TM1–TM2 loop segment strongly diminished La3+ block of CRAC channels, while also causing modest changes in ion selectivity (32, 33, 43). More recently, several groups have reported that CRAC channels are formed from the homomeric assembly of four Orai1 subunits (59–61). Collectively, these studies provided the foundation to understand the molecular determinants of ion selectivity and permeation in CRAC channels. Current models of the tertiary structure of the channel posit that residues in the TM1 and TM3 segments line the ion conduction pathway, with the acidic residues identified from the mutations, in particular E106 and E190, constituting key components of the CRAC channel selectivity filter (33, 62, 63).
To understand additional features of the pore that govern ion selectivity, measurements of permeability ratios for a series of ions that differ in size, charge, and chemical properties such as hydration energy are often employed in studies of ion channels. For CRAC channels, however, obtaining information on the selectivity of various ions has been difficult, because the removal of Ca2+ causes CRAC channels to lose activity by a process referred to as depotentiation (51, 64). Thus, measurements of the divalent ion selectivity from steady-state currents have led to the mistaken notion that the order of divalent ion conductance is Ca2 + > Sr2 + > Ba2+ (16, 18). However, more recent measurements of peak currents immediately following solution exchange indicate that the conductance series is, in fact, the reverse: Ba2 + > Sr2 + > Ca2+ (53, Prakriya and Lewis, unpublished data). This series is identical to the selectivity series seen in Cav channels and suggests that despite the strikingly different molecular compositions of CRAC and Cav channels, the core mechanisms of ion permeation and selectivity are similar in the two channel types.
The Orai1 mutagenesis studies also provided insight into a puzzling hallmark of CRAC channels: their unusually low permeability to Cs+. CRAC channels are practically impermeable to Cs+ (PCs/PNa approximately 0.1) (50–52, 54), which stands in sharp contrast to L-type Cav channels that readily pass Cs+ under divalent-free conditions (PCs/PNa approximately 0.6) (65) as well as other Ca2+-selective channels such as TRPV6 channels (PCs/PNa approximately 0.6) (66). One clue to explain this difference has come from estimates of the minimal pore diameter of these channels. By examining the channel permeability for a series of monovalent organic cations of varying sizes, the diameter of Cav channels has been estimated to approximately 6 Å (67, 68) and that of the TRPV6 channels to be approximately 5.5 Å (69). By comparison, the narrowest region of the CRAC channel pore is only approximately 3.8 Å (43, 52). Given that the diameter of a naked Cs+ ion is approximately 3.6 Å, the finding that CRAC channels have a narrow pore suggests that the low permeability to Cs+ reflects steric hindrance to its permeation. In agreement with this hypothesis, mutations of key acidic residues such as E106 and E190 in Orai1 significantly widen the pore and concomitantly increase the Cs+ permeability of mutant CRAC channels (43). Intriguingly, decreases in Ca2+ selectivity caused by these mutations correlate well with increases in Cs+ permeability, suggesting that pore geometry has a powerful influence on both Ca2+ and Cs+ selectivity (43).
Altogether, the mutagenesis studies provided a first glimpse into the structural elements controlling Ca2+ selectivity, although this work has also contributed to its share of outstanding questions. For example, it is clear that the acidic residues identified in these studies do not by themselves account for all aspects of permeation in CRAC channels, such as its low conductance (see below). What other parts of the channel are involved, and how do they interact with the conducting ions? Additionally, why are mutations of E106 are so poorly tolerated for channel function? While a conservative substitution of E106 to aspartate, as described above, is tolerated and produces channels with diminished Ca2+ selectivity, other substitutions including alanine, glutamine, or cysteine produce non-functional CRAC channels (31, McNally and Prakriya, unpublished data). This is in sharp contrast to the quadruple alanine or glutamine mutations of the EEEE locus in Cav channels, which exhibit robust monovalent permeation (57). Exactly how these mutations exert their profound effects on ion permeation remains unknown. An additional area of uncertainty is whether the residues implicated in these studies truly line the pore (as hypothesized) or whether the mutations alter ion selectivity because of indirect, allosteric effects. This concern may be especially valid for E190: although a glutamine substitution of this residue alters the ion selectivity of mutant channels, substitutions to alanine or aspartate have no detectable effect (31). These specific questions more broadly reflect the scarcity of information on the pore architecture, including the identity, number, and locations of the ion-binding sites. Ultimately, progress in understanding these issues will require elucidation of the structural characteristics of the pore.
