

Abstract
Introduction
Smad3, a component of the TGFβ signaling cascade, contributes to G1 arrest in breast cancer cells. Cyclin D1/CDK4 promotes G1/S-phase transition, and CDK phosphorylation of Smad3 has been associated with inhibition of Smad3 activity. We hypothesized that overexpression of cyclin D1 exerts tumorigenic effects in breast cancer cells through CDK4-mediated phosphorylation and inhibition of Smad3 and release of G1 arrest.
Methods
Real-time quantitative RT-PCR and immunoblotting were used to evaluate expression of study proteins in cyclin D1-overexpressing breast cancer cells. Smad3 transcriptional activity and cell cycle control were examined in cells transfected with wild type (WT) Smad3 or Smad3 with single or multiple CDK phosphorylation site mutations (M), in the presence or absence of CDK4 inhibitor or co-transfection with cdk4 siRNA.
Results
Transfection of the Smad3 5M construct resulted in decreased c-myc and higher p15INK4B expression. Compared with WT Smad3, overexpression of the Smad3 T8, T178, 4M, or 5M mutant constructs resulted in higher Smad3 transcriptional activity. Compared with cells transfected with WT Smad3, Smad3 transcriptional activity was higher in cells overexpressing Smad3 mutant constructs and treated with CDK4 inhibitor or transfected with cdk4 siRNA. Cells transfected with Smad3 T8 or T178 and treated with CDK4 inhibitor showed an increase in the G1 cell population.
Conclusions
Inhibition of CDK-mediated Smad3 phosphorylation released cyclin D1-regulated blockade of Smad3 transcriptional activity and recovered cell cycle arrest in breast cancer cells. Targeted inhibition of CDK4 activity may have a role in the treatment of cyclin D-overexpressing breast cancers.
Keywords: Smad3, breast cancer, cyclin D, CDK4, TGFβ
Introduction
The TGFβ superfamily consists of a large group of secreted polypeptide growth factors that bind transmembrane serine-threonine kinase receptor complexes (1, 2). TGFβ/activin signaling is transduced through receptor-mediated phosphorylation of intracellular Smad2 and Smad3, which form a complex with Smad4, translocate to the nucleus, and, along with co-activators and cell-specific DNA-binding factors, regulate target gene expression relevant to several aspects of cell growth and differentiation (3-5). The significance of TGFβ signaling in breast cancer has been widely studied, though less is understood about the relevance of Smad-mediated transcriptional activity in cancer cell growth control (6).
Several lines of evidence suggest that Smad3 may be involved in cell cycle arrest. The Smad3 co-factors E2F4/5 and p107, along with Smad4, bind to a Smad-E2F site on c-myc, causing repression of this cell cycle mitogen (7). Furthermore, a nuclear Smad complex that includes Smad3 and the transcription factor Sp1 is thought to mediate transcription of the cyclin-dependent kinase (CDK) inhibitors p15 and p21, whose promoters contain an Sp1 binding site (8, 9). Additionally, Smad3/4 complexes, along with the forkhead box O protein, bind promoters responsible for transcription of p15 and p21 (10). Conversely, c-myc overexpression can inhibit the Smad-dependent transcription of p15 and p21 (11). Thus, working in conjunction with several different critical growth-regulatory mechanisms, Smad3 may act in a dual capacity to both inhibit c-myc expression and stimulate CDK inhibitor transcription to help actualize G1-phase cell cycle arrest. Consequently, the loss of Smad3 function could induce a potent cell cycle release allowing for the uncontrolled cell growth characteristic of malignancy.
A relationship between Smad signaling and both normal and malignant mammary cell growth has been shown. Differential expression of activin and TGFβ receptors and Smads has been found in the mammary gland during pregnancy and lactation (12-14). Also, in a breast cancer tissue microarray study, Xie et al demonstrated that the loss of Smad4 correlated with axillary lymph node involvement, and the loss of phosphorylated Smad2 correlated with decreased overall survival (15). Subsequent work examining a panel of MCF-10A pre-malignant and transformed malignant mammary cell lines showed that Smad2/3 signaling conferred both tumor suppressant and oncogenic effects, dependent upon the primary or metastatic environment (16). Decreased levels of nuclear Smad3 have also been associated with larger tumor size, higher tumor grade, and estrogen receptor (ER)-negative breast cancers (17). These data point toward a dynamic role for Smad signaling in breast cancer favoring a tumor suppressant function in well differentiated, earlier stage disease.
The potential mechanisms responsible for circumvention of Smad-mediated cell growth control are being explored. Matsuura et al have found Smad3 activity to be negatively regulated by CDK4 and CDK2 phosphorylation in fibroblasts (18, 19). Several Smad3 inhibitory CDK phosphorylation sites have been identified, primarily within the linker region of the molecule (5, 18). Cyclin D exerts its action via CDK4, and in Mv1Lu mink lung epithelial cells, cyclin D overexpression was found to induce Smad3 linker phosphorylation via CDK4, which led to inhibition of wild type (WT) Smad3 activity (18, 19). The cyclin D-CDK4/6 complex is critical to cell cycle progression, as it induces phosphorylation inhibition of the Rb protein. Rb protein phosphorylation permits E2F-mediated transcription of genes responsible for cell cycle mitogenesis (20, 21). Thus, as cyclin D regulates one of the key initiating factors for cell cycle progression, the overexpression of this protein may render cells vulnerable to malignant transformation.
It follows that overexpression of cyclin D has been found in aggressive breast cancers, and this overexpression is associated with a poor prognosis. Cyclin D overexpression is pervasive in human breast cancers, including heritable breast cancers with BRCA2 mutations (22-24). It has been shown that Smad3 and BRCA2 can synergize to affect their tumor suppressant functions (25). Potentially, the mutation of BRCA2 found in some tumors overexpressing cyclin D contributes to the inhibition of Smad3 cell cycle control in these cancers (25). Additionally, both the Ras and human epidermal growth factor receptor 2 (HER2) oncogenes have been linked to cyclin D1 promoter activation in breast cancer. This finding implies that the known pathologic repercussions of Ras and neu overexpression in certain breast cancers involve cyclin D1/CDK4 (23, 26). Furthermore, patients with highly proliferative cancers, such as cancers that overexpress cyclins, tend to initially respond well to chemotherapy, though many of these patients ultimately recur. These poor outcomes point to critical resistant flaws in cell cycle control inherent to cancers that overexpress cyclins. The current study examines the impact of cyclin D overexpression and CDK4 phosphorylation on Smad3 action in breast cancer cell lines. We hypothesize that the oncogenic effects of cyclin D1 overexpression in breast cancer are induced, in part, through CDK4-mediated phosphorylation inhibition of Smad3 and loss of cell cycle arrest.
Results
Characterization of cyclin D1-overexpressing MCF7 and T47D breast cancer cell lines
The study panel of cells included MCF7 and T47D parental, vector control, and cyclin D1-overexpressing (CD1) breast cancer cell lines. The MCF7 cell line is known to be sensitive to both activin and TGFβ, whereas the T47D cell line is responsive only to activin (27). Immunoblotting for cyclin D1 confirmed that the MCF7-CD1 and T47D-CD1 lines expressed the highest amount of this protein relative to the parental and control cells (Figure 1A). Protein expression of Smad3, phosphorylated Smad3 (phospho-Smad3), CDK2, and CDK4 were similar across the parental, vector control, and CD1 lines. Kinase assays were performed to measure the relative amounts of CDK4 and CDK2 activity in the study cells. As expected, the MCF7-CD1 and T47D-CD1 cells showed the highest amount of CDK4 activity relative to the parental and vector control transfected cells (Figure 1B). No difference in CDK2 activity was found among the study cells (Figure 1C). To determine the subcellular localization of Smad3 in the study panel of cells, immunofluorescence was performed using an anti-Smad3 or anti phospho-Smad3 antibody. Immunofluorescent staining for Smad3 and phospho-Smad3 showed that the MCF7 (Figures 2A and 2B) and T47D (Figures 2C and 2D) study cells contained both cytoplasmic and nuclear Smad3. Importantly, the study cells designed to overexpress cyclin D demonstrated the presence of cytoplasmic and nuclear Smad3 helping to exclude compromise of nucleocytoplasmic shuttling in these cells as a cause for the decreased Smad3 activity found in this setting. While variations in nucleocytoplasmic shuttling of Smad3 may contribute to the level of Smad3 activity in a given cell line, overall, each of the 6 cell lines in the study panel demonstrated the capacity for nuclear translocation of Smad3.
FIGURE 1.
