

Abstract
Adeno-associated virus is a promising vector for gene therapy. In the current study, the binding site on AAV serotype 3B for the heparan sulfate proteoglycan (HSPG) receptor has been characterized. X-ray diffraction identified a disaccharide binding site at the most positively charged region on the virus surface. The contributions of basic amino acids at this and other sites were characterized using site-directed mutagenesis. Both heparin and cell binding are correlated to positive charge at the disaccharide binding site, and transduction is significantly decreased in AAV-3B vectors mutated at this site to reduce heparin binding. While the receptor attachment sites of AAV-3B and AAV-2 are both in the general vicinity of the viral spikes, the exact amino acids that participate in electrostatic interactions are distinct. Diversity in the mechanisms of cell attachment by AAV serotypes will be important considerations for the rational design of improved gene therapy vectors.
Keywords: Adeno-associated virus, heparin, heparan sulfate, receptor
Adeno-associated virus (AAV) is a small (~25nm) parvovirus, with a ~4.7kb ssDNA genome packaged inside a non-enveloped capsid of T=1 icosahedral (60-fold) symmetry (Caspar and Klug, 1962; Xie et al., 2002). As a non-pathogenic virus, AAV has become a leading candidate vector for human gene therapy (Hildinger and Auricchio, 2004). Several naturally occurring serotypes of AAV have been identified, each having broad, but distinct tissue specificity (Buning, Braun-Falco, and Hallek, 2004; Mitchell et al., 2010). In addition to having broad tropism, AAV vectors are often neutralized in individuals previously exposed to virus or vector (Zaiss and Muruve, 2005). Knowledge of the structure and infectious pathway of AAV serotype capsids provides a template to engineer vectors that more specifically target diseased tissues, and to engineer neutralization escape variants that remain viable in cell entry (Buning et al., 2003; Flotte, 2004; Mitchell et al., 2010).
Prior to cell entry, AAV serotype 3 (AAV-3) attaches to target cells by binding heparan sulfate proteoglycan (HSPG) (Handa et al., 2000; Rabinowitz et al., 2002), but few details are known. In contrast, the binding site on AAV-2 for HSPG (or its analog heparin) has been well characterized, and is centered at Arg585 and Arg588 on the sides of the 3-fold proximal spikes (Kern et al., 2003; O’Donnell, Taylor, and Chapman, 2009; Opie et al., 2003). Intriguingly, these residues are not conserved in AAV-3, and the determinants of receptor binding by AAV-3 remain unknown.
An understanding of the diversity in AAV-heparin interactions will advance our fundamental understanding of receptor attachment. AAV-3 is of particular interest because of its ability to transduce hematopoietic cells (Handa et al., 2000) and liver cancer cells (Glushakova et al., 2009) relatively efficiently. However, AAV-3 transduction levels are low for most cell types (Van Vliet et al., 2008). For AAV-2, heparin binding correlates closely with tissue specificity (Asokan et al., 2010; Grimm et al., 2008). In addition, the heparin binding site on AAV-2 can be replaced with peptide ligands to efficiently re-target vectors to desired tissues (Perabo et al., 2006; Shi and Bartlett, 2003; Shi et al., 2006). Similarly, detailed knowledge of receptor interactions by this serotype could increase its therapeutic potential.
We recently determined the crystal structure of AAV-3B (Lerch, Xie, and Chapman, 2010), a minor variant of AAV-3. The overall capsid structure is similar to that of other AAV serotypes which all have spike-like protrusions surrounding the 3-fold axes. Despite the structural similarity, the electrostatic surface potential of AAV-3B is quite different from that of other serotypes in the region corresponding to the AAV-2 HSPG-binding site. This has functional implications, as HSPG and heparin are negatively charged and typically form ionic interactions with basic regions on the surface of heparan-binding proteins (Conrad, 1998). Two regions near the spikes that are positively charged and unique to AAV-3B (Lerch, Xie, and Chapman, 2010), could, we hypothesized, facilitate receptor interactions in AAV-3B.
In the current study, interactions between AAV-3B and heparan sulfate analogs were investigated. The location of the receptor binding site was determined from crystallographic data from a complex of AAV-3B and an HSPG analog. AAV-3B capsid mutants were then used to (1) confirm the structural identification of the heparin binding site, (2) correlate heparin and cell binding to positive charge on the capsid surface, and (3) demonstrate the requirement of the heparin binding site for cellular transduction.
Results
Prediction of potential heparin binding residues
From the 2.6Å crystal structure of AAV-3B (Lerch, Xie, and Chapman, 2010), candidate receptor binding sites were identified. Specifically, the electrostatic surface potential shows positively-charged regions near the 3-fold proximal spikes that are unique to AAV-3B (Figure 1A). Heparin binding proteins typically interact with their ligands through one or a cluster of basic residues. One of the positively charged regions is centered on Arg447. Arg447 is conserved in several other AAV serotypes, many of which do not bind heparin. In AAV-2, Arg447 is not involved in heparin interactions. In fact, its charge is neutralized by a salt-bridge with Glu499 (Figure 1B). In AAV-3B, Glu499 is replaced by Asn500, so the region is more positively charged. A second positively charged region is centered on Arg594, the only surface-exposed basic amino acid in AAV-3B that is not conserved in other serotypes. Three Arg594 residues from adjacent subunits cluster at the 3-fold icosahedral axis to form the region of strongest positive charge on the AAV-3B surface (Figure 1A).
Figure 1.

Identification of potential heparin binding residues on AAV-3B. Two views near the 3-fold promixal spikes of AAV-3B are shown. (A) The spike-like protrusions of AAV-3B viewed down a 3-fold symmetry axis. A ribbon representation of the AAV-3B capsid can be seen beneath the translucent molecular surface, which is colored by electrostatic surface potential (blue = positive; red = negative). Two regions near residues Arg447 and Arg594 have strong positive surface charge and were identified as candidate receptor binding sites. (B) Structural overlay of a single spike from AAV-3B (green) and AAV-2 (magenta). Arg447 is conserved in AAV-2, but forms a salt bridge with Glu499 (dashed line). Asn500 is the equivalent residue in AAV-3B and, as a neutral amino acid, does not pair with Arg447, leaving a stronger positive surface charge at this site. On the left, differences in the AAV-2 HSPG site are highlighted with Arg585 & Arg588 of AAV-2 replaced by Ser586 and Thr589 in AAV-3B.
Crystallographic identification of a receptor binding locus on AAV-3B
Diffraction data were collected from AAV-3B crystals grown in the presence of the heparin analog sucrose octasulfate (SOS). SOS has been used previously in structural studies of heparin-binding proteins (Innis and Hyvonen, 2003). The best crystal diffracted X-rays to 6.5Å resolution and belongs to space group F4132. Resolution of 6.5Å is not sufficient to build atomic models, but it suffices to identify a ligand-binding site on a structure determined at 2.6Å resolution, especially with the high quality maps available following 5-fold non-crystallographic symmetry averaging (Badger et al., 1988). The 2.6Å native AAV-3B structure (Lerch, Xie, and Chapman, 2010) could be superimposed accurately on the SOS complex map by alignment of their icosahedral symmetry axes with no degrees of freedom (see Materials and Methods). In fact, without any atomic refinement (which would be susceptible to over-fitting at 6.5Å resolution), the native AAV-3B structure yields Rcryst/Rfree of 0.27/0.28 when compared to the diffraction data of the SOS complex (Table 1).
Table 1.
Summary of diffraction data processing and refinement statistics
| Parameter | Data Processing |
|---|---|
| Space group | F4132 |
| Unit cell dimensions (Å) | a = b = c = 608.58 |
| Resolution range (Å)a | 80-6.5 (6.61-6.50) |
| # Observations | 370,209 |
| Unique Reflections | 19,447 |
| Redundancy | 19.0 (19.6) |
| Rmerge (%)b | 16.9 (53.5) |
| <I>/<σI> | 30.2 (7.5) |
| Completeness (%) | 99.9 (100) |
| cRcryst / Rfree | 0.278 / 0.269 |
Numbers in parentheses are values in highest resolution shell
Rmerge = ∑hkl ∑i ∣Ii(hkl) - <I(hkl)>∣/∑hkl ∑i Ii(hkl), where Ii(hkl) is the ith observation of a symmetry equivalent of reflection hkl.
Rfree was calculated with 1953 reflections (10% of data) selected randomly
A minimally biased maximum likelihood difference map, averaged according to the 5-fold non-crystallographic symmetry (NCS), was calculated using Fourier coefficients of mFo-DFc and model phases, φc, where Fo and Fc are the observed and model structure amplitudes, m is the figure of merit weight, and D accounts for model errors (Read, 1986). A strong positive peak, 10σ above the mean density, signified something present in the crystals of SOS complex, but absent from the atomic model of uncomplexed virus. It was positioned between the spikes above the outer surface of the capsid. The density is centered on a viral 3-fold axis above the cluster of three Arg594 residues from neighboring capsid subunits (Figure 2) that form the most positive region on the viral surface (see above). The peak is centered ~7Å from the Nω atoms of Arg594 and can accommodate one SOS molecule. Manually modeling with SOS places several sulfate groups within 3-5Å of Arg594, i.e. close enough for an ionic interaction. The density is located on a 3-fold symmetry axis, and therefore represents a mixture of 3 SOS orientations, precluding detailed modeling. Thus, corroborating experimental evidence would be sought (following sections) to characterize independently the functional significance of residues implicated by the low resolution crystallography.
Figure 2.
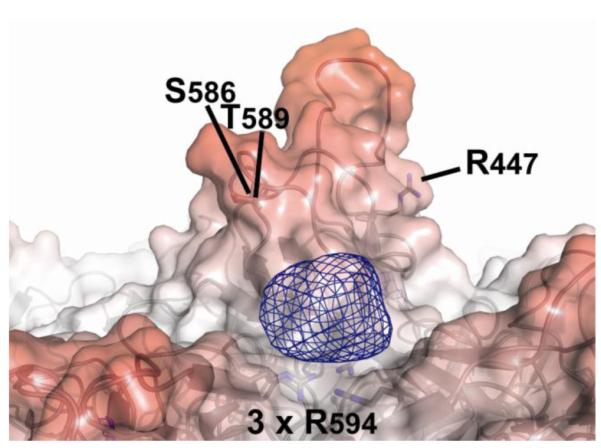
The SOS binding site on AAV-3B. Difference (mFo-DFc) electron density (blue mesh, contoured at 5σ) indicates SOS bound where it can interact electrostatically with Arg594 in maps calculated using data from the AAV-3B:SOS co-crystals. The density can accommodate a single SOS molecule bound to the capsid on the 3-fold symmetry axis. A ribbon representation of the 2.6Å AAV-3B structure (Lerch, Xie, and Chapman, 2010) is overlayed with a translucent molecular surface.
Other features in the difference map were considered. A 10σ peak in the difference map near the tip of the spikes is unlikely to be SOS because: (1) it has an elongated shape, (2) modeling with SOS results in clashes with protein atoms, and (3) the contact surface is not positively charged. Locally high B-factors in all AAV structures indicate that the region is among the most disordered (Govindasamy et al., 2006; Lerch, Xie, and Chapman, 2010; Nam et al., 2007; Ng et al., 2010; Xie et al., 2002). The SOS complex and native AAV-3B crystal forms have distinct packing interactions at this exposed region of the surface that affect some, but not all of the NCS-related subunits, likely resulting in different distributions of conformers that could give rise to difference peaks at this site. The next strongest peak (8σ) is on the inner surface on a 5-fold symmetry axis. With noise proportional to the square root of the local symmetry, the signal/noise of this peak is just 60% of the Arg594 site (Arnold and Rossmann, 1986). The inner surface is clearly not the site of receptor binding, and this peak was interpreted as the highest level of noise. Thus, only the peak near Arg594 is likely to be SOS.
AAV-3B mutants targeted to disrupt heparin binding
The role of Arg594 and other positively charged residues near AAV-3B’s surface spikes was further investigated by site-directed mutagenesis. To decrease the surface charge on the side of the AAV-3B spike, Arg447 was substituted with alanine. Separately, Asn500 was substituted with glutamate in an attempt to mimic the neutralizing salt bridge between Glu499 and Arg447 in AAV-2. Targeting the SOS site observed crystallographically, Arg594 was substituted with either alanine or glutamate. These AAV-3B mutations (R447A, R594A, R594E, and N500E) were anticipated to reduce heparin binding.
Heparin binding measurements of wild-type (WT) AAV-3B and AAV-2 showed that the AAV-3B elutes from a heparin column at ~200mM lower NaCl than AAV-2 (see Figure 4A), consistent with prior observations that AAV-3B has a weaker affinity than AAV-2 (Lerch, Xie, and Chapman, 2010; Rabinowitz et al., 2002). The salt concentration at which heparin binding proteins elute from a heparin column is correlated with their heparin binding affinity (Thompson, Pantoliano, and Springer, 1994). Wild-type AAV-3B eluted with the addition of 125-150mM NaCl (i.e. at 262-287mM total [NaCl], including the PBS running buffer; Figure 3B). R447A and N500E mutants showed only slightly decreased affinity, eluting with the addition of 75-125mM NaCl at a total [NaCl] of 212-262 mM, indicating at most a minor role in heparin binding. WT, R447A and N500E were not found in the flow-through and column wash fractions. By contrast, mutants R594A and R594E were found only in the flow through and column washes (Figure 3A), indicating that these capsids did not bind to heparin at physiological ionic strength, and signaling an important role for Arg594 in heparin binding.
Figure 4.

Heparin affinity chromatography of AAV-3B mutants with enhanced binding. The heparin binding residues from AAV-2 (Arg585 & Arg588) were introduced into the equivalent positions in AAV-3B (replacing Ser586 and Thr589). (A) The heparin binding affinity of the single S586R and T589R mutants was increased over that of AAV-3B, and was comparable to that of AAV-2. AAV-3B S586R/T589R shows a striking increase in heparin binding affinity, presumably due to the combination of heparin binding sites from two serotypes. (B) AAV-3B S586R/T589R, N500E/S586R/T589R and R447A/S586R/T589R mutants all bound heparin with high affinity, while R594A/S586R/T589R showed weaker binding, comparable to that of AAV-2.
Figure 3.

Heparin-affinity chromatography for AAV-3B mutants designed for diminished binding. Samples were applied to a heparin column, washed, and eluted in PBS with increasing NaCl concentrations. Capsids were assayed by ELISA, using a capsid-specific monoclonal antibody (Grimm et al., 1999). (A) Capsids were detected in the flow-through and 1st wash fraction only for AAV-3B mutants R594A and R594E, while all other capsids retained at least some affinity. (B) Elution profiles. The [NaCl] added is shown, and the total [NaCl], including the 137mM of the PBS running buffer, is shown in parentheses. AAV-3B bound most tightly, and N500E and R447A eluted in slightly lower NaCl concentrations, suggesting that these residues might play a minor role in heparin binding. R594A or R594E capsid mutants were not detected in any of the elution fractions, indicating that mutation of Arg594 abrogates heparin binding by AAV-3B.
Gain of function AAV-3B mutants
To generate capsids with enhanced heparin binding, Arg585 and Arg588 (the strongest determinants of heparin binding for AAV2 (Kern et al., 2003; Opie et al., 2003)) were introduced into the equivalent positions in AAV-3B. Mutation at either of the two sites, with S586R or T589R, was sufficient to raise heparin affinity to AAV-2-like levels (Figure 4A), suggesting that other sites (such as Arg594) contribute in AAV-3B. Introduction of both arginines in a double mutant (S586R/T589R) resulted in a strong increase in heparin affinity over AAV-2, the capsids eluting with the addition of ~750mM NaCl at a total [NaCl] of ~900 mM (Figure 4).
New constructs were now made with AAV-3B mutations R447A, R594A, and N500E in the S586R/T589R background. R447A and N500E as triple mutants had affinities that were unchanged relative to the S586R/T589R double mutant (Figure 4B). The triple mutant, R594A/S586R/T589R, eluted with the addition of 400-600mM NaCl (total [NaCl] ≈ 650 mM), which is indistinguishable from AAV-2. This confirms that the presence of Arg594 in AAV-3B compensates for the absence of the arginine at 586 and 589 that would be present in AAV-2.
Cell binding and transduction
While heparin affinity is often used as a proxy, measurement of cell binding and transduction, where possible, would offer more functionally relevant characterization. Using methods previously established for AAV-2 (Kern et al., 2003), HeLa cell binding was first measured for WT AAV-3B and AAV-2 capsids (Figure 5A) using a range of virus concentrations from 20ng/mL to 10μg/mL (or 3 × 108 to 3 × 1011 particles/mL). The concentrations of AAV capsids were determined by ELISA, using a monoclonal antibody (A20) specific for assembled AAV-2 or -3B capsids (Grimm et al., 1999). Equivalent amounts of AAV capsids were immobilized on a monolayer of HeLa cells. Capsid particles that remained bound after washing with PBS were measured by a cell-based ELISA (Kern et al., 2003). WT AAV-2 exhibited stronger binding than WT AAV-3B (Figure 5A), consistent with their heparin-binding affinities. Many of the AAV-3B mutants were available only at concentrations less than the detection limit for this assay. However, mutants prepared in the high affinity S586R/T589R background were amenable to measurement. S586R/T589R exhibited stronger cell binding than WT AAV-3B (Figure 5B). Triple mutants R447A/S586R/T589R and N500E/S586R/T589R bound cells comparably to the S586R/T589R double mutant. R594A/S586R/T589R bound cells less efficiently than the S586R/T589R mutant, and was comparable to that of AAV-2. All of these cell-binding results are consistent with the measured heparin affinities and indicate that Arg594 is a key contributor to cell binding in AAV-3B.
Figure 5.

Cell binding by AAV-3B mutants. AAV capsids bound to HeLa cells were detected by a cell-based ELISA (Kern et al., 2003). (A) AAV-2 capsids bind cells to a greater extent than WT AAV-3B at similar concentrations. (B) Comparison of cell attachment for the enhanced-affinity AAV-3B mutants. S586R/T589R, N500E/S586R/T589R and R447A/S586R/T589R all bound cells comparably and at higher levels than AAV-2. The R594A/S586R/T589R mutant, however, showed decreased cell binding over the S586R/T589R mutant. As observed for heparin, R594A/S586R/T589R bound cells at levels comparable to AAV-2.
Finally, to establish the role of Arg594 in the infectious pathway of AAV-3B, transduction assays were performed using WT and R594A AAV-3B vectors, which package a GFP reporter gene. After 48 hours, HeLa cells transduced with 250 rAAV-3B vector genome-containing particles (vg) per cell showed strong expression of GFP (Figure 6B), with a transduction efficiency of 3-4 × 103 vector genome-containing particles per transduction event. In contrast, transduction with R594A rAAV vectors at 250 vg/cell resulted in no GFP expression (Figure 6C). At 4-fold higher titer (1000 vg/cell), faint GFP expression was observed in a small number of cells transduced with R594A vectors (Figure 6D), but the transduction efficiency was 100 x lower than wild type at ~5 × 105 vg / transduction. Thus, low levels of cell entry are measurable even with the R594A mutation, either due to residual binding or use of an alternative entry pathway (Opie et al., 2003). However, sharply reduced transduction efficiency confirms a role for Arg594 in cell entry and show that disruption of the heparin binding site on AAV-3B results in a decrease in transduction by two orders of magnitude.
Figure 6.

Arg594 is for a key determinant of AAV-3B transduction. HeLa cells (shown at 200x magnification) were treated with WT or R594A AAV-3B vectors carrying a GFP gene. GFP expression was not observed in control cells (A), whereas strong expression was observed in cells transduced with AAV-3B vectors (B). AAV-3B R594A vectors did not transduce cells at 250 vg/cell (C), but GFP expression was observed in a few cells after treatment with 1000 vg/cell (D, arrow).
Discussion
A combination of structural and biochemical approaches have been used to identify a major heparin binding determinant on the surface of AAV-3B. An absence of negative density in the difference map of magnitude comparable to the positive SOS peak indicates that large conformational changes in the capsid are not induced by SOS. Similarly, there was no evidence of heparin-induced conformational changes in 8Å cryo-EM structure of the heparin-AAV2 complex (O’Donnell, Taylor, and Chapman, 2009). Both studies stand in stark contrast to the inference of large induced changes from subtle differences in maps of AAV-2 at much lower (18Å) resolution (Levy et al., 2009). Unlike the higher resolution studies, the 18Å study had to rely on map features commensurate with experimental noise, a risky endeavor. However, if the proposed triggering mechanism were a reality, one would expect it to be conserved across AAV serotypes. The finding here that the principal determinants of heparin binding in AAV-2 and AAV-3B are ~20 Å apart makes the proposed triggering mechanism even less plausible.
In the case of AAV-2, Arg585 and Arg588 are core to receptor interactions, but additional neighboring residues contribute to heparin binding, as evidenced by less tightly bound heparin polymer that can be seen at lower contour levels in the AAV-2 complex structure (O’Donnell, Taylor, and Chapman, 2009). Several of these residues are also conserved in AAV-3B (including Arg475, Arg485, Arg488, and Lys533). The current study uses a disaccharide heparin analog and therefore reveals only the site of tightest binding. It is quite plausible that a polymer could extend from this site to interact with the conserved basic amino acids that are part of the AAV-2 binding site. However, our binding studies show that the cluster at Arg594 can account for most of the observed binding under physiological salt concentrations, and that other residues play a more minor role. What is clear is that the core residues for AAV-2 and AAV-3B are distinct. In AAV-3B, they are at the mid-point between three adjacent spikes, while in AAV-2 they occur on the side of each spike.
The gain-of-function mutants in AAV-3B have some interesting implications. The S586R/T589R mutant combines the heparin binding determinants of AAV-2 with those of AAV-3B, creating a chimeric capsid with a broad receptor binding site and heparin binding affinity that is significantly greater than any naturally occurring AAV serotype. The apparent absence of such a “super-binding” variant in nature suggests that selective pressure is not unimodally in the direction of stronger receptor-binding. One might speculate that overly tight binding might be a selective disadvantage in vivo if either virus becomes non-productively sequestered in, for example, heparanoid-rich connective tissue.
While the AAV-2 and AAV-3B heparin-binding sites can be contrasted at a detailed level, there are fundamental similarities in binding at sites in the vicinity of the three-fold spikes. Proximity to the spikes could be providing partial protection of the binding site from immune surveillance (Rossmann, 1989). Alternatively, generally similar locations might, conceivably, result from a need to be near a site important for a later step in cell entry or trafficking. It is intriguing that within the constrained proximity to the spikes, the serotypes have selected different positively charged amino acids to anchor their core receptor interactions. One possible rationalization is that these sites remain under at least some immune surveillance, and that there has continued to be selective pressure to change residues within an essential binding site where antibody binding might have strong neutralizing potential.
The identification of the receptor binding site on AAV-3B provides new potential for its use as a therapeutic vector. Improved gene therapy vectors based on other AAV serotypes have been engineered rationally, using insights from AAV structures and receptor binding properties (Asokan et al., 2010; Shi and Bartlett, 2003; Shi et al., 2006). Through its heparin binding site, it has been demonstrated that the AAV-3B capsid can be modified to either to enhance or reduce receptor binding, providing a novel template on which next generation gene therapy vectors can be designed.
Materials and Methods
Cell Culture
HeLa cells were maintained in Joklik’s Modified Essential Medium (JMEM, Sigma-Aldrich) at 37°C in 5% CO2. Medium was supplemented with 10% Cosmic Calf serum (HyClone, Inc.), 10mg/L gentimicin, and an antimicrobial cocktail containing 10μg/mL penicillin, 50mg/L streptomycin, and 25mg/L amphotericin (Gibco, Inc.). SF9 cells were maintained in SF900-II medium (Invitrogen) in suspension at a cell density of 0.5-2.0 × 106 cells/mL.
Plasmids and mutagenesis
Mutations were introduced into the AAV-3B capsid gene in the pRepCap3B plasmid (Rutledge, Halbert, and Russell, 1998) using the QuikChange kit (Strategene) according to the manufacturer’s protocol. pRepCap3B contains the Rep and Cap genes of AAV-3B , but lacks the inverted terminal repeats necessary for genome packaging.
AAV-3B virus-like particles (VLPs) produced in SF9 cells were used as an ELISA standard. The AAV-3B capsid sequence was amplified from pRepCap3B, introducing 5′ BglII and 3′ XbaI restriction sites. Additional mutations to the 5′ end of the capsid gene were introduced using the forward amplification primer to improve the capsid protein ratio in VLPs, as described (Urabe, Ding, and Kotin, 2002). The insert was digested with BglII and XbaI (New England Biolabs) and sub-cloned into the backbone of the pFBDVPm11 plasmid (obtained from Robert Kotin and as previously described (Urabe, Ding, and Kotin, 2002)), digested with BamHI and XbaI. The sequences of all plasmids used were confirmed by sequencing at the DNA Sequence Analysis Shared Resource in the Molecular Microbiology and Immunology Department at OHSU.
Preparation of AAV
Production of infectious AAV-2 and AAV-3B was performed as previously described (Lerch et al., 2009; Xie et al., 2004). Production of AAV-3B VLPs from SF9 cells followed methods used for production of AAV-2 (Urabe, Ding, and Kotin, 2002) using the Bac-to-Bac Baculovirus Expression Vector System (Invitrogen). VLPs were harvested from SF9 cells by three rounds of freeze-thawing and purified by three consecutive CsCl gradients, as described for infectious AAV purification (Xie et al., 2004).
For transfection of the pRepCap3B plasmids containing capsid mutations, HeLa cells were plated at half confluence in 30mL JMEM with 10% FCS per T-75 flask one day prior to transfection. 30μg of each pRepCap3B plasmid per T-75 flask was transfected with 75μL Lipofectamine 2000 in OptiMEM medium (Invitrogen) according to the manufacturer’s protocol. Six hours after transfection, medium was replaced and adenovirus type 2 (Ad-2) at an MOI of 1 was added to the cells to provide helper functions in AAV gene expression. 72 hours post-transfection, cells were harvested by scraping and pelleted by centrifugation at 112,400 × g (25,000 rpm) for 2 hours using a Beckman SW28 ultracentrifuge rotor. Cell pellets were suspended in 1mL PBS and subjected to 3 rounds of freeze-thawing to lyse the cells. Cell debris was clarified by centrifugation at 5,000 × g for 10 min, and clarified lysates containing AAV-3B and mutant capsids were stored at −20°C. Wild-type and mutant AAV-3B capsid expression was analyzed by Western blot, probing with a monoclonal antibody (MAb) specific for denatured capsids (B1, American Research Products, Inc.), and the appropriate ratio of capsid proteins was confirmed. Proper assembly of capsids was confirmed by dot blot, probed with MAb A20, which is specific for intact capsids (Wobus et al., 2000).
Transducing vectors based on WT and the R594A mutant AAV-3B capsid were prepared in the Vollum Viral Core at OHSU using the triple transfection method described previously (Ayuso et al.). Vectors containing a GFP reporter gene (driven by a CMV promoter and flanked by AAV-2 inverted terminal repeats) were purified from the cell medium by PEG precipitation, followed by a single centrifugation gradient in CsCl. Vectors were exchanged into PBS in micro-concentrators (Amicon) and titers (vector genomes/mL (vg/mL)) were determined by quantitative PCR.
Crystallization and data collection
Crystals of infectious AAV-3B in complex with the heparin analog sucrose octasulfate (SOS) were grown by the hanging-drop vapor diffusion method at room temperature. 2μL of purified infectious AAV-3B (concentrated to 6mg/mL in 100mM HEPES, 50mM MgCl2, pH7.4) was mixed with 2μL of a well solution containing 1.3% PEG 6000, 225mM NaCl and 1mM SOS. Crystals grew to ~300μm in 3-4 weeks. Crystals were harvested by adding a harvest buffer (5% PEG 6000, 200mM NaCl, 100mM HEPES pH7.5, 50mM MgCl2, 1mM SOS) to the crystallization drop and subsequently cryo-protected by soaking for 1-5 minutes in harvest buffer that contained 10% PEG 6000 (total concentration) and 30% glycerol prior to flash-freezing in liquid nitrogen.
Diffraction data were collected at the BioCARS 14-BM-C beamline at the Advanced Photon Source, Argonne, IL. Crystals were exposed to 0.979Å (12,668eV) synchrotron radiation for 15 s with 0.5° oscillations. To obtain adequate diffraction spot separation, the detector was positioned 600mm from the crystal, with a beam stop 100mm from the crystal and the X-ray source was focused 400mm from the sample. Reflection intensities were indexed, integrated and scaled using HKL 2000 (Otwinowski and Minor, 1997).
Diffraction data to 6.5Å resolution were collected from the best crystal. 187 frames were obtained, and the data were 99.9% complete with 19-fold redundancy. Despite strong reflections (<I>/<σI> = 7.5) in the highest resolution shell (6.61-6.5Å), the intensities of reflections at higher resolution decreased sharply, preventing their use in structure determination. The data were processed in the space group F4132 (which is different from the native AAV3B crystals) with an Rmerge of 16.9%. The 2.6Å structure of AAV-3B (PDB ID 3KIC; (Lerch, Xie, and Chapman, 2010)) was used to determine the position and orientation of the virus in the unit cell. A pentamer of viral subunits (1/12th of the capsid) is contained in the asymmetric unit. The position and orientation of the virus are fixed, as the 32 crystallographic and icosahedral symmetry axes are co-incident. Thus, the virus particle, centered at the origin of the unit cell, was positioned by aligning one 3-fold and two 2-fold icosahedral symmetry axes with those of the unit cell. Bulk solvent scaling was performed in Phenix (Adams et al., 2002), and electron density map averaging was performed using Rave (Kleywegt et al., 2004).
Capsid titer
The concentration of AAV capsids in clarified HeLa cell lysates was determined by sandwich ELISA (Grimm et al., 1999). Capsid ELISAs were performed as described, except that 0.5% BSA (Jackson ImmunoResearch) was used as a blocking agent, and streptavidin-conjugated horse radish peroxidase (R&D Systems, Inc) and 1-Step Ultra TMB-ELISA Substrate (Pierce) were used in development. AAV-3B VLPs purified from SF9 cells were used as a standard for concentration determination.
Heparin binding
Heparin binding was analyzed by affinity chromatography using a 1mL HiTrap Heparin HP column (GE Healthcare). Purified infectious AAV-2 or AAV-3B or clarified HeLa cell lysates containing 200-500ng AAV mutant capsids were loaded onto the column after first washing with 5M NaCl and equilibrating with 10 column volumes (CV) of PBS. Unbound capsids were washed from the column with 5 CV PBS, collected over 3 fractions. Capsids were eluted from the column in 1 CV steps using PBS supplemented with additional NaCl. For capsid mutants with greater affinity than WT AAV-3B, 100mM NaCl increments were used (PBS + 0.1-1.0M NaCl). For mutants that bound to the column more weakly than WT AAV-3B, a fine step-gradient of 25mM NaCl increments was used (PBS + 25-500mM NaCl). Flow-through (FT), wash, and elution fractions were analyzed by an AAV capsid ELISA (Grimm et al., 1999).
Cell binding and transduction
Cell binding assays followed methods previously used for AAV-2 (Kern et al., 2003). HeLa cells were plated in 96-well culture plates (BD Falcon) at half-confluence one day before cell binding was measured. AAV capsids were diluted in ice-cold PBS and incubated with cells that had also been cooled to 4°C. Infectious AAV-3B and AAV-2 were measured in 2-fold serial dilutions from 10μg/mL-20ng/mL, and AAV-3B mutant capsids were compared at 150-300ng/mL. Incubation of capsids on cells was carried out at 4°C for 1 hour, and cells were washed twice with chilled PBS. Cells were fixed in methanol for 20 minutes at −20°C and washed in PBS + 0.05% Tween-20. Bound capsids were detected with the AAV capsid ELISA procedure described above (Grimm et al., 1999).
For cell transduction assays, HeLa cells were plated in 48-well culture plates (BD Falcon) at half-confluence one day before transduction. AAV-3B vectors were diluted in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 2% FBS, and added to cells at 100-1000vg/cell. 48 hours after addition of vectors, cells were analyzed for GFP expression. At least 6 independent fields of cells expressing GFP were counted, and transduction efficiencies (calculated as the number of vector particles required for one transduction event) were determined by correcting for the number of genome-containing particles and total number of cells.
Acknowledgements
The authors would like to thank Robert Kotin and David Russell for providing plasmids, Eric Washburn at the Vollum Viral Core for assistance in vector production and Vukica Srajer for assistance with data collection. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science under Contract No. DE-AC02-06CH11357. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant number RR00707. This research was supported by the National Institutes of Health R01-GM66875 (MSC). TFL was supported by a fellowship from the American Heart Association, Pacific Mountain Affiliate.
Abbreviations and symbols
- HSPG
heparan sulfate proteoglycan
- AAV
adeno-associated virus
- SOS
sucrose octasulfate
- NCS
non-crystallographic symmetry
- WT
wild type
- ELISA
enzyme-linked immunosorbant assay
- GFP
green fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 11):1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Arnold E, Rossmann MG. Effect of errors, redundancy, and solvent content in the molecular replacement procedure for the structure determination of biological macromolecules. Proceedings of the National Academy of Sciences, USA. 1986;83:5489–93. doi: 10.1073/pnas.83.15.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asokan A, Conway JC, Phillips JL, Li C, Hegge J, Sinnott R, Yadav S, DiPrimio N, Nam HJ, Agbandje-McKenna M, McPhee S, Wolff J, Samulski RJ. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol. 2010;28(1):79–82. doi: 10.1038/nbt.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso E, Mingozzi F, Montane J, Leon X, Anguela XM, Haurigot V, Edmonson SA, Africa L, Zhou S, High KA, Bosch F, Wright JF. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 17(4):503–10. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- Badger J, Minor I, Kremer M, Oliveira M, Smith TJ, Griffith JP, Guerin DM, Krishnaswarmy S, Luo M, Rossmann MG, McKinlay M, Diana G, Dutko FJ, Fancher M, Rueckert R, Heinz BA. Structural Analysis of a series of antiviral agents complexed with human rhinovirus 14. Proceedings of the National Academy of Sciences, USA. