

HisC2 from M. tuberculosis was cloned, overexpressed, purified and crystallized and the crystals were characterized.
Keywords: HisC2, Mycobacterium tuberculosis, Mycobacterium smegmatis
Abstract
HisC2 from Mycobacterium tuberculosis was overexpressed in M. smegmatis and purified to homogeneity using nickel–nitrilotriacetic acid metal-affinity and gel-filtration chromatography. Diffraction-quality crystals were grown using the hanging-drop vapour-diffusion technique from a condition consisting of 7 mg ml−1 HisC2 (in 20 mM Tris pH 8.8, 50 mM NaCl and 5% glycerol), 1 M succinic acid pH 7.0, 0.1 M HEPES pH 7.0 and 1%(w/v) polyethylene glycol monomethyl ether 2000. The crystals belonged to the orthorhombic space group P21212, with unit-cell parameters a = 255.98, b = 77.09, c = 117.97 Å. X-ray diffraction data were recorded to 2.45 Å resolution from a single crystal using the in-house X-ray facility.
1. Introduction
The genome sequence of Mycobacterium tuberculosis (Mtb), the debilitating human pathogen that causes the chronic infectious disease tuberculosis (TB) in humans, revealed that this bacterium has its own machinery for the manufacture of various amino acids (Cole et al., 1998 ▶). Among these, the histidine-biosynthetic pathway, which leads to the enzymatic synthesis of histidine from 5-phosphoribosyl-1-pyrophosphate in ten steps, is conserved in archaea, bacteria, fungi and plants but is absent in mammals (Ames et al., 1960 ▶; Alifano et al., 1996 ▶; Stepansky & Leustek, 2006 ▶). Moreover, a high-density mutagenesis study showed that this pathway is among the essential pathways required for optimal growth of Mtb (Sassetti et al., 2003 ▶); the enzymes of this pathway are therefore potential drug targets.
This pathway has been extensively studied genetically and biochemically in Salmonella typhimurium and Escherichia coli (Brenner & Ames, 1971 ▶). Importantly, in the last decade the structures of enzymes of this pathway from various organisms such as Mtb (Cho et al., 2003 ▶; Javid-Majd et al., 2008 ▶; Due et al., 2011 ▶), E. coli (Sivaraman et al., 2001 ▶; Barbosa et al., 2002 ▶; Rangarajan et al., 2006 ▶), Methanobacterium thermoautotrophicum (Sivaraman et al., 2005 ▶), Thermotoga maritima (Schwarzenbacher et al., 2004 ▶), Filobasidiella neoformans (Sinha et al., 2004 ▶), Arabidopsis thaliana (Glynn et al., 2005 ▶) and Saccharomyces cerevisiae (Quevillon-Cheruel et al., 2006 ▶) have contributed to understanding the enzymatic mechanisms at the molecular level; however, complete understanding of the molecular basis of histidine biosynthesis in a particular organism requires structural characterization of all the relevant enzymes from that organism. In this respect, we have initiated structure–function relationship studies of enzymes of this pathway that have not yet been characterized from Mtb. In the present study, we report the molecular biology and preliminary X-ray diffraction studies of histidinol phosphate aminotransferase (HisC2), one of the enzymes involved in the seventh step that leads to the production of l-histidinol phosphate from imidazole acetol phosphate (Fig. 1 ▶).
Figure 1.

A schematic representation of the various enzymatic steps that lead to the synthesis of histidine in M. tuberculosis. The step that is catalysed by HisC2 is shown in the blue box.