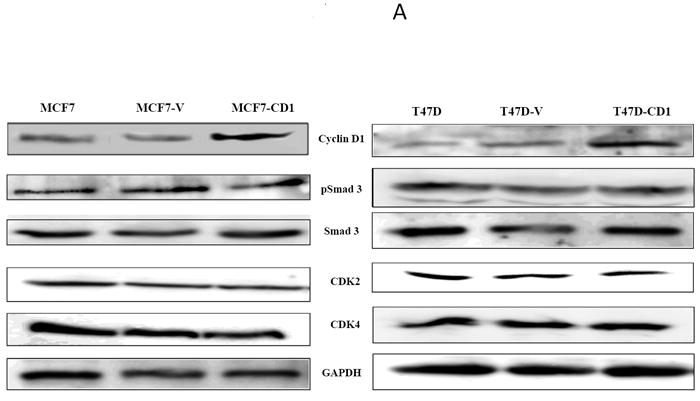
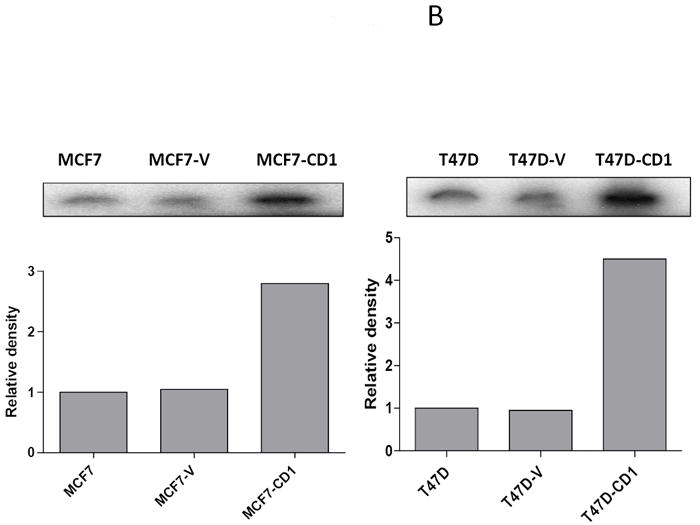
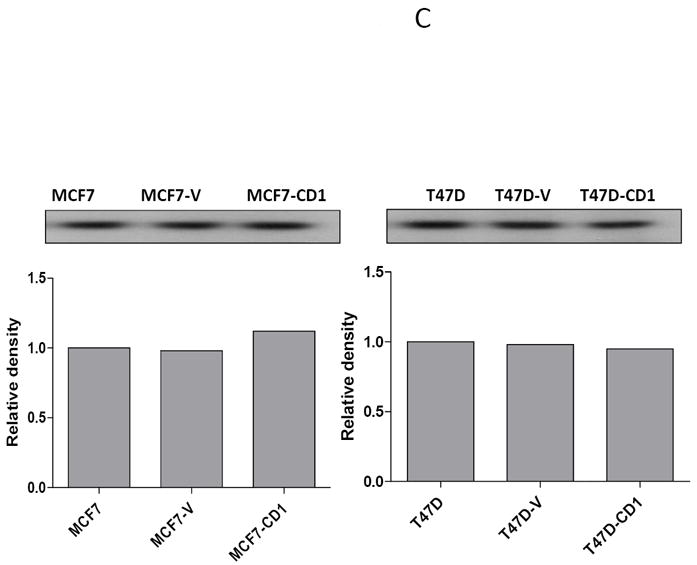
Cyclin D expression and activity in MCF7 and T47D stably transfected cells. (A) Protein extracts from stably transfected MCF7 or T47D clones were separated (30 μg/lane) on SDS/PAGE and subjected to immunoblot analysis with the indicated antibodies. (B) CDK4 kinase assay radiographs and densitometric quantification in MCF7 and T47D study cell lines.
FIGURE 2.
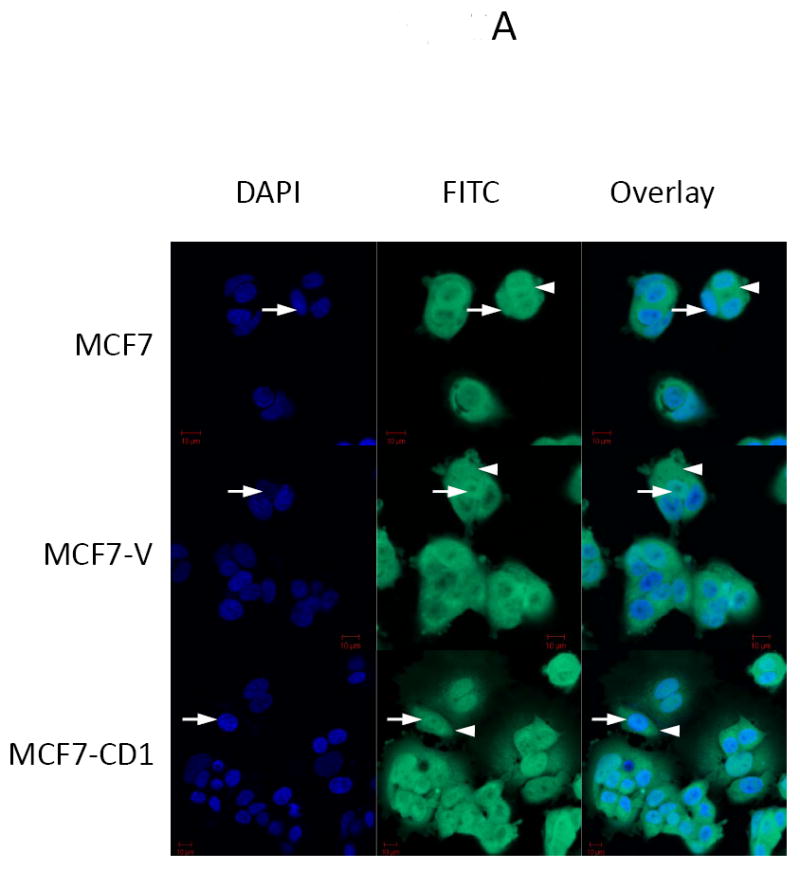
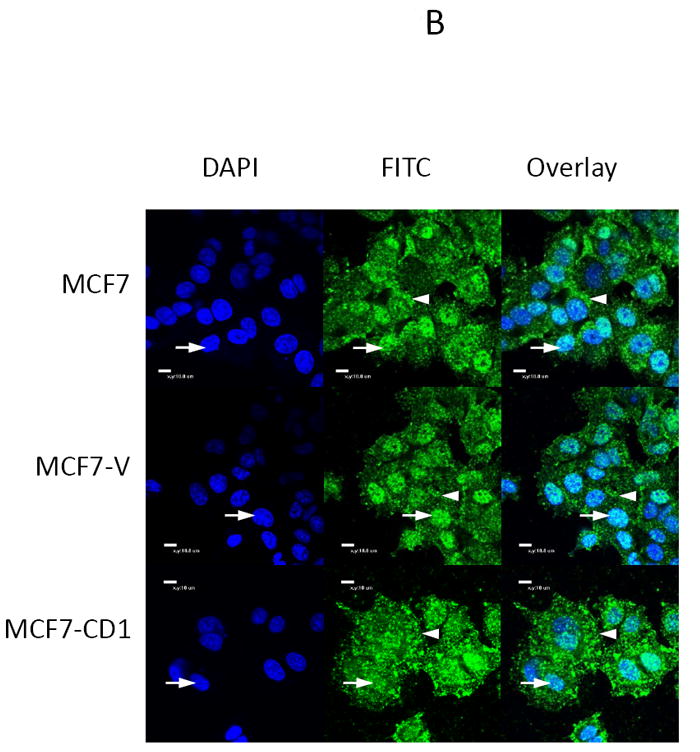
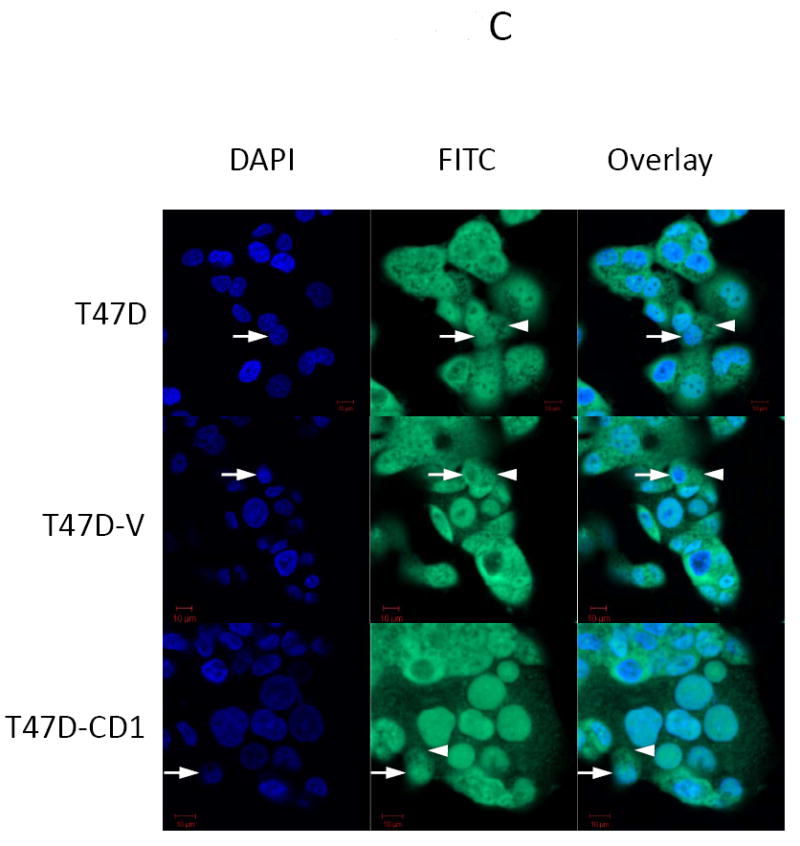
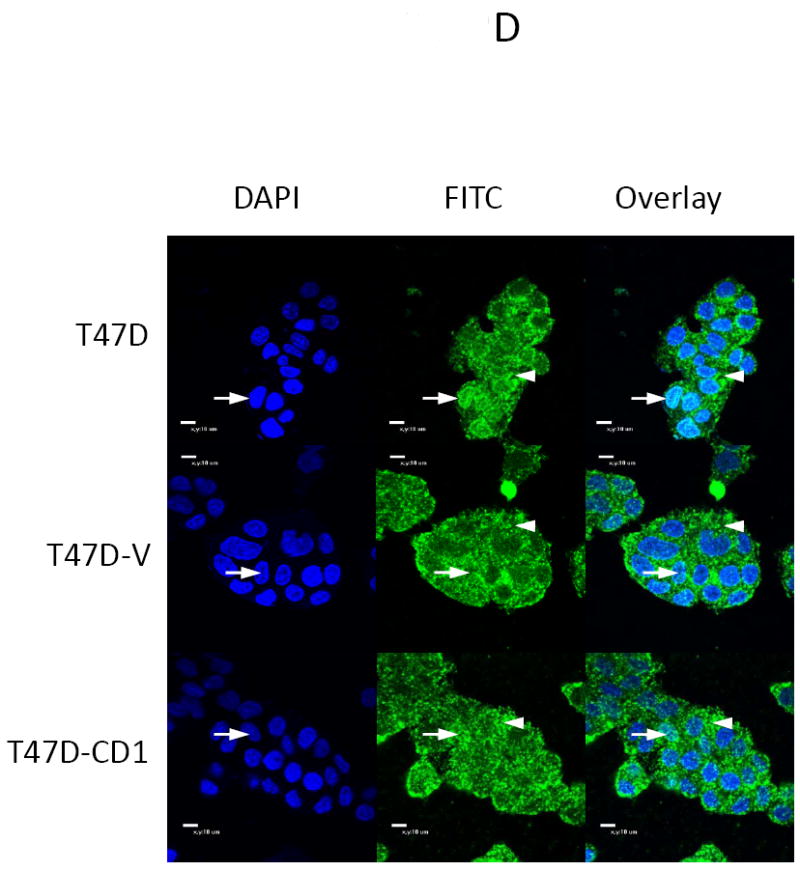
Smad3 and phospho-Smad3 localization in cyclin D-overexpressing MCF7 and T47D cells. Smad3 localized to the nucleus in MCF7 (A, B) and T47D (B, C) parental, vector control (V), and cyclin D-overexpressing (CD1) cell lines. Endogenous Smad3 and phospho-Smad3 were visualized by immunofluorescence microscopy with an anti-Smad3 polyclonal antibody and FITC-conjugated secondary antibody. Cell nuclei were counterstained with DAPI. Scale bar is 10 μm. Arrows denote nuclei. Arrowheads denote cytoplasmic Smad3.
Cyclin D modulates Smad3 transcriptional activity
The MCF7 and T47D breast cancer cell lines were transfected with WT Smad3, vector control, or the Smad3 5M CDK phosphorylation site mutant, and the expression levels of p15INK4B, p21, and c-myc were determined by real-time quantitative RT-PCR (Figures 3A and 3B). Parental and control MCF7 and T47D cells transfected with the WT Smad3 or Smad3 5M constructs showed lower levels of c-myc transcripts compared with study cells transfected with the vector control. Conversely, parental and control MCF7 and T47D cells transfected with the WT Smad3 and Smad3 5M constructs had higher amounts of p15INK4B and p21 transcripts when compared with cells transfected with the vector control. As expected, the cyclin D-overexpressing lines transfected with the control vector had the highest amounts of c-myc and lowest amounts of p15INK4B transcripts. When these cells were transfected with WT Smad3 or Smad3 5M, c-myc mRNA levels were lower and p15INK4B mRNA levels were higher than in the controls. Of note, the impact of WT Smad3 or 5M Smad3 overexpression on c-myc and p15INK4B transcript levels was more pronounced in T47D cells than in MCF7 cells. In addition, p21 transcript levels remained low in MCF7-CD1 cells and high in T47D-CD1 cells, regardless of WT Smad3 or Smad3 5M overexpression. These data suggest that in parental breast cancer cells, restoration of Smad3 function by overexpression of either WT Smad3 or the Smad3 5M CDK phosphorylation site mutant resulted in lower c-myc expression and higher CDK inhibitor expression. In breast cancer cells that overexpress cyclin D1, restoration of Smad3 function also resulted in relatively lower c-myc and higher p15INK4B expression; however, expression levels of p21 were not affected.
FIGURE 3.
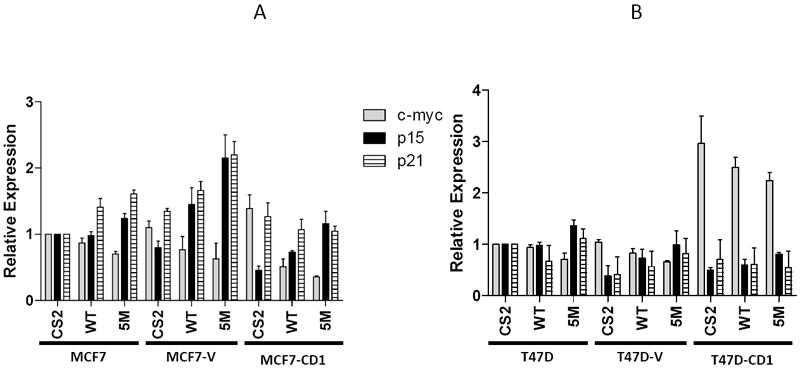
Expression of Smad3-regulated genes in study cell lines. MCF7 (A) and T47D (B) cell lines were transfected with wild type Smad3 (WT) or the 5M Smad3 CDK phosphorylation site mutant, and transcript levels of c-myc, p15, and p21 were measured by real-time quantitative RT-PCR.
Abrogation of Smad3 phosphorylation by CDK increases transcriptional activity in MCF7 and T47D cells
Transfection experiments were performed to determine the transcriptional activity of the various Smad3 CDK phosphorylation site mutant constructs in the MCF7 (Figure 4A) and T47D (Figure 4B) study cells. An additional ER-negative breast cancer cell line, Hs578T (Figure 4C), was also examined. The Hs578T cell line is known to be sensitive to TGFβ signaling (27). Study cells were co-transfected with each of the Smad3 expression plasmids, the Smad-responsive CAGA-luc reporter, and a pRenilla luciferase construct. Compared with WT Smad3, overexpression of the Smad3 T178 mutant in the MCF7 and Hs578T cell lines, and the Smad3 T8 mutant in the T47D cell lines, resulted in relatively higher CAGA-luc reporter activity. For all the study cell lines, overexpression of the Smad3 4M and Smad3 5M constructs also showed high reporter activity. Based on these experiments, the Smad3 T178 mutant was chosen for further study in the MCF7 and Hs578T cell lines and the Smad3 T8 mutant was chosen for further study in the T47D cell lines. The Smad3 5M construct was also further examined in all study cell lines.
FIGURE 4.
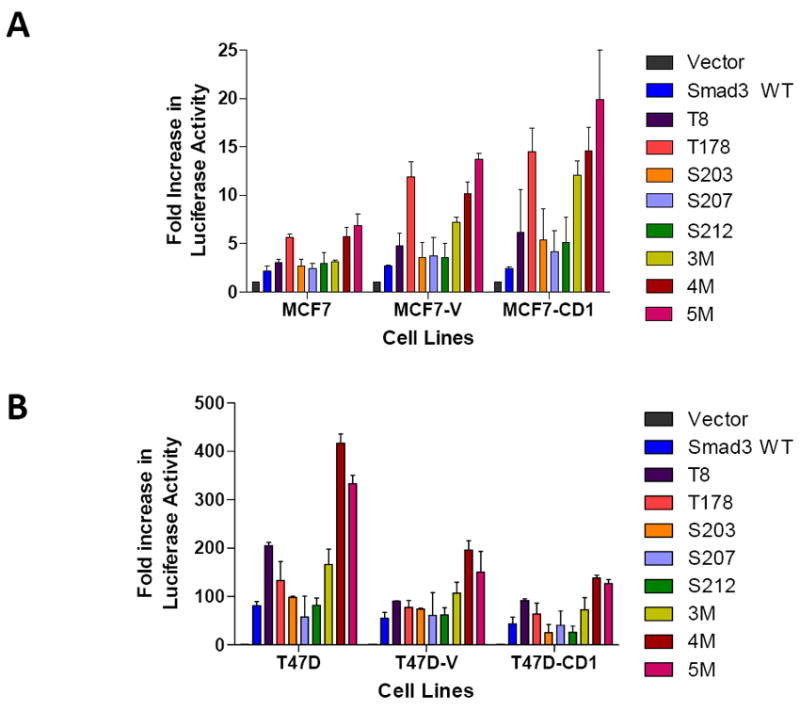
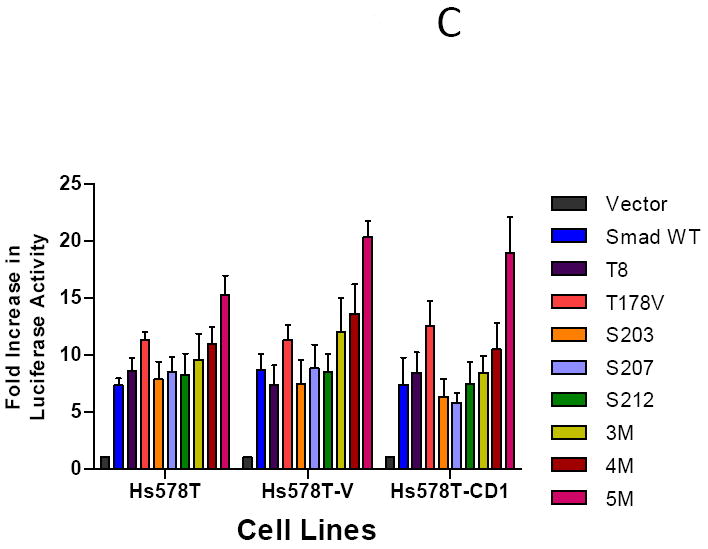
Relative transcriptional activity of various Smad3 constructs in cyclin D1 overexpressing MCF7 (A), T47D (B), and Hs578T (C) cell lines. Cells were co-transfected with the Smad-responsive CAGA-luc reporter construct and Renilla luciferase reporter, in addition to the indicated Smad3 expression constructs. Firefly and Renilla luciferase activities were determined. Data are shown as fold increase in normalized luciferase activity (firefly/Renilla) compared with empty vector-transfected MCF7, T47D, or Hs578T cells. Error bars indicate standard deviation from the mean of normalized luciferase activity for each study condition.
CDK4 inhibition restores Smad3 activity in breast cancer cell lines
To examine the direct impact of CDK inhibition on Smad3-responsive reporter activity in all MCF7 (Figure 5A) and T47D (Figure 5B) cell lines, the cells were transfected with WT Smad3 expression vector and the CAGA-luc and pRenilla reporter constructs. The cells were then treated with increasing concentrations of CDK2 and CDK4 inhibitors. Treatment with escalating doses of the CDK2 inhibitor resulted in a small increase in Smad3 reporter activity for the MCF7 cells, though no significant increase in reporter activity was found for the T47D treated cells. Furthermore, we did not see a great increase in Smad3 reporter activity when we tested higher concentrations of the CDK2 inhibitor (400 and 800 nM), though a slight increase found in cells treated with 800 nM of inhibitor may be attributed to the ability of this CDK2 inhibitor to also act against CDK1, which has an IC50 value of 760 nM and would be affected at this concentration (data not shown). A dose-dependent increase in Smad3 transcriptional activity was seen in all 6 MCF7 and T47D cell lines treated with increasing concentrations of the CDK4 inhibitor. Thus, Smad3 transcriptional activity could be enhanced through inhibition of CDK4, including cells overexpressing cyclin D1.
FIGURE 5.
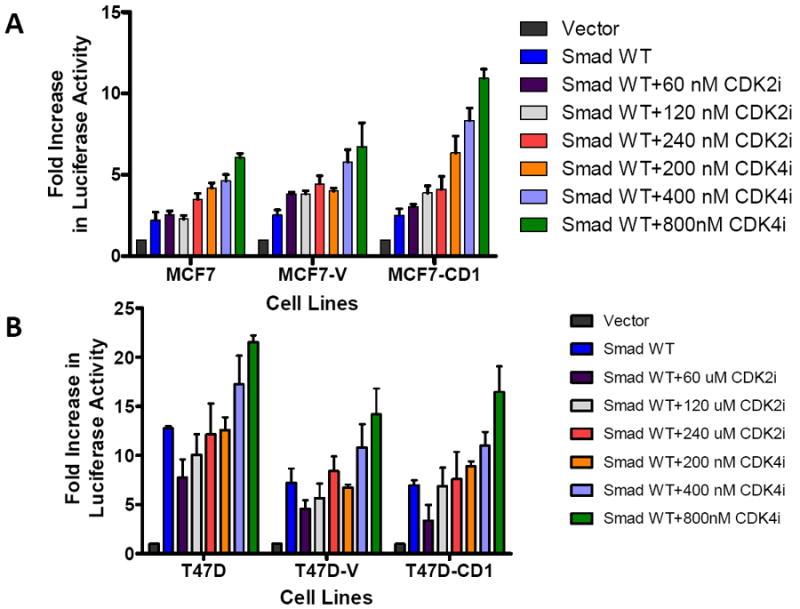
Dose-dependent increase in Smad3 transcriptional activity in MCF7 and T47D cells treated with increasing concentrations of CDK2 or CDK4 inhibitors. MCF7 (A) or T47D (B) study cell lines were transfected with the Smad-responsive CAGA-luc reporter construct, Renilla luciferase reporter, and wild type (WT) Smad3 expression vector, then treated with the indicated concentrations of CDK2 inhibitor (CDK2i) or CDK4 inhibitor (CDK4i). Firefly and Renilla luciferase activities were determined. Data are shown as fold increase in normalized luciferase activity (firefly/Renilla) compared with empty vector-transfected MCF7 or T47D cells. Error bars indicate standard deviation from the mean of normalized luciferase activity for each study condition.
To further examine the combined impact of CDK4 inhibition and abrogation of CDK phosphorylation of Smad3 on Smad3 transcriptional activity, MCF7 (Figure 6A), T47D (Figure 6B), and Hs578T (Figure 6C) parental, vector control, and cyclin D1-overexpressing cells were transfected with Smad3 WT or Smad3 mutant constructs, the CAGA-luc reporter, and the pRenilla reporter, and then treated with vehicle or 800 nM CDK4 inhibitor. As seen previously, when compared with WT Smad3, transfection with the Smad3 T178, Smad3 T8, or Smad3 5M CDK phosphorylation site mutants resulted in higher CAGA-luc reporter activity. Cells transfected with the Smad3 T178 or Smad3 T8 mutants and treated with CDK4 inhibitor further increased CAGA-luc reporter activity, including the cyclin D1-overexpressing cell lines. Interestingly, transfection with the Smad3 5M mutant and treatment with the CDK4 inhibitor resulted in decreased CAGA-luc reporter activity in MCF7 and Hs578T cells and abolished CAGA-luc reporter activity in T47D cells. This finding suggests that some amount of CDK4 activity may be required for Smad3 signaling.
FIGURE 6.
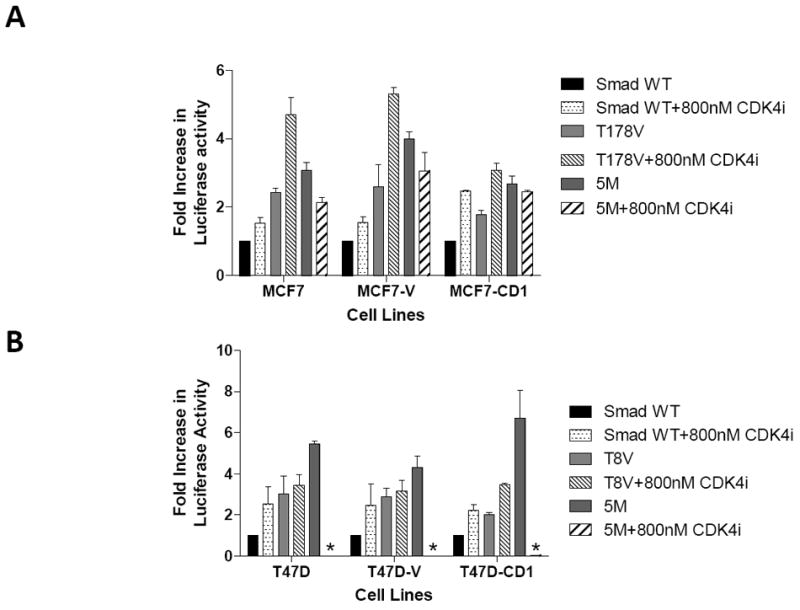
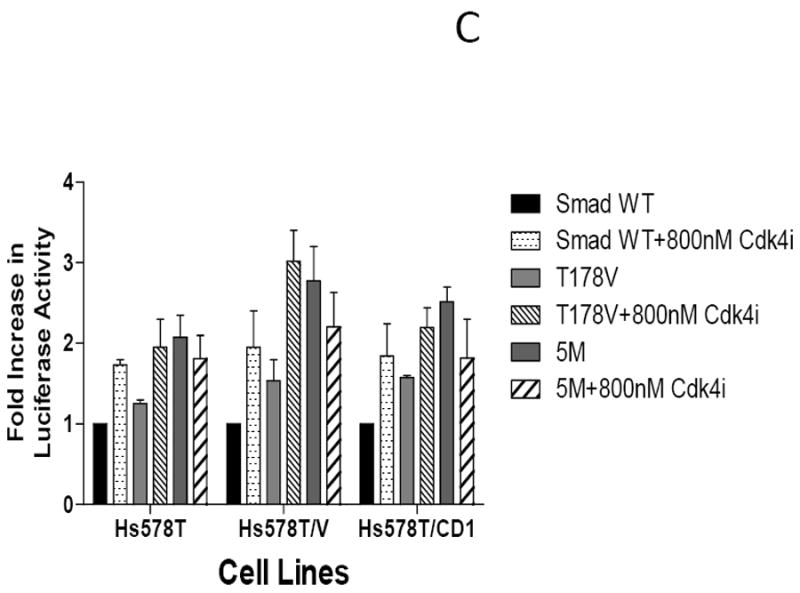
Restoration of Smad3 transcriptional activity with inhibition of CDK4 activity. MCF7 (A), T47D (B), and Hs578T (C) study cell lines were transfected with the Smad-responsive CAGA-luc reporter construct, Renilla luciferase reporter, and either wild type (WT) Smad3, 5M Smad3, T178 Smad3 (MCF7 and Hs578T), or T8 Smad3 (T47D only) expression vectors. The cells were treated with vehicle or 800 nM CDK4 inhibitor (CDK4i). Firefly and Renilla luciferase activities were determined. Data are shown as fold increase in normalized luciferase activity (firefly/Renilla) compared with empty vector-transfected MCF7, T47D, or Hs578T cells. Error bars indicate standard deviation from the mean of normalized luciferase activity for each study condition. For Figure 6B * denotes value too low to graphically represent.
siRNA knockdown of CDK4 restores Smad3 activity in MCF7 cell lines
To further explore the impact of CDK4 on Smad3 transcriptional activity, MCF7 study cells were transfected with scrambled or cdk4-specific siRNA. The effect of CDK4 inhibition was similar in both sets of study cells; thus, MCF7 cells were selected for subsequent knockdown experiments. MCF7 cells transfected with cdk4 siRNA demonstrated a 70% decrease in CDK4 levels compared with cells treated with a control vector or scrambled siRNA, thus showing the specificity of the cdk4 siRNA knockdown (Figure 7A). Subsequent co-transfection of the MCF7 study panel of cells with the cdk4 siRNA, WT Smad3, and the Smad3-responsive CAGA-luc reporter resulted in increased reporter activity in the MCF7 parental and vector control cells (Figure 7B). This increase was not seen in the cyclin D-overexpressing MCF7-CD1 cells, where transfection of cdk4 siRNA was not sufficient to enhance Smad3 activity. Co-transfection of the study cells with cdk4 siRNA and the Smad3 T178 CDK phosphorylation site mutant also resulted in an increase in CAGA-luc reporter activity above that seen in MCF7 parental and vector control cells containing control vector or scrambled siRNA. Though less pronounced, an increase in reporter activity was also seen in the MCF7-CD1 cells co-transfected with cdk4 siRNA and the Smad3 178V construct, suggesting that the high cyclin D1 levels in these cells maintained suppression of Smad3 activity. These data support a direct role of CDK4 in the inhibition of Smad3 action in breast cancer cells.
FIGURE 7.
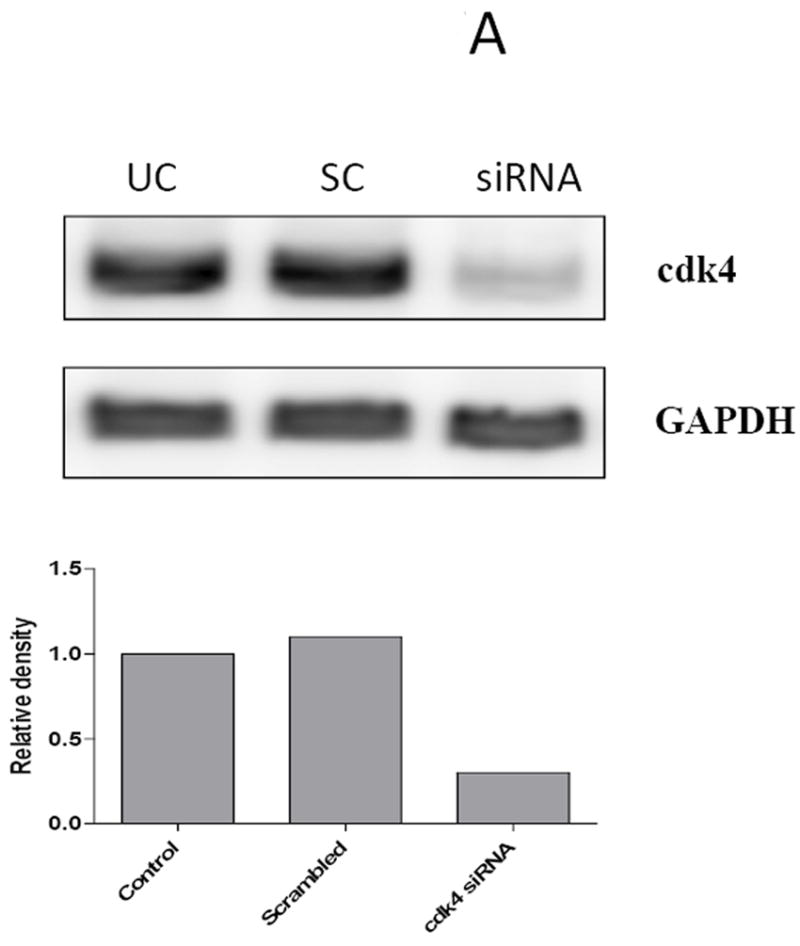
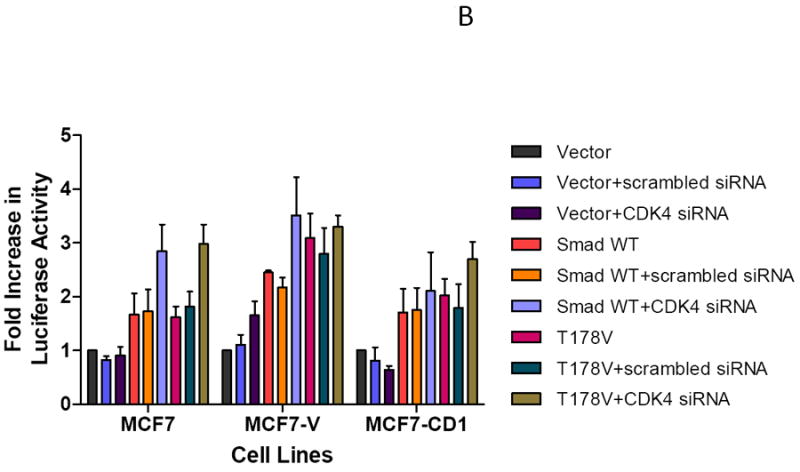
Restoration of Smad3 transcriptional activity with siRNA knockdown of CDK4. (A) MCF7 cells were transfected with scrambled (SC) or cdk4 siRNA (siRNA) for 48 hours, then the cells were lysed and the level of CDK4 protein was determined in untransfected cells (UC) and transfected cells by immunoblot analysis using anti-CDK4 antibody. GAPDH was used as loading control. (B) MCF7 study cell lines were co-transfected with the Smad-responsive CAGA-luc reporter construct and Renilla luciferase reporter; siRNA or cdk4 siRNA; and either empty vector, wild type (WT) Smad3, or T178 Smad3 mutant expression vectors. Firefly and Renilla luciferase activities were determined. Data are shown as fold increase in normalized luciferase activity (firefly/Renilla) compared with empty vector-transfected MCF7 or T47D cells. Error bars indicate standard deviation from the mean of normalized luciferase activity for each study condition.
Smad3 overexpression and CDK4 inhibition increase G1 cell cycle distribution
To study the impact of CDK4 inhibition on Smad3 on cell cycle control, the MCF7 study cell lines were transfected with either WT Smad3 or the Smad3 T178 CDK phosphorylation site mutant and treated with CDK4 inhibitor or vehicle alone. T47D study cell lines were transfected with either WT Smad3 or the Smad3 T8 mutant and treated with CDK4 inhibitor or vehicle control. Cells were harvested and cell cycle profiles were assessed. MCF7 parental cells had a baseline G1 population of 82.2% (Figure 8A). This increased to 88.6% after treatment with CDK4 inhibitor, irrespective of WT Smad3 or Smad3 T178 overexpression. By comparison, MCF7-CD1 cells had a lower baseline G1 population (55.1%) compared with the parental line, and had the highest number of cells in the G1 population upon Smad3 T178 mutant overexpression and treatment with CDK4 inhibitor (79.0%).
FIGURE 8.
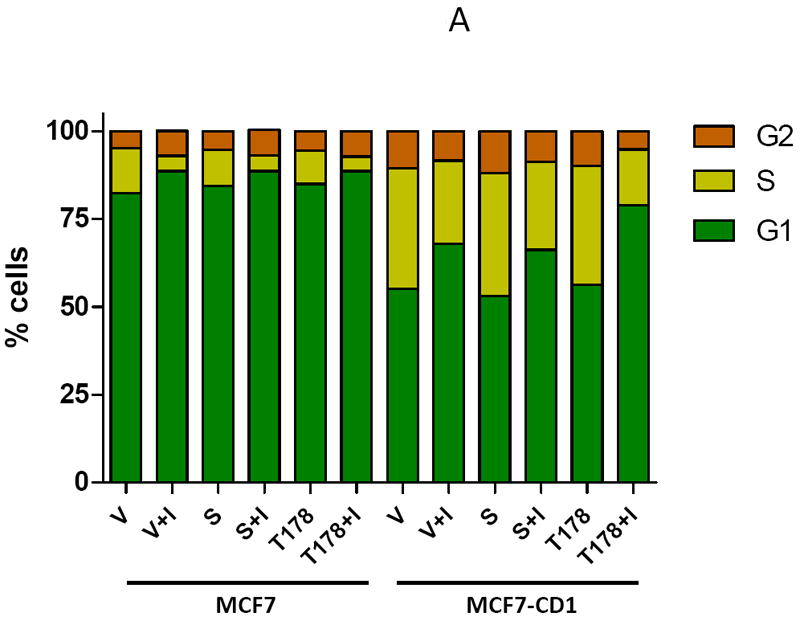
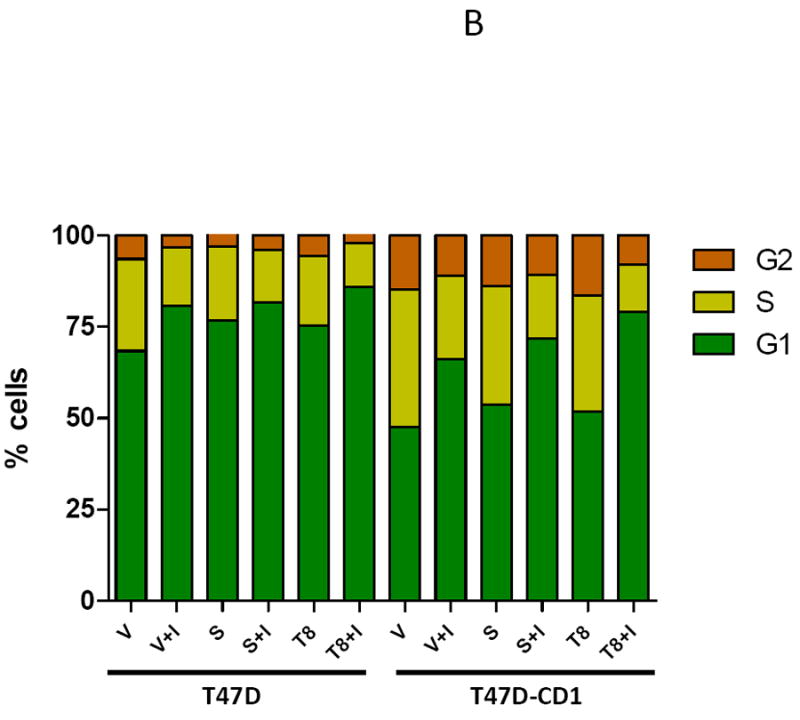
Inhibition of CDK4-mediated phosphorylation of Smad3 leads to G1 arrest in MCF7 parental and cyclin D-overexpressing cell lines. (A) MCF7 and MCF7-CD1 cells were transfected with empty vector (V), wild type Smad3 (S), or the T178 Smad3 mutant expression vectors. (B) T47D and T47D-CD1 cells were transfected with empty vector (V), wild type Smad3 (S), or the T8 Smad3 mutant expression vectors. The cells were then treated with vehicle or CDK4 inhibitor (I). Cells were harvested and the cell cycle profile was assessed by FACS analysis. Figures show one representative experiment from 3 separate data sets.
Parental T47D cells had a baseline G1 population of 68.4%, which increased to 85.8% in T47D cells overexpressing the Smad3 T8 mutant and treated with the CDK4 inhibitor (Figure 8B). T47D-CD1 cells had a baseline G1 population of 47.6% as compared to 79.0% for T47D-CD1 cells transfected with the Smad3 T8 mutant and treated with the CDK4 inhibitor. These data support the hypothesis that inhibition of CDK4-mediated phosphorylation of Smad3, by eliminating the Smad3 phosphorylation sites and through treatment with CDK4 inhibitor, restores Smad3 activity and cell cycle control in breast cancer cells, including those overexpressing cyclin D1.
Discussion
The goal of this work was to examine the impact of cyclin D1 overexpression and CDK4 phosphorylation on Smad3 activity in breast cancer cells. A correlation between CDK4 inhibition and restoration of Smad3 function was established. Overexpression of cyclin D1 repressed WT Smad3 transcriptional activity, and this activity was rescued with the overexpression Smad3 containing single or multisite mutations at CDK phosphorylation sites. Direct inhibition of CDK4 also restored Smad3 transcriptional activity, and this effect was further augmented by overexpressing Smad3 containing mutated CDK phosphorylation sites. Transfection of cyclin D1-overexpressing breast cancer cells with Smad3 T178 and T8 CDK phosphorylation site mutants and treatment with a CDK4 inhibitor resulted in a significant increase in the G1 cell cycle population. Taken together, these data support our hypothesis that cell cycle control is regulated, in part, by Smad3, and that the growth suppressive effect of Smad3 is lost upon CDK4 phosphorylation of Smad3 in cyclin D1-overexpressing breast cancer cells.
Previous work has examined the role of Smad3 in cell cycle regulation. Smad3, acting in conjunction with both cytoplasmic and nuclear co-factors, has been linked to the transcription of CDK inhibitors p15 and p21 and the inhibition of mitogenic c-myc in several different cell lines, including breast cancer cells (7, 8, 28). Furthermore, Smad3 overexpression in T47D breast cancer cells was shown to induce cell cycle arrest and expression of the CDK inhibitor p15, as well as repress cyclin A expression and Rb phosphorylation (29). The current study adds to the mechanistic understanding of these observations through the examination of Smad3 constructs with CDK mutation sites in breast cancer cell lines overexpressing cyclin D1. Notably, in the parental cells used in the study, c-myc transcript levels decreased after transfection with WT Smad3 and this decrease was significantly augmented by transfection of the 5M Smad3 construct. In the study cells overexpressing cyclin D1, the impact of transfection of the 5M Smad3 construct on c-myc repression and p15 transcription was even more pronounced. This finding supports a repressive effect of cyclin D1 overexpression on Smad3 action in the breast cancer cell lines, and also confirms previous findings of the role of Smad3 in the regulation of c-myc repression and p15 transcription (10, 30).
In contrast to p15 expression, p21 transcript levels were not affected by overexpression of the 5M Smad3 CDK phosphorylation site mutant in cyclin D1-overexpressing breast cancer cells in our study. While both TGFβ and BMP ligand subgroups within the TGFβ superfamily facilitate p21 transcription, BMP-related Smad signaling is associated with higher p21 expression levels than signaling through TGFβ/activin (31). On the other hand, while the BMP-specific Smads induce p21 mRNA and protein expression, this induction does not result in epithelial growth inhibition as seen with TGFβ/activin-specific Smad signaling (31). Importantly, these data suggest that while transcription of p21 is associated with several members of the TGFβ superfamily, additional co-factors or signaling targets must also be involved to facilitate cell growth arrest (31). Serum response factor (SRF) has recently been associated with cancer progression and metastasis, specifically as an inhibitor of Smad3-mediated transcription of p15 and p21 (32). It has been proposed that SRF acts at the nuclear level through association with the Smad3/4 complex to inhibit binding to and transcription of Smad-responsive genes (32). Potentially, the presence or absence of SRF in certain breast cancer settings may contribute to the loss of Smad-mediated cell cycle control potentiated by CDK phosphorylation. The role of additional co-factors involved in cross-talk with TGFβ/activin-specific Smad signaling to regulate cell cycle entry will be examined in future studies.
Of note, Smad3 activity was restored most effectively with overexpression of the 5M Smad3 CDK phosphorylation site mutant in the MCF7-CD1 and Hs578T-CD1 cell lines, and by overexpression of the 4M or 5M Smad3 mutants in T47D-CD1 cells. Thus, cyclin D/CDK-mediated Smad3 repression is most effectively overcome through mutation of most or all CDK phosphorylation sites within the protein. In light of these findings, CDK inhibitors that block CDK2 and CDK4 phosphorylation at the Smad3 mutation sites were examined for their ability to restore Smad3 action. In both MCF7 and T47D study cell lines, CDK2 inhibition resulted in a slight, if any, increase in Smad3 activity above that seen with overexpression of WT Smad3, while CDK4 inhibition resulted in a significant increase in Smad3 transcriptional activity. Thus, CDK2 phosphorylation appears to play a minor role in Smad3 inhibition in MCF7 cells and no appreciable role in T47D cells. Our subsequent experiments silencing CDK4 activity, either through overexpression of Smad3 mutants containing phosphorylation site mutations or through treatment with CDK4 inhibitor, confirmed the impact of CDK4 inhibition on restoring Smad3 transcriptional activity. We expected that maximal inhibition of the CDK4 effect on Smad3 activity would be achieved with transfection of the 5M Smad3 mutant in addition to treatment with a CDK4 inhibitor. However, this led to reporter activity that was slightly lower than that seen in MCF7-CD1 and Hs578T-CD1 cells transfected with the 5M construct alone, and to near complete abrogation of reporter activity in the T47D-CD1 cells.
The transcriptional activity of Smad3 is modulated by the binding of co-factors like p300/CBP (33). Competitive binding of p300/CBP between Stat1α a component of the Jak-Stat pathway, and Smad3 has been described as a mechanism involved in regulation of collagen transcription in fibroblasts (34). In conjunction with p300/CBP, the PIAS3 protein, from the Protein Inhibitor of Activated Stat family, may also modulate TGFβ-mediated Smad3 transcriptional activity (35). p300/CBP binding to Smad3 takes place within the linker region of the Smad3 molecule and may be affected by CDK phosphorylation in this region (36). Likewise, the presence or absence of CDK phosphorylation within the Smad3 linker region may affect the capacity for binding of Smad3 to various co-repressors or co-activators, including Stat1α or PIAS3; this may represent a mechanism for cyclin-mediated phosphorylation inhibition of Smad3 activity (19). On the other hand, p300/CBP binding to Smad3 may require some level of CDK activity in T47D cells, which may explain why Smad3 transcriptional activity was completely abolished by overexpression of the 5M Smad3 mutant and treatment with CDK4 inhibitor. Recent data from myeloma cells found that phosphorylation of Smad2 at the T8 site by CDK2 prevented Smad2 from linking with the co-Smad, Smad4, thus inhibiting Smad-dependent transcriptional activity (37). In our breast cancer cell studies, however, mutation of the T8 site proved significant for restoring Smad3 activity in T47D cells, though less so in MCF7 and Hs578T cells. At minimum, these data point toward a dual significance of CDK phosphorylation of Smads in different malignant settings and the potential for certain CDK sites to be critical for Smad interaction with co-factors and co-regulators important for transcriptional activity (37).
Previous work has shown that the mean tumor size in patients with moderate to intense nuclear Smad3 breast cancer tissue staining was almost 1.5 cm smaller than those patients with low or no nuclear Smad3 staining (17). The correlation between nuclear Smad3 and smaller tumor size implicates activated and functional nuclear Smad3 in the inhibition of breast tumor cell proliferation. Tumor size, a component of the American Joint Committee on Cancer Staging System for breast cancer, is one of the most powerful predictors of breast cancer prognosis, with smaller tumors having a more favorable outcome. As nuclear Smad3 staining intensity has shown an independently significant correlation with smaller tumor size, it may also have favorable prognostic significance (17). Our data support the use of Smad3 as a prognostic marker in conjunction with cyclin expression, as tumors with activated nuclear Smad3, but with high cyclin activity, may have lost Smad3-mediated cell cycle control as a consequence of CDK phosphorylation. Tian et al have demonstrated in a mouse model that tumors formed from MCF-10A cells overexpressing Smad3 grew more slowly than MCF-10A cells overexpressing a Smad3 dominant negative mutant (16). Likewise, the current study demonstrated that breast cancer cells overexpressing Smad3 with CDK phosphorylation site mutations could restore cell cycle control and G1 arrest, even in the presence of high levels of cyclin D1. The significance of these findings further reveal how Smad3-mediated cell cycle control can be undermined in breast cancers that overexpress cyclin D and restored by blockade of Smad3 CDK phosphorylation.
The possibility of targeting CDKs for therapeutic benefit is being pursued. Three classes of CDK inhibitors have been designed: agents that affect several CDKs simultaneously, agents that target individual CDKs, and agents that inhibit both CDK activity as well as other kinase targets (38). Phase 1 and 2 trials have been pursued to study the impact of CDK2 and CDK4 inhibitors, such as flavopiridol, which blocks CDKs 1, 2, 4, 6, 7, and 9, on solid and hematogenous malignancies (39, 40). Early results from breast cancer studies examining the impact of flavopiridol in combination with trastuzumab, histone deacetylase inhibitors, and docetaxel appear promising, with the treatment regimen leading to decreases in tumor cell proliferation and tumor size (39, 41, 42). Additionally, work on the blockade of AP-1 in MCF7 breast cancer cells has resulted in cell growth arrest in the G1 phase of the cell cycle and was associated with a reduced expression of cyclins D and E, decreased CDK2/4 activity and increased p27 expression (43). These data point toward a potential role for targeted CDK inhibitors in breast cancer therapy (43). To date, these agents have not been studied in the context of cyclin-overexpressing breast cancers or Smad3 signaling. In this work, treatment of cyclin D1-overexpressing cells with a CDK4 inhibitor resulted in a dose-dependent increase in Smad3 transcriptional activity; in this setting, CDK4 inhibitor treatment appeared to be more effective at restoring Smad3 activity than CDK2 inhibitor treatment. This finding supports a more selective approach to the development of therapeutic CDK inhibitors to maximize treatment effectiveness and minimize co-morbidity.
This study focuses on the impact of cyclin D1 overexpression on Smad3 action in breast carcinogenesis. Breast cancers that overexpress cyclins have been associated with poor prognosis (44-46). A clear understanding of the mechanisms underlying Smad3-mediated cell cycle arrest is crucial to efforts aimed at preventing or interrupting oncogenic insults to the Smad3 signaling pathway that favor breast cancer growth and dedifferentiation. Furthermore, knowledge of the mechanisms responsible for cell cycle deregulation characteristic of cancers with high cyclin activity are critical to the discovery of improved treatment strategies for the patients with these tumors. These efforts will ultimately allow for the further development of individualized prognostic markers to help actualize patient-specific treatment strategies. Specifically, strategies that target inhibition of CDKs or block CDK/cyclin activity may hold promise for patients with cyclin-overexpressing breast cancers that currently have a disease sub-type with a poor prognosis.
Materials and Methods
Cell Culture and Antibodies
MCF7, Hs578T, and T47D cells were obtained from the American Type Culture Collection (Manassas, VA) and were maintained in DMEM-F12, DMEM, or RPMI medium, respectively. Culture media was supplemented with antibiotics, 10% FBS, and 0.01 mg/ml bovine insulin (Hs578T cells only). The study cells stably expressing empty vector or cyclin D1 were maintained in the same fashion as the parental lines. Anti-cyclin D1, anti-CDK2, anti-CDK4, anti-GAPDH, and anti-β-actin antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); anti-Smad3 and anti-phospho-Smad antibodies were purchased from Cell Signaling Technology (Danvers, MA).
Generation of Stable Cell Lines
Human cyclin D1 cDNA was inserted into the cytomegalovirus-driven pcDNA-DEST40 expression vector (Invitrogen, Carlsbad, CA) containing a neomycine resistance gene to obtain the pEXPR-Cyclin D1 expressing plasmid. To create the vector control (V) and cyclin D1-overexpressing (CD1) cell lines, cells were stably transfected with empty vector (pcDNA-DEST40) or pEXPR-Cyclin D1 plasmids. Two days post-transfection, stable clones were selected in media containing 500 mg/mL of G418 (Invitrogen). The resistant clones were maintained in G418 for 21 days. Cyclin D1 overexpression was verified by immunoblot analysis as shown in Figure 1A.
Immunoblotting
Briefly, cells were scraped, pelleted, and rinsed with ice-cold HEPES-buffered saline (pH 7.0), then lysed in an ice-cold cell lysis buffer containing protease and phosphatase inhibitors. Cellular lysates were spun, supernatants were recovered, and the total protein concentration was determined by protein assay (Bio-Rad, Hercules, CA). For immunoblotting, 30 μg or 50 μg total lysate in 2x SDS-PAGE sample buffer (1:1, v/v) was electrophoresed and transferred to a nitrocellulose membrane. Membranes were incubated with the appropriate primary antibody in TBS-T (pH 7.5) and 5% skim milk or 5% BSA at 4°C overnight. After rising, the membrane was incubated with secondary antibody in TBS-T buffer for 1 hour. Protein bands were visualized by SuperSignal (Thermo Scientific, Rockford, IL). For reblotting, filters were agitated with the stripping buffer (62.5 mM Tris pH 6.8, 2% SDS, 100 mM β-mercaptoethanol) for 30 minutes at 60°C. After 2 washes with TBS-T buffer, filters were treated with specific primary antibody followed by the corresponding secondary antibody, and the signal was detected by SuperSignal (Thermo Scientific). All immunoblot experiments were conducted 3 times, and representative results are shown.
Immunofluorescence
Subcellular localization of Smad3 and phospho-Smad3 in parental and cyclin D1 overexpressing cells was determined by indirect immunofluorescence using an anti-Smad3 or anti-phospho-Smad3 antibody. After fixation with 4% paraformaldehyde, slides were incubated with anti-Smad3 antibody at 4°C for 16 hours. Slides were washed 3 times with TNT buffer (0.1 M Tris-HCl, pH 7.5, 0.15 M NaCl, 0.05% Tween 20). After 30 minutes of incubation at room temperature with secondary anti-rabbit-HRP conjugated antibody, slides were incubated with Fluorophore Tyramide (PerkinElmer, Boston, MA) for 10 minutes. After mounting the slides with Vectashield Mounting Medium (Vector Laboratories, Inc., Burlingame, CA) containing a DAPI nuclear stain, the cells were observed with a Leica microscope (Leica Microsystems, Inc, Bannockburn, IL) equipped with an oil immersion 40x lens. Digital images were collected using filters for fluorescein and DAPI.
CDK Kinase Assays
Cell extracts were obtained by lysing cells in lysis buffer [50 mM HEPES pH 7.0, 250 mM NaCl, 5 mM EDTA pH 8.0, 0.5% NP-40, 1 mM PMSF, 1 mM sodium vanadate, 1X Halt Protease Inhibitor Cocktail (Pierce, Rockford, IL)] on ice for 30 minutes with occasional agitation. Cell debris was removed by centrifugation at 14,000 g for 10 minutes at 4°C. Protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad). 50 μg of protein lysate was incubated with 2 μg of anti-CDK4 or anti-CDK2 IgG (Santa Cruz, CA) at 4°C for 1 hour followed by immunoprecipitation with protein A/G plus agarose conjugate (Santa Cruz) at 4°C overnight. Beads were washed 3 times with kinase buffer (8 mM MOPS pH 7.0, 0.2 mM EDTA). The CDK4 or CDK2 kinase reactions were performed at 30°C for 30 minutes in kinase buffer containing 2 μg recombinant Rb protein or Histone H1 (Millipore, Billerica, MA) as substrate for CDK4 and CDK2 respectively, and 2 μCi of 32P- γATP. The reaction was stopped by adding 5x SDS loading buffer and boiled for 5 minutes before loading on a 10% SDS-PAGE gel. The gel was then exposed to a phosphor screen for 24 hours and analyzed using a Storm 860 Molecular Imager (GE Healthcare, Piscataway, NJ). Quantitative analysis was performed with MultiGauge software (FujiFilm, Valhalla, NY).
Transfection and Luciferase Assays
Smad3 expression plasmids have been described previously and contain individual site mutations (T8, T178, S203, S207 or S212) or multiple site mutations [3M (T8/T178/S212), 4M (T178/S203/S207/S212), 5M (T8/T178/S203/S207/S212)] (18). The Smad3 mutants were a gift from Dr. Fang Liu (Rutgers, State University of New Jersey, Piscataway, NJ). DNA transfection experiments were performed using Lipofectamine 2000 (Invitrogen). Approximately 5.5×104 cells were seeded into 48-well plates 24 hours before transfection. 200 ng of a Smad3-responsive promoter firefly luciferase-reporter construct (CAGA-luc) and 10 ng of a control reporter construct (pRenilla) were co-transfected with 200 ng of Smad expression plasmid. The cells were lysed with passive lysis buffer (Promega, Madison, WI) for dual-luciferase assay 48 hours post-transfection. Luciferase activity was measured by a Synergy2 luminometer (BioTeck Instruments Inc, Winooski, Vermont) and firefly luciferase activity was normalized to Renilla luciferase activity. Representative means ± SD were obtained from 3 independent experiments.
Real-time RT-PCR
Levels of mRNA for c-myc, p15INK4, and p21 were determined by real-time quantitative RT-PCR using PerfeCTa SYBR Green Fast Mix (Quanta Bioscience, Gaithersburg, MD). Total RNA was isolated from cells using the RNeasy mini kit (Qiagen, Valencia, CA). Amplification of the samples (1 μg of total RNA per reaction) was carried out using qScript cDNA Super Mix (Quanta Bioscience, Gaithersburg, MD). All amplifications were run on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) using the following conditions: initial denaturation, 95°C for 15 minutes, followed by 40 cycles at 95°C for 1 second and 60°C for 30 seconds. Gene expression was analyzed in terms of the threshold cycle (Ct) normalized to GAPDH (ΔCt). ΔCt values were then compared between control samples and transfected cells to calculate ΔΔCt. Final comparison of transcript ratios between samples was given as 2-ΔΔCt. Representative means ± SD were obtained from 3 replicates of 3 independent experiments.
RNA Interference
Transfection of cdk4 siRNA (ThermoScientific, Waltham, MA) was carried out 24 hours after plating cells in a 96-well plate. Cells were incubated overnight with the transfection mix containing 12.5 pM siRNA and 0.25 μl DharmaFECT Transfection Reagent (ThermoScientific) in Opti-MEM medium (Invitrogen). Transfection of reporter constructs and Smad expression plasmids were carried out as described above. The cells were lysed with passive lysis buffer (Promega, Madison, WI) for dual-luciferase assay at 48 hours post-transfection. Luciferase activities were measured as described above. Means ± SD were obtained from 3 replicates of a representative experiment.
Treatment with CDK Inhibitors
After transfection, the cells were placed in media and treated with CDK4 inhibitor II (Catalog #219477; IC50 value of 200 nM) or CDK2 inhibitor II [Catalog #219445; IC50 value of 60 nM; (Calbiochem, San Diego, CA)] for 48 hours. Based on the IC50 for each inhibitor, the concentrations of the CDK2 and CDK4 inhibitors used in our proposed studies were selected to induce specific CDK2 and CDK4 inhibition. Control cells were maintained in complete medium and were treated with solvent alone. All experiments were done in triplicate.
FACS and Cell Cycle Analysis
For flow cytometric analysis, cells were plated at a density of 8 × 105 cells per well in 6-well plates. Cells were harvested 48 hours after transfection, washed once with PBS, fixed in 70% cold ethanol, and incubated for 30 minutes at 4°C in PBS containing 100 μg/ml of RNase A and 50 μg/ml of propidium iodide. Cells were loaded into a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ) for cell cycle analysis. Flow cytomeric analysis was done for at least 20,000 individual events per reaction. Distribution of cells in the 3 phases of the cell cycle is represented by percentage. Representative results reflect the trends observed in 3 separate experiments.
Statistical Analysis
Statistical procedures for p-value analysis were performed by Student’s t-test with 2 populations to determine the significance of comparisons. P ≤ 0.05 was considered statistically significant.
Acknowledgments
We thank Dr. Fang Liu (Rutgers, State University of New Jersey, Piscataway, NJ) for providing the mutant Smad3 constructs.
Support: JSJ is a Lynn Sage Scholar supported by the American Cancer Society-Illinois Division, the Dixon Foundation, and an NIH KL2 research grant.
References
- 1.Brown KA, Pietenpol JA, Moses HL. A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-beta signaling. J Cell Biochem. 2007;101(1):9–33. doi: 10.1002/jcb.21255. [DOI] [PubMed] [Google Scholar]
- 2.Yue J, Mulder KM. Transforming growth factor-beta signal transduction in epithelial cells. Pharmacol Ther. 2001;91(1):1–34. doi: 10.1016/s0163-7258(01)00143-7. [DOI] [PubMed] [Google Scholar]
- 3.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–91. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 4.Nakao A, Imamura T, Souchelnytskyi S, et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. Embo J. 1997;16(17):5353–62. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuura I, Chiang KN, Lai CY, et al. Pin1 promotes TGF-beta-induced migration and invasion. J Biol Chem. 2009 doi: 10.1074/jbc.M109.063826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumont N, Arteaga CL. Transforming growth factor-beta and breast cancer: Tumor promoting effects of transforming growth factor-beta. Breast Cancer Res. 2000;2(2):125–32. doi: 10.1186/bcr44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110(1):19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 8.Feng XH, Liang YY, Liang M, Zhai W, Lin X. Direct interaction of c-Myc with Smad2 and Smad3 to inhibit TGF-beta-mediated induction of the CDK inhibitor p15(Ink4B) Mol Cell. 2002;9(1):133–43. doi: 10.1016/s1097-2765(01)00430-0. [DOI] [PubMed] [Google Scholar]
- 9.Pardali K, Kurisaki A, Moren A, ten Dijke P, Kardassis D, Moustakas A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-beta. J Biol Chem. 2000;275(38):29244–56. doi: 10.1074/jbc.M909467199. [DOI] [PubMed] [Google Scholar]
- 10.Millet C, Zhang YE. Roles of Smad3 in TGF-beta signaling during carcinogenesis. Crit Rev Eukaryot Gene Expr. 2007;17(4):281–93. doi: 10.1615/critreveukargeneexpr.v17.i4.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol Lett. 2002;82(1-2):85–91. doi: 10.1016/s0165-2478(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 12.Jeruss JS, Santiago JY, Woodruff TK. Localization of activin and inhibin subunits, receptors and SMADs in the mouse mammary gland. Mol Cell Endocrinol. 2003;203(1-2):185–96. doi: 10.1016/s0303-7207(02)00291-5. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen AV, Pollard JW. Transforming growth factor beta3 induces cell death during the first stage of mammary gland involution. Development. 2000;127(14):3107–18. doi: 10.1242/dev.127.14.3107. [DOI] [PubMed] [Google Scholar]
- 14.Robinson SD, Silberstein GB, Roberts AB, Flanders KC, Daniel CW. Regulated expression and growth inhibitory effects of transforming growth factor-beta isoforms in mouse mammary gland development. Development. 1991;113(3):867–78. doi: 10.1242/dev.113.3.867. [DOI] [PubMed] [Google Scholar]
- 15.Xie W, Mertens JC, Reiss DJ, et al. Alterations of Smad signaling in human breast carcinoma are associated with poor outcome: a tissue microarray study. Cancer Res. 2002;62(2):497–505. [PubMed] [Google Scholar]
- 16.Tian F, DaCosta Byfield S, Parks WT, et al. Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res. 2003;63(23):8284–92. [PubMed] [Google Scholar]
- 17.Jeruss JS, Sturgis CD, Rademaker AW, Woodruff TK. Down-regulation of activin, activin receptors, and Smads in high-grade breast cancer. Cancer Res. 2003;63(13):3783–90. [PubMed] [Google Scholar]
- 18.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430(6996):226–31. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 19.Liu F. Smad3 phosphorylation by cyclin-dependent kinases. Cytokine Growth Factor Rev. 2006;17(1-2):9–17. doi: 10.1016/j.cytogfr.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 20.Ewen ME, Lamb J. The activities of cyclin D1 that drive tumorigenesis. Trends Mol Med. 2004;10(4):158–62. doi: 10.1016/j.molmed.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 21.Tashiro E, Tsuchiya A, Imoto M. Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression. Cancer Sci. 2007;98(5):629–35. doi: 10.1111/j.1349-7006.2007.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy PG, Thompson AM. Cyclin D1 and breast cancer. Breast. 2006;15(6):718–27. doi: 10.1016/j.breast.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411(6841):1017–21. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- 24.Honrado E, Benitez J, Palacios J. The molecular pathology of hereditary breast cancer: genetic testing and therapeutic implications. Mod Pathol. 2005;18(10):1305–20. doi: 10.1038/modpathol.3800453. [DOI] [PubMed] [Google Scholar]
- 25.Preobrazhenska O, Yakymovych M, Kanamoto T, et al. BRCA2 and Smad3 synergize in regulation of gene transcription. Oncogene. 2002;21(36):5660–4. doi: 10.1038/sj.onc.1205732. [DOI] [PubMed] [Google Scholar]
- 26.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–66. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 27.Kalkhoven E, Roelen BA, de Winter JP, et al. Resistance to transforming growth factor beta and activin due to reduced receptor expression in human breast tumor cell lines. Cell Growth Differ. 1995;6(9):1151–61. [PubMed] [Google Scholar]
- 28.Chen CR, Kang Y, Massague J. Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program. Proc Natl Acad Sci U S A. 2001;98(3):992–9. doi: 10.1073/pnas.98.3.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burdette JE, Jeruss JS, Kurley SJ, Lee EJ, Woodruff TK. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res. 2005;65(17):7968–75. doi: 10.1158/0008-5472.CAN-04-3553. [DOI] [PubMed] [Google Scholar]
- 30.Liu F, Matsuura I. Inhibition of Smad antiproliferative function by CDK phosphorylation. Cell Cycle. 2005;4(1):63–6. doi: 10.4161/cc.4.1.1366. [DOI] [PubMed] [Google Scholar]
- 31.Pardali K, Kowanetz M, Heldin CH, Moustakas A. Smad pathway-specific transcriptional regulation of the cell cycle inhibitor p21(WAF1/Cip1) J Cell Physiol. 2005;204(1):260–72. doi: 10.1002/jcp.20304. [DOI] [PubMed] [Google Scholar]
- 32.Lee HJ, Yun CH, Lim SH, et al. SRF is a nuclear repressor of Smad3-mediated TGF-beta signaling. Oncogene. 2007;26(2):173–85. doi: 10.1038/sj.onc.1209774. [DOI] [PubMed] [Google Scholar]
- 33.Inoue Y, Itoh Y, Abe K, et al. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene. 2007;26(4):500–8. doi: 10.1038/sj.onc.1209826. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh AK, Yuan W, Mori Y, Chen S, Varga J. Antagonistic regulation of type I collagen gene expression by interferon-gamma and transforming growth factor-beta. Integration at the level of p300/CBP transcriptional coactivators. J Biol Chem. 2001;276(14):11041–8. doi: 10.1074/jbc.M004709200. [DOI] [PubMed] [Google Scholar]
- 35.Long J, Wang G, Matsuura I, He D, Liu F. Activation of Smad transcriptional activity by protein inhibitor of activated STAT3 (PIAS3) Proc Natl Acad Sci U S A. 2004;101(1):99–104. doi: 10.1073/pnas.0307598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G, Long J, Matsuura I, He D, Liu F. The Smad3 linker region contains a transcriptional activation domain. Biochem J. 2005;386(Pt 1):29–34. doi: 10.1042/BJ20041820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baughn LB, Di Liberto M, Niesvizky R, et al. CDK2 phosphorylation of Smad2 disrupts TGF-beta transcriptional regulation in resistant primary bone marrow myeloma cells. J Immunol. 2009;182(4):1810–7. doi: 10.4049/jimmunol.0713726. [DOI] [PubMed] [Google Scholar]
- 38.Malumbres M, Pevarello P, Barbacid M, Bischoff JR. CDK inhibitors in cancer therapy: what is next? Trends Pharmacol Sci. 2008;29(1):16–21. doi: 10.1016/j.tips.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 39.Fornier MN, Rathkopf D, Shah M, et al. Phase I dose-finding study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors. Clin Cancer Res. 2007;13(19):5841–6. doi: 10.1158/1078-0432.CCR-07-1218. [DOI] [PubMed] [Google Scholar]
- 40.Lin TS, Ruppert AS, Johnson AJ, et al. Phase II Study of Flavopiridol in Relapsed Chronic Lymphocytic Leukemia Demonstrating High Response Rates in Genetically High-Risk Disease. J Clin Oncol. 2009 doi: 10.1200/JCO.2009.22.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nahta R, Trent S, Yang C, Schmidt EV. Epidermal growth factor receptor expression is a candidate target of the synergistic combination of trastuzumab and flavopiridol in breast cancer. Cancer Res. 2003;63(13):3626–31. [PubMed] [Google Scholar]
- 42.Mitchell C, Park MA, Zhang G, et al. Extrinsic pathway- and cathepsin-dependent induction of mitochondrial dysfunction are essential for synergistic flavopiridol and vorinostat lethality in breast cancer cells. Mol Cancer Ther. 2007;6(12 Pt 1):3101–12. doi: 10.1158/1535-7163.MCT-07-0561. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Lu C, Shen Q, Munoz-Medellin D, Kim H, Brown PH. AP-1 blockade in breast cancer cells causes cell cycle arrest by suppressing G1 cyclin expression and reducing cyclin-dependent kinase activity. Oncogene. 2004;23(50):8238–46. doi: 10.1038/sj.onc.1207889. [DOI] [PubMed] [Google Scholar]
- 44.Naidu R, Wahab NA, Yadav MM, Kutty MK. Expression and amplification of cyclin D1 in primary breast carcinomas: relationship with histopathological types and clinico-pathological parameters. Oncol Rep. 2002;9(2):409–16. [PubMed] [Google Scholar]
- 45.Keyomarsi K, Tucker SL, Buchholz TA, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med. 2002;347(20):1566–75. doi: 10.1056/NEJMoa021153. [DOI] [PubMed] [Google Scholar]
- 46.Yang C, Trent S, Ionescu-Tiba V, et al. Identification of cyclin D1- and estrogen-regulated genes contributing to breast carcinogenesis and progression. Cancer Res. 2006;66(24):11649–58. doi: 10.1158/0008-5472.CAN-06-1645. [DOI] [PubMed] [Google Scholar]