Unitary conductance
A key distinctive feature of the CRAC channel is its extremely small apparent unitary conductance. Whole-cell ICRAC generates little visible current noise and the activity of single CRAC channels is yet to be directly measured. Estimates from noise measurements of whole-cell ICRAC in T cells indicate a unitary conductance of 9–24 fS in 2–110 mM Ca2+ (18, 51). Measurements in mast cells similarly resulted in a conductance estimate of < 1 pS based on the low level of visible current noise in whole-cell recordings of ICRAC (17). As expected from the increased macroscopic Na+ currents in divalent-free solutions, the monovalent conductance of Na+-conducting CRAC channels is estimated to be at least an order of magnitude larger, or approximately 0.7 pS (52). The unitary Na+ current amplitude (0.11 pA at −110 mV) corresponds to an ion flux rate of approximately 7 × 105 Na+/s, much faster than most ion carriers but compatible with a channel-like transport mechanism (52).
By comparison, measurements of the single channel conductance of Cav L-type channels yield values of 4–10 pS for Ca2+ and 85 pS for Na+ (65), indicating a > 100-fold larger rate of ion flux. This difference is all the more striking in light of the many similarities in the core mechanisms of Ca2+ selectivity and permeation profile of the two channel types noted above. Interestingly, we find that the association rate of Ca2+ for its pore blocking site is approximately 4 × 106 M−1 s−1 in the CRAC channel (43, 52). By contrast, the corresponding rate determined for L-type Cav channels from single channel recordings is approximately 4 × 108 M−1 s−1 (56), a value close to the diffusion limit. The sharply lower association rate of Ca2+ for its blocking site in CRAC channels indicates that Ca2+ access from the extracellular space to the selectivity filter is severely limited, a factor that could explain the small conductance of CRAC channels. Although we do not yet know the reasons for this difference, the narrower pore of the CRAC channel noted above may be an underlying factor, especially if it presents a rate-limiting barrier for the flux of ions through the pore.
The small conductance and high Ca2+ selectivity of CRAC channels have important implications for their function. Given that the kinetics of activation and deactivation of CRAC channels occurs on a slow time scale of seconds to tens of seconds, the high efficiency of Ca2+ entry with little or no accompanying Na+ influx prevents membrane depolarization that could compromise the driving force for Ca2+ entry over such long durations. At the same time, the small conductance of CRAC channels ensures strong specificity for the activation of Ca2+-dependent downstream functions. A striking example of this specificity is seen in the gene expression of c-fos by Ca2+ oscillations triggered by stimulation of mast cells with the leukotriene LTC4 (70). c-fos gene expression was observed only following Ca2+ influx through CRAC channels, even when Ca2+ oscillations of the same amplitude could be artificially maintained in the absence of CRAC channel-mediated Ca2+ entry (70). Likewise, activation of Ca2+-dependent enzymes and the production of leukotrienes in mast cells are selectively coupled to Ca2+ influx through CRAC channels (71). These studies directly illustrate a key point: that the role of CRAC channels for cellular Ca2+ signaling extends significantly beyond the mere maintenance of ER Ca2+ store homeostasis.
Rectification
Another prominent hallmark of ICRAC noted in nearly all electrophysiological studies of CRAC channels is the steep inward rectification of the whole-cell ICRAC. The rectification is not simply because most patch-clamp studies of CRAC channels employ Cs+, which is largely impermeable through CRAC channels, as the main charge carrier in internal solutions. The sharp inward rectification is also observed under a range of conditions with various permeant ions in internal solutions, including in symmetrical Na+ ions (52, 54). Currently, little is known of the mechanisms underlying this feature and the inability to resolve single CRAC channel currents has severely hampered efforts to determine whether it arises because of block, intrinsic channel gating, or inherent asymmetry in the ion conduction pore. Rectification is unaffected by intracellular Mg2+ or polyamines suggesting that internal blockade by these charged compounds is not the cause (51, 54, 72, 73). Inward rectification of ICRAC is also seen in Orai1 mutants with altered Ca2+ and Cs+ selectivity under divalent-free conditions (43), indicating that the molecular determinants of this feature are unrelated to the elements involved in ion selectivity. Interestingly, rectification is strongly reduced when ammonium derivatives such as hydroxyl ammonium or methyl ammonium are employed as charge carriers (52), suggesting that ion-pore interactions may contribute to this feature. The molecular underpinnings of inward rectification and its consequences for cellular function remain obscure.
Molecular mechanisms of channel gating
Activation by store depletion
The identification of STIM1 and Orai1 provided the molecular tools to illuminate the nature of the signal linking ER Ca2+ store depletion to channel activation. As noted above, STIM1 senses the depletion of ER Ca2+ through its luminal EF-hand domain. STIM1 is distributed diffusely throughout the ER membrane in resting cells. ER Ca2+ store depletion triggers a coordinated series of events beginning with STIM1 oligomerization followed by its redistribution into discrete puncta at the junctional ER sites where the ER tubules and the plasma membrane are within 20 nm of each other (30, 36, 38, 74). The puncta of over-expressed STIM1 accumulate at the sites of Ca2+ influx through active CRAC channels (75). Orai1, which is also diffusely distributed in the plasma membrane at rest, aggregates and colocalizes with STIM1 at the same puncta following store depletion (75–79). Collectively, these studies revealed a fascinating complexity of signaling in the activation of CRAC channels involving coclustering of STIM1 in the junctional ER directly opposite to Orai1 in the plasma membrane.
The finding that STIM1 and Orai1 accumulate in overlapping clusters in closely apposed membranes raised a key molecular question central to resolving the historical debate on the nature of the CRAC channel activation mechanism: does STIM1 directly interact with Orai1? Support for a direct physical interaction between STIM1 and Orai1 was provided by findings showing that overexpressed STIM1 and Orai1 co-immunoprecipitate with each other (32, 33) and that the redistribution and formation of overlapping STIM1–Orai1 puncta occurs in parallel with increases in fluorescence resonance energy transfer (FRET) between these proteins (78, 79). An amphipathic domain in the C-terminus of Orai1 was identified as the likely STIM1 interaction site (76), and two hydrophobic Orai1 residues, L273 (78) and L276 (79), were determined to be critical for this interaction. More recently, evidence supporting a direct physical interaction of STIM1 and Orai1 has come from structure-function studies that have discovered a minimal domain of STIM1 that can activate CRAC channels independently of ER store depletion. Several groups have identified an approximately 107 amino acid region in a coiled-coiled domain of the C-terminus of STIM1 that activates CRAC channels independently of ER Ca2+ store depletion when over-expressed in HEK293 cells (80–83). Park et al. (80) tested whether this minimal activation region of STIM1 directly binds Orai1 using GST-pulldown assays and size-exclusion chromatography. Their results indicated that the GST-tagged STIM1 domain coprecipitates Orai1 and co-elutes with Orai1, indicating that this region of STIM1 directly binds to Orai1 in vitro.
These results indicate that the cytoplasmic portion of STIM1 activates CRAC channels by direct binding to Orai1 and argue against the hypothesis that a diffusible messenger released by ER Ca2+ store depletion activates CRAC channels. The ability of the entire C-terminus of STIM1 to activate CRAC channels was found to be significantly lower than that of the 107-amino acid peptide region in several studies, suggesting that this region of STIM1 is normally hidden and that a conformational change, possibly caused by Ca2+ unbinding at the luminal EF-hand domain, is needed to expose the activation domain of STIM1 (80, 82, 83). Thus, CRAC channel activation occurs via a conformational coupling mechanism involving direct physical associations between the channel and its sensor in closely apposed membranes.
Although these studies have established the framework for understanding the signal linking ER Ca2+ store depletion to the opening of CRAC channels, how STIM1 binding subsequently causes the gating conformational changes that culminate in the opening of the pore remains unclear. Elucidating the nature and identity of these gating movements is critical for understanding the many modes of regulation of CRAC channels: by ER Ca2+ stores, second messengers, extracellular Ca2+, and pharmacological agents. To address this issue, we have identified a conformational rearrangement in Orai1 triggered by store depletion by monitoring FRET between cyan fluorescence protein (CFP) and yellow fluorescence protein (YFP) probes attached to the C- or N-termini of Orai1 (79). ER Ca2+ store depletion causes a small but significant decline in FRET between the probes of adjacent Orai1 subunits, suggesting that STIM1 binding to Orai1 leads to conformational changes in the intracellular C- and N-termini of Orai1 (79). These rearrangements may be coupled to movements of the gate and provide the energy required for channel opening.
Other modes of CRAC channel regulation
In addition to ER Ca2+ store depletion and refilling, the CRAC channel exhibits various other modes of channel regulation. These include (i) Ca2+-dependent potentiation (CDP) of CRAC channels in store-depleted cells after the readdition of extracellular Ca2+ and the reverse process of depotentiation after removal of divalent cations, (ii) fast inactivation, arising from feedback inhibition of ICRAC during brief hyperpolarizing steps by the high local [Ca2+] around individual CRAC channels, and (iii) enhancement of channel activity by low doses of 2-APB (<5 μM) and inhibition by higher doses (>10 μM). These forms of regulation and our understanding of their underlying mechanisms are discussed below.
CDP
CDP was first described by Zweifach and Lewis (64, 84) and is indicative of a process wherein the activity of CRAC channels is strongly facilitated by extracellular Ca2+. Conversely, the removal of extracellular Ca2+ causes the CRAC channel activity to drop, a process referred to as depotentiation (51). The mechanism of CDP, including the location of the Ca2+-binding site, remains unknown. Our recent studies indicate that CDP is directly dependent on the degree of pore occupancy by divalent ions, and ions with higher permeability support a greater degree of potentiation (Prakriya and Lewis, unpublished data). These results are compatible with the idea that the CDP binding site is located within the CRAC channel pore. However, how Ca2+ binding is coupled to changes in channel activity remains unclear. Noise analysis suggests that CDP occurs from the recruitment of CRAC channels from a ‘silent’ state to one of high open probability (52). A similar stepwise recruitment of silent channels to a high open-probability mode also occurs during the slow activation of CRAC channels by store depletion (52), raising the possibility that the conformational changes underlying these events are identical. In support of this idea, examination of Orai1 conformational changes by FRET reveal that readdition of extracellular Ca2+ to store depleted cells causes a qualitatively similar change in Orai1–Orai1 FRET as ER Ca2+ store depletion (79). Thus, there may be common gating steps in the process by which store depletion and CDP increase channel activity.
Fast inactivation
Fast inactivation is a prominent hallmark of CRAC channels and is observed as a decline in ICRAC over 200–500 ms during hyperpolarizing voltage steps. Fast inactivation is thought to be mediated by feedback inhibition of channel activity by the high local [Ca2+]i around individual CRAC channels (17, 85, 86). Key aspects of the fast inactivation mechanism such as the identities of the Ca2+ binding sites, the nature of the inactivation gate, and whether inactivation occurs by a change in open probability or unitary Ca2+ conductance remain unknown. Past experiments have shown that the Ca2+ binding to the inactivation site is sensitive to chelation of intracellular Ca2+ by fast buffers such as BAPTA, suggesting that the Ca2+ binding site is probably located on the intracellular face of the channel (17, 85, 86), but whether the binding site resides directly on Orai1 or on another protein is unclear. Interestingly, mutations in the putative selectivity filter of Orai1 that alter the ion selectivity of CRAC channels also strongly diminish fast inactivation, suggesting that the structural elements regulating ion selectivity and inactivation gating may be coupled (43). The nature of this coupling will require elucidation of critical structural elements of the pore such the pore-lining residues and the location of the channel gate.
Modulation by 2-APB
Of the various modes of CRAC channel regulation, the modulation of channel activity by the pharmacological agent 2-aminoethyldiphenyl borate (2-APB) has perhaps been the most puzzling and garnered widest attention. 2-APB elicits complex effects including a two-to fivefold persistent enhancement of ICRAC at low concentrations (1–5 μM) and transient enhancement followed by strong inhibition of ICRAC at higher concentrations (>10 μM) (87). The multiplicity of 2-APB effects has long intrigued SOC investigators, and many explanations have been invoked over the years for its likely mode of action. Some early studies concluded that the inhibition of SOC activity by 2-APB occurs through the drug’s known ability to inhibit IP3 receptors, and this effect was cited in strong support for a role for IP3 receptors in transducing the activation of SOCs (88, 89). However, subsequent studies indicated that 2-APB inhibits CRAC channels directly and even in cells lacking IP3 receptors, casting doubts about this interpretation (87, 90, 91). More recently, studies in HEK293 cells overexpressing STIM1 found that 2-APB (50–100 μM) reverses STIM1 puncta in store-depleted cells (92, 93). However, this effect is largely eliminated when Orai1 is co-expressed together with STIM1, suggesting that disruption of the STIM1–Orai1 aggregates is not the underlying mechanism of CRAC channel inhibition by 2-APB (79, 93).
Noise analysis suggests that the potentiation of ICRAC seen at low concentrations of 2-APB arises from the recruitment of CRAC channels from a silent state to one of high open probability (52). As noted earlier, a similar stepwise recruitment of silent channels to a high open-probability mode also occurs during the slow activation of CRAC channels by store depletion (52). Thus, it is tempting to speculate that the conformational changes in CRAC channels occurring during 2-APB-induced potentiation of ICRAC and store depletion-induced channel activation may be identical. In support of this idea, two studies employing FRET have found that 2-APB facilitates the association between STIM1 and Orai1 (79, 94). Thus, 2-APB-induced potentiation of ICRAC and the opening of CRAC channels by store depletion may share common mechanisms, both involving increased binding of STIM1 to Orai1 culminating in channel activation. By contrast, the inhibition of ICRAC by high concentrations of 2-APB does not appear to involve obvious decreases in STIM1–Orai1 interaction. In fact, by monitoring conformational changes in CRAC channels with Orai1–Orai1 FRET, a recent study has found that high (but not low) concentrations of 2-APB produce conformational changes in Orai1 (79). Based on these findings, one model of 2-APB action proposes that the drug induced potentiation of ICRAC arises from strengthening of STIM1–Orai1 contacts, whereas the inhibition, which is specific to high drug concentrations, arises from alterations in CRAC channel gating (79). A direct effect of high concentrations of 2-APB on CRAC channel gating is also supported by electrophysiological measurements of ICRAC revealing that 2-APB-induced current inhibition is accompanied by the loss of fast Ca2+-dependent inactivation, a gating process probably mediated by conformational changes in the channel (87).
Admittedly, these descriptions of the molecular mechanisms of channel modulation are still rudimentary and many puzzling questions remain. However, the studies of CDP and 2-APB described above project a dynamic picture of channel regulation wherein the interaction of STIM1 with Orai1 and the downstream conformational changes in Orai1 can be independently modulated to tune CRAC channel activity. These checkpoints might also be targets for modulation in vivo by second messengers and may be attractive targets for designing drugs to alter CRAC channel activity in diseased cells.
Future perspectives
The past few years have seen a remarkable transformation in our understanding of the molecular basis of store-operated Ca2+ entry. Two key molecular components of the CRAC channel pathway have been identified and the nature of the signal linking store depletion to channel activation has been elucidated. A large body of knowledge also exists about the fundamental biophysical properties of CRAC channels, providing a firm foundation to explore the molecular mechanisms of Ca2+ selectivity and channel gating. Thus, a major goal of future efforts will be the illumination of the molecular and structural basis of the intriguing properties of the CRAC channel. What is the structure of the CRAC channel pore? Where is the channel gate? How is STIM1 binding to Orai1 coupled to the opening of the pore? These questions will require a combination of approaches, from X-ray crystallography to solve the channel structure to traditional molecular and electrophysiological approaches to determine the functions of particular protein domains. There is great anticipation that the inner workings of this important and enigmatic channel will finally be understood.
Acknowledgments
Work in the author’s laboratory was supported by grants from the NIH and AHA. The author is grateful to the members of the laboratory for helpful discussions and Dr. T. Hornell for a critical reading of the manuscript.
References
- 1.Berridge MJ. Capacitative calcium entry. Biochem J. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 3.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 4.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- 5.Penner R, Matthews G, Neher E. Regulation of calcium influx by second messengers in rat mast cells. Nature. 1988;334:499–504. doi: 10.1038/334499a0. [DOI] [PubMed] [Google Scholar]
- 6.Matthews G, Neher E, Penner R. Second messenger-activated calcium influx in rat peritoneal mast cells. J Physiol. 1989;418:105–130. doi: 10.1113/jphysiol.1989.sp017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thastrup O, et al. Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions. 1989;27:17–23. doi: 10.1007/BF02222186. [DOI] [PubMed] [Google Scholar]
- 9.Takemura H, Hughes AR, Thastrup O, Putney JW., Jr Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. J Biol Chem. 1989;264:12266–12271. [PubMed] [Google Scholar]
- 10.Mason MJ, Garcia-Rodriguez C, Grinstein S. Coupling between intracellular Ca2+ stores and the Ca2+ permeability of the plasma membrane. Comparison of the effects of thapsigargin, 2,5-di-(tert-butyl)-1,4-hydroquinone, and cyclopiazonic acid in rat thymic lymphocytes. J Biol Chem. 1991;266:20856–20862. [PubMed] [Google Scholar]
- 11.Sarkadi B, Tordai A, Homolya L, Scharff O, Gardos G. Calcium influx and intracellular calcium release in anti-CD3 antibody-stimulated and thapsigargin-treated human T lymphoblasts. J Membr Biol. 1991;123:9–21. doi: 10.1007/BF01993958. [DOI] [PubMed] [Google Scholar]
- 12.Cheek TR, Thastrup O. Internal Ca2+ mobilization and secretion in bovine adrenal chromaffin cells. Cell Calcium. 1989;10:213–221. doi: 10.1016/0143-4160(89)90004-3. [DOI] [PubMed] [Google Scholar]
- 13.Ohuchi K, Takahashi C, Hirasawa N, Watanabe M, Fujiki H, Tsurufuji S. Stimulation of histamine release and arachidonic acid metabolism in rat peritoneal mast cells by thapsigargin, a non-TPA-type tumor promoter. Biochim Biophys Acta. 1989;1003:9–14. doi: 10.1016/0005-2760(89)90091-x. [DOI] [PubMed] [Google Scholar]
- 14.Brayden DJ, Hanley MR, Thastrup O, Cuthbert AW. Thapsigargin, a new calcium-dependent epithelial anion secretagogue. Br J Pharmacol. 1989;98:809–816. doi: 10.1111/j.1476-5381.1989.tb14609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gouy H, Cefai D, Christensen SB, Debre P, Bismuth G. Ca2+ influx in human T lymphocytes is induced independently of inositol phosphate production by mobilization of intracellular Ca2+ stores. A study with the Ca2+ endoplasmic reticulum-ATPase inhibitor thapsigargin. Eur J Immunol. 1990;20:2269–2275. doi: 10.1002/eji.1830201016. [DOI] [PubMed] [Google Scholar]
- 16.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 17.Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung SC, McDonald TV, Gardner P. Inhibition by SK&F 96365 of Ca2+ current, IL-2 production and activation in T lymphocytes. Br J Pharmacol. 1994;113:861–868. doi: 10.1111/j.1476-5381.1994.tb17072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aussel C, Marhaba R, Pelassy C, Breittmayer JP. Submicromolar La3+ concentrations block the calcium release-activated channel, and impair CD69 and CD25 expression in CD3- or thapsigargin-activated Jurkat cells. Biochem J. 1996;313:909–913. doi: 10.1042/bj3130909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fanger CM, Hoth M, Crabtree GR, Lewis RS. Characterization of T cell mutants with defects in capacitative calcium entry: genetic evidence for the physiological roles of CRAC channels. J Cell Biol. 1995;131:655–667. doi: 10.1083/jcb.131.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol. 2001;2:316–324. doi: 10.1038/86318. [DOI] [PubMed] [Google Scholar]
- 23.Feske S, et al. Amutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 24.Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem. 1994;269:32327–32335. [PubMed] [Google Scholar]
- 25.Le Deist F, et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood. 1995;85:1053–1062. [PubMed] [Google Scholar]
- 26.Feske S, Prakriya M, Rao A, Lewis RS. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J Exp Med. 2005;202:651–662. doi: 10.1084/jem.20050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 28.Lewis RS. Store-operated calcium channels. Adv Second Messenger Phosphoprotein Res. 1999;33:279–307. doi: 10.1016/s1040-7952(99)80014-7. [DOI] [PubMed] [Google Scholar]
- 29.Roos J, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 32.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vig M, et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang SL, et al. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Picard C, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360:1971–1980. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang SL, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vig M, et al. CRACM1 is a plasma membrane protein essential for store-operated ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peinelt C, et al. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mercer JC, et al. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 43.Yamashita M, Navarro-Borelly L, McNally BA, Prakriya M. Orai1 mutations alter ion permeation and Ca2+-dependent inactivation of CRAC channels: evidence for coupling of permeation and gating. J Gen Physiol. 2007;130:525–540. doi: 10.1085/jgp.200709872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gwack Y, et al. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 45.Lis A, et al. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17:794–800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gross SA, Wissenbach U, Philipp SE, Freichel M, Cavalie A, Flockerzi V. Murine ORAI2 splice variants form functional Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem. 2007;282:19375–19384. doi: 10.1074/jbc.M701962200. [DOI] [PubMed] [Google Scholar]
- 47.Wissenbach U, Philipp SE, Gross SA, Cavalie A, Flockerzi V. Primary structure, chromosomal localization and expression in immune cells of the murine ORAI and STIM genes. Cell Calcium. 2007;42:439–446. doi: 10.1016/j.ceca.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 48.Schneggenburger R, Zhou Z, Konnerth A, Neher E. Fractional contribution of calcium to the cation current through glutamate receptor channels. Neuron. 1993;11:133–143. doi: 10.1016/0896-6273(93)90277-x. [DOI] [PubMed] [Google Scholar]
- 49.Neher E. The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- 50.Lepple-Wienhues A, Cahalan MD. Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophys J. 1996;71:787–794. doi: 10.1016/S0006-3495(96)79278-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prakriya M, Lewis RS. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J Gen Physiol. 2002;119:487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prakriya M, Lewis RS. Regulation of CRAC channel activity by recruitment of silent channels to a high open-probability gating mode. J Gen Physiol. 2006;128:373–386. doi: 10.1085/jgp.200609588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su Z, Shoemaker RL, Marchase RB, Blalock JE. Ca2+ modulation of Ca2+ release-activated Ca2+ channels is responsible for the inactivation of its monovalent cation current. Biophys J. 2004;86:805–814. doi: 10.1016/S0006-3495(04)74156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bakowski D, Parekh AB. Monovalent cation permeability and Ca2+ block of the store-operated Ca2+ current ICRAC in rat basophilic leukemia cells. Pflugers Arch. 2002;443:892–902. doi: 10.1007/s00424-001-0775-8. [DOI] [PubMed] [Google Scholar]
- 55.Sather WA, McCleskey EW. Permeation and selectivity in calcium channels. Annu Rev Physiol. 2003;65:133–159. doi: 10.1146/annurev.physiol.65.092101.142345. [DOI] [PubMed] [Google Scholar]
- 56.Lansman JB, Hess P, Tsien RW. Blockade of current through single calcium channels by Cd2+, Mg2+, and Ca2+ Voltage and concentration dependence of calcium entry into the pore. J Gen Physiol. 1986;88:321–347. doi: 10.1085/jgp.88.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ellinor PT, Yang J, Sather WA, Zhang JF, Tsien RW. Ca2+ channel selectivity at a single locus for high-affinity Ca2+ interactions. Neuron. 1995;15:1121–1132. doi: 10.1016/0896-6273(95)90100-0. [DOI] [PubMed] [Google Scholar]
- 58.Yang J, Ellinor PT, Sather WA, Zhang JF, Tsien RW. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature. 1993;366:158–161. doi: 10.1038/366158a0. [DOI] [PubMed] [Google Scholar]
- 59.Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 2008;586:419–425. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Penna A, et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ji W, et al. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci USA. 2008;105:13668–13673. doi: 10.1073/pnas.0806499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cai X. Molecular evolution and structural analysis of the Ca2+ release-activated Ca2+ channel subunit, Orai. J Mol Biol. 2007;368:1284–1291. doi: 10.1016/j.jmb.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 63.Hogan PG, Rao A. Dissecting ICRAC, a store-operated calcium current. Trends Biochem Sci. 2007;32:235–245. doi: 10.1016/j.tibs.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 64.Zweifach A, Lewis RS. Calcium-dependent potentiation of store-operated calcium channels in T lymphocytes. J Gen Physiol. 1996;107:597–610. doi: 10.1085/jgp.107.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hess P, Lansman JB, Tsien RW. Calcium channel selectivity for divalent and monovalent cations. Voltage and concentration dependence of single channel current in ventricular heart cells. J Gen Physiol. 1986;88:293–319. doi: 10.1085/jgp.88.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Voets T, et al. CaT1 and the calcium release-activated calcium channel manifest distinct pore properties. J Biol Chem. 2001;276:47767–47770. doi: 10.1074/jbc.C100607200. [DOI] [PubMed] [Google Scholar]
- 67.Cataldi M, Perez-Reyes E, Tsien RW. Differences in apparent pore sizes of low and high voltage-activated Ca2+ channels. J Biol Chem. 2002;277:45969–45976. doi: 10.1074/jbc.M203922200. [DOI] [PubMed] [Google Scholar]
- 68.McCleskey EW, Almers W. The Ca channel in skeletal muscle is a large pore. Proc Natl Acad Sci USA. 1985;82:7149–7153. doi: 10.1073/pnas.82.20.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Voets T, Janssens A, Droogmans G, Nilius B. Outer pore architecture of a Ca2+-selective TRP channel. J Biol Chem. 2004;279:15223–15230. doi: 10.1074/jbc.M312076200. [DOI] [PubMed] [Google Scholar]
- 70.Di Capite J, Ng SW, Parekh AB. Decoding of cytoplasmic Ca2+ oscillations through the spatial signature drives gene expression. Curr Biol. 2009;19:853–858. doi: 10.1016/j.cub.2009.03.063. [DOI] [PubMed] [Google Scholar]
- 71.Chang WC, Di Capite J, Singaravelu K, Nelson C, Halse V, Parekh AB. Local Ca2+ influx through Ca2+ release-activated Ca2+ (CRAC) channels stimulates production of an intracellular messenger and an intercellular pro-inflammatory signal. J Biol Chem. 2008;283:4622–4631. doi: 10.1074/jbc.M705002200. [DOI] [PubMed] [Google Scholar]
- 72.Kozak JA, Kerschbaum HH, Cahalan MD. Distinct properties of CRAC and MIC channels in RBL cells. J Gen Physiol. 2002;120:221–235. doi: 10.1085/jgp.20028601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kozak JA, Matsushita M, Nairn AC, Cahalan MD. Charge screening by internal pH and polyvalent cations as a mechanism for activation, inhibition, and rundown of TRPM7/MIC channels. J Gen Physiol. 2005;126:499–514. doi: 10.1085/jgp.200509324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Z, Lu J, Xu P, Xie X, Chen L, Xu T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. 2007;282:29448–29456. doi: 10.1074/jbc.M703573200. [DOI] [PubMed] [Google Scholar]
- 77.Xu P, Lu J, Li Z, Yu X, Chen L, Xu T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem Biophys Res Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- 78.Muik M, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- 79.Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383–5401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Park CY, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Muik M, et al. A cytosolic homomerization and a modulatory domain within STIM1 C-terminus determine coupling to ORAI1 channels. J Biol Chem. 2009;284:8421–8426. doi: 10.1074/jbc.C800229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009;385:49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Christian EP, Spence KT, Togo JA, Dargis PG, Patel J. Calcium-dependent enhancement of depletion-activated calcium current in Jurkat T lymphocytes. J Membr Biol. 1996;150:63–71. doi: 10.1007/s002329900030. [DOI] [PubMed] [Google Scholar]
- 85.Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J Gen Physiol. 1995;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fierro L, Parekh AB. Fast calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. J Membr Biol. 1999;168:9–17. doi: 10.1007/s002329900493. [DOI] [PubMed] [Google Scholar]
- 87.Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Rossum DB, Patterson RL, Ma HT, Gill DL. Ca2+ entry mediated by store depletion, S-nitrosylation, and TRP3 channels. Comparison of coupling and function. J Biol Chem. 2000;275:28562–28568. doi: 10.1074/jbc.M003147200. [DOI] [PubMed] [Google Scholar]
- 89.Ma HT, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- 90.Gregory RB, Rychkov G, Barritt GJ. Evidence that 2-aminoethyl diphenylborate is a novel inhibitor of store-operated Ca2+ channels in liver cells, and acts through a mechanism which does not involve inositol trisphosphate receptors. Biochem J. 2001;354:285–290. doi: 10.1042/0264-6021:3540285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma HT, et al. Assessment of the role of the inositol 1,4,5-trisphosphate receptor in the activation of transient receptor potential channels and store-operated Ca2+ entry channels. J Biol Chem. 2001;276:18888–18896. doi: 10.1074/jbc.M100944200. [DOI] [PubMed] [Google Scholar]
- 92.Peinelt C, Lis A, Beck A, Fleig A, Penner R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J Physiol. 2008;586:3061–3073. doi: 10.1113/jphysiol.2008.151365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dehaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW., Jr Complex actions of 2-aminoethyldiphenyl borate (2-APB) on store-operated calcium entry. J Biol Chem. 2008;283:19265–19273. doi: 10.1074/jbc.M801535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y, et al. STIM protein coupling in the activation of Orai channels. Proc Natl Acad Sci USA. 2009;106:7391–7396. doi: 10.1073/pnas.0900293106. [DOI] [PMC free article] [PubMed] [Google Scholar]