2. Materials and methods
2.1. Molecular cloning
The gene (Rv3772) encoding HisC2 was cloned in the M. smegmatis (Msg)–E. coli shuttle expression vector pYUB1062, which was itself constructed from the pET30a plasmid and the NarI/EcoRV DNA fragment of pMV206 (Wang et al., 2010 ▶). The protocol used for cloning involved initial PCR amplification of the gene using two gene-specific primers, Phusion polymerase (Finnzymes), dNTPs, MgCl2 and Mtb H37Rv genomic DNA. The forward primer was 5′-CACC CATATGGTGACCGCCCGCCTGCGACC-3′, which contains the first four directional cloning specific nucleotides (shown in bold) and an NdeI restriction site (underlined) followed by the first 20 nucleotides of the open reading frame (ORF); the reverse primer was 5′-TAT AAGCTTTTGGTCGCTCCGCCAGCGGC-3′, which contains three arbitrarily chosen nucleotides (shown in bold) to assist in digestion at the HindIII restriction site (underlined) followed by the reverse complement of the last 20 nucleotides of the ORF but lacking the stop codon to allow the addition of a His tag at the C-terminus of the recombinant protein. The amplified gene was then directionally cloned into the entry vector pENTR (Invitrogen) as per the manufacturer’s protocol to obtain the recombinant plasmid pENTR-3772. The entry clone was digested with NdeI and HindIII and the insert was purified using a gel-extraction kit (Qiagen). The expression clone was obtained by ligating the insert into similarly digested and purified pYUB1062 vector using T4 DNA ligase. The reaction mixture was transformed into DH5α cells and spread onto Luria–Bertani (LB) plates containing 150 µg ml−1 hygromycin B. The colonies containing the clone were identified by colony PCR followed by an insert ‘fall-out’ observation on an agarose gel after restriction digestion of positive clones using the same restriction enzymes. The sequence and the directionality of the clone were confirmed by DNA sequencing (Ocimum Biosolutions, India). A construct with the following expression sequence, consisting of the vector-derived start codon-encoded methionine (bold), HisC2 (starting with a valine instead of a methionine), seven extra residues derived from the vector (italics) and a hexahistidine tag (underlined) at the C-terminus of HisC2, was overexpressed in Msg: MVTARLRPELAGLPVYVPGKTVPGAIKLASNETVFGPLPSVRAAIDRATDTVNRYPDNGCVQLKAALARHLGPDFAPEHVAVGCGSVSLCQQLVQVTASVGDEVVFGWRSFELYPPQVRVAGAIPIQVPLTDHTFDLYAMLATVTDRTRLIFVCNPNNPTSTVVGPDALARFVEAVPAHILIAIDEAYVEYIRDGMRPDSLGLVRAHNNVVVLRTFSKAYGLAGLRIGYAIGHPDVITALDKVYVPFTVSSIGQAAAIASLDAADELLARTDTVVAERARVSAELRAAGFTLPPSQANFVWLPLGSRTQDFVEQAADARIVVRPYGTDGVRVTVAAPEENDAFLRFARRWRSDQKLAAALE HHHHHH.
2.2. Overexpression
The expression clone was electroporated into Msg cells at 2500 V, 1000 Ω and 25 µF using a 2 mm electroporation cuvette (Bio-Rad, USA). The transformed cells were plated onto 7H10 agar plates supplemented with 10% OADC (oleic acid–albumin–dextrose–catalase), 0.5% Tween 80, 100 µg ml−1 hygromycin B and 25 µg ml−1 kanamycin. Colonies appeared after about 72 h. A single colony was inoculated in LB broth (primary culture) supplemented with 0.2% glycerol, 0.05% Tween 80, 100 µg ml−1 hygromycin B and 25 µg ml−1 kanamycin. When the optical density at 600 nm (OD600) reached 0.8–1.0, the primary culture was diluted about 32-fold into a 5 l secondary culture. At an OD600 of ∼0.7, the secondary culture was induced with 0.2% acetamide for 24 h at 310 K and 200 rev min−1. The cells were harvested by spinning down the culture at 10 000g and 277 K for 25 min. The cell pellet was frozen at 193 K until further use.
2.3. Purification
All purification steps were carried out at 277 K. The cell pellet was thawed on ice and resuspended in a buffer consisting of 20 mM Tris pH 8.8, 50 mM NaCl, 5% glycerol (buffer A) supplemented with 1 mM DTT and one Complete Mini EDTA-free protease inhibitor cocktail tablet (Roche). The cells were lysed by passing them through a cell disrupter twice at 138 MPa (Constant Systems Ltd, UK). The lysate was clarified by centrifugation at 10 000g for 40 min. The recombinant protein was purified by affinity chromatography using a ÄKTAexplorer liquid-chromatographic system (GE Healthcare Life Sciences, USA). Briefly, the clarified supernatant was loaded onto a 5 ml HisTrap FF column and nonspecifically bound proteins were removed by washing the column first with buffer A and then with buffer A supplemented with 2 M NaCl and 10 mM imidazole. Subsequently, HisC2 was eluted from the column using buffer A containing 300 mM imidazole. HisC2 was further purified by gel-filtration chromatography on a HiLoad 16/60 Superdex 200 pg column (GE Healthcare Life Sciences, USA). The purity of the protein was assessed by 12% Coomassie-stained SDS–PAGE (Fig. 2 ▶). The identity of the protein was confirmed by mass-spectrometric analysis (Technoconcept, India).
Figure 2.
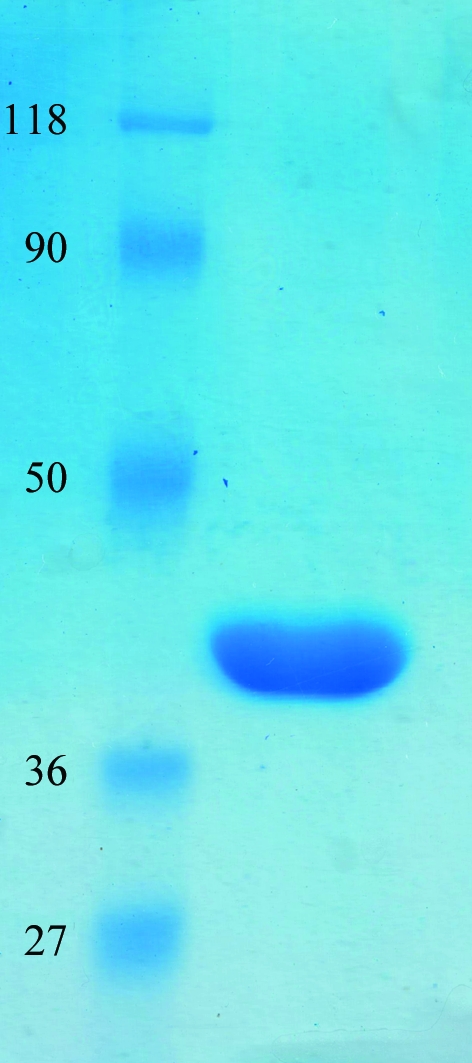
SDS–PAGE profile of the purified recombinant HisC2. The gel was stained with Coomassie Brilliant Blue R-250. Lane 1, molecular-mass marker (labelled in kDa); lane 2, purified HisC2. 30 µg protein was loaded onto the gel.
2.4. Crystallization and data collection
The purified HisC2 in 20 mM Tris pH 8.8, 50 mM NaCl, 5% glycerol buffer was divided into two aliquots and concentrated to 7 and 10 mg ml−1. Separate crystallization trials were performed for these aliquots as follows.
Crystallization experiments were carried out in a 96-well plate using the hanging-drop vapour-diffusion technique, the commercially available crystallization screens Crystal Screen, Crystal Screen 2 and Index Screen (Hampton Research, USA) and a Mosquito robot (TTP LabTech, England). Droplets of 1 µl in volume (with a 1:1 protein:precipitant ratio) were equilibrated against 100 µl reservoir solution at 298 K. Diffraction-quality crystals were obtained in Index Screen condition No. 34 [1 M succinic acid pH 7.0, 0.1 M HEPES pH 7.0, 1%(w/v) polyethylene glycol monomethyl ether 2000] using the 7 mg ml−1 HisC2 stock after two weeks.
A single crystal was mounted on a CryoLoop supported on a magnetic CrystalCap, rinsed in cryoprotectant solution [30%(v/v) glycerol in reservoir solution], placed onto the magnetic base of the goniometer head and aligned in the Cu Kα X-ray beam generated by the in-house X-ray generator, a Rigaku FR-E+ SuperBright microfocus rotating-anode generator. Test diffraction images were recorded at various angles on an R-AXIS IV++ detector. Based on the data-collection strategy, a complete native diffraction data set was collected at 100 K. The data set was processed using the program HKL-2000 (Otwinowski & Minor, 1997 ▶). The data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Space group | P21212 |
| Unit-cell parameters (Å, °) | a = 255.98, b = 77.09, c = 117.97, α = β = γ = 90 |
| Matthews coefficient† (Å3 Da−1) | 3.67, 2.94, 2.45, 2.10 |
| Solvent content† (%) | 66.48, 58.09, 49.71, 41.33 |
| Temperature (K) | 100 |
| Detector | R-AXIS IV++ |
| Wavelength (Å) | 1.5418 |
| Crystal-to-detector distance (mm) | 200 |
| Resolution (Å) | 50.0–2.45 (2.54–2.45) |
| Unique reflections | 86797 (8078) |
| Multiplicity | 5.0 (4.6) |
| 〈I/σ(I)〉 | 13.3 (4.4) |
| Completeness (%) | 99.1 (93.6) |
| Rmerge‡ (%) | 10.5 (42.8) |
| Overall B factor from Wilson plot (Å2) | 43.93 |
Values are given for four, five, six and seven molecules in the crystal asymmetric unit, respectively.
R
merge(I) = 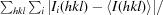
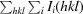 . 〈I(hkl)〉 is the average intensity of the i observations of reflection hkl.
. 〈I(hkl)〉 is the average intensity of the i observations of reflection hkl.
3. Results and discussion
Overexpression of HisC2 [cloned in vectors pDEST15 (GST tag) and pDET17 (His tag)] in an E. coli BL21 (DE3) host was not successful. However, we were able to produce milligram quantities of soluble HisC2 using the Msg expression system. This result, together with previously published data (Bashiri et al., 2007 ▶; Goldstone et al., 2008 ▶; Ahangar et al., 2011 ▶), suggests that Msg is a better alternative host for the expression of Mtb proteins. HisC2 was purified to homogeneity by Ni–NTA metal-affinity and gel-filtration chromatography (Fig. 2 ▶). Diffraction-quality crystals (Fig. 3 ▶) were obtained by the hanging-drop vapour-diffusion technique. The crystals diffracted to 2.45 Å resolution (Fig. 4 ▶) and a complete native data set was collected. Assuming the presence of four monomeric molecules (with a calculated molecular weight of 39 658 Da each) per crystal asymmetric unit, the calculated Matthews coefficient and solvent content are 3.67 Å3 Da−1 and 66.48%, respectively (Matthews, 1968 ▶). The structure of HisC2 was solved by the molecular-replacement method using the crystal structure (a dimer) of its Listeria innocua counterpart (PDB entry 3ffh; Midwest Center for Structural Genomics, unpublished work) as the search model. The search model shares 29% sequence identity with Mtb HisC2 (Fig. 5 ▶). The program Phaser (McCoy et al., 2007 ▶) from CCP4 (Winn et al., 2011 ▶) was used to solve the structure and yielded a model comprised of four subunits (two dimers). The values of R work and R free after 50 cycles of rigid-body refinement followed by 100 cycles of positional refinement using the program REFMAC5 (Murshudov et al., 2011 ▶) from CCP4 were 0.412 and 0.474, respectively. At this stage, substitution of HisC2-specific amino acids into the electron density was begun using the program Coot (Emsley & Cowtan, 2004 ▶). After every round of model building, simulated-annealing and positional refinement was carried out. After substitution of 70% of the sequence of the L. innocua molecular-replacement model with the corresponding Mtb sequence, the values of R work and R free were 33.5% and 37.3%, respectively. Substitution of the remaining L. innocua sequence with the correct Mtb sequence and further refinement are in progress.
Figure 3.

Crystals of HisC2. The approximate dimensions of the HisC2 crystals were 300 × 50 × 50 µm.
Figure 4.

A representative diffraction image collected from a single HisC2 crystal (0.5° oscillation range). The concentric red circles show the resolution limits of the frame.
Figure 5.

Amino-acid sequence alignment of Mtb HisC2 and its counterpart from L. innocua. The secondary-structural elements (helices represented by coils, β-strands by arrows and turns by letter Ts) corresponding to histidinol phosphate aminotransferase from L. innocua are shown. Conserved residues are highlighted with a red background. The sequence alignment was performed by the program MultAlin (Corpet, 1988 ▶) and the figure was generated using the program ESPript (Gouet et al., 1999 ▶).
Acknowledgments
We thank Professor William R. Jacobs of the Department of Microbiology and Immunology and the Howard Hughes Medical Institute, Albert Einstein College of Medicine, Bronx, New York, USA for providing us with the M. smegmatis expression system. Mtb H37Rv genomic DNA was obtained through the Biodefense and Emerging Infections Research Resources Repository (BEI Resources), NIAID, NIH. The work was supported by a start-up grant (to BKB) from the National Institute of Immunology (NII), New Delhi, India. The in-house X-ray diffraction facility used for data collection was established with financial support from the Department of Biotechnology (DBT), Government of India. We acknowledge Ravikant Pal for his help during data collection. NN is a junior research fellow of the Council of Science and Industrial Research, Government of India, India.
References
- Ahangar, M. S., Khandokar, Y., Nasir, N., Vyas, R. & Biswal, B. K. (2011). Acta Cryst. F67, 1451–1456. [DOI] [PMC free article] [PubMed]
- Alifano, P., Fani, R., Liò, P., Lazcano, A., Bazzicalupo, M., Carlomagno, M. S. & Bruni, C. B. (1996). Microbiol. Rev. 60, 44–69. [DOI] [PMC free article] [PubMed]
- Ames, B. N., Garry, B. & Herzenberg, L. A. (1960). J. Gen. Microbiol. 22, 369–378. [DOI] [PubMed]
- Barbosa, J. A., Sivaraman, J., Li, Y., Larocque, R., Matte, A., Schrag, J. D. & Cygler, M. (2002). Proc. Natl Acad. Sci. USA, 99, 1859–1864. [DOI] [PMC free article] [PubMed]
- Bashiri, G., Squire, C. J., Baker, E. N. & Moreland, N. J. (2007). Protein Expr. Purif. 54, 38–44. [DOI] [PubMed]
- Brenner, M. & Ames, B. N. (1971). In Metabolic Pathways, Vol. 5, edited by J. Vogel. New York: Academic Press.
- Cho, Y., Sharma, V. & Sacchettini, J. C. (2003). J. Biol. Chem. 278, 8333–8339. [DOI] [PubMed]
- Cole, S. T. et al. (1998). Nature (London), 393, 537–544.
- Corpet, F. (1988). Nucleic Acids Res. 16, 10881–10890. [DOI] [PMC free article] [PubMed]
- Due, A. V., Kuper, J., Geerlof, A., von Kries, J. P. & Wilmanns, M. (2011). Proc. Natl Acad. Sci. USA, 108, 3554–3559. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Glynn, S. E., Baker, P. J., Sedelnikova, S. E., Davies, C. L., Eadsforth, T. C., Levy, C. W., Rodgers, H. F., Blackburn, G. M., Hawkes, T. R., Viner, R. & Rice, D. W. (2005). Structure, 13, 1809–1817. [DOI] [PubMed]
- Goldstone, R. M., Moreland, N. J., Bashiri, G., Baker, E. N. & Lott, J. S. (2008). Protein Expr. Purif. 57, 81–87. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Javid-Majd, F., Yang, D., Ioerger, T. R. & Sacchettini, J. C. (2008). Acta Cryst. D64, 627–635. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Quevillon-Cheruel, S., Leulliot, N., Graille, M., Blondeau, K., Janin, J. & van Tilbeurgh, H. (2006). Protein Sci. 15, 1516–1521. [DOI] [PMC free article] [PubMed]
- Rangarajan, E. S., Proteau, A., Wagner, J., Hung, M. N., Matte, A. & Cygler, M. (2006). J. Biol. Chem. 281, 37930–37941. [DOI] [PubMed]
- Sassetti, C. M., Boyd, D. H. & Rubin, E. J. (2003). Mol. Microbiol. 48, 77–84. [DOI] [PubMed]
- Schwarzenbacher, R. et al. (2004). Proteins, 54, 801–805. [DOI] [PubMed]
- Sinha, S. C., Chaudhuri, B. N., Burgner, J. W., Yakovleva, G., Davisson, V. J. & Smith, J. L. (2004). J. Biol. Chem. 279, 15491–15498. [DOI] [PubMed]
- Sivaraman, J., Li, Y., Larocque, R., Schrag, J. D., Cygler, M. & Matte, A. (2001). J. Mol. Biol. 311, 761–776. [DOI] [PubMed]
- Sivaraman, J., Myers, R. S., Boju, L., Sulea, T., Cygler, M., Davisson, V. J. & Schrag, J. D. (2005). Biochemistry, 44, 10071–10080. [DOI] [PubMed]
- Stepansky, A. & Leustek, T. (2006). Amino Acids, 30, 127–142. [DOI] [PubMed]
- Wang, F., Jain, P., Gulten, G., Liu, Z., Feng, Y., Ganesula, K., Motiwala, A. S., Ioerger, T. R., Alland, D., Vilchèze, C., Jacobs, W. R. & Sacchettini, J. C. (2010). Antimicrob. Agents Chemother. 54, 3776–3782. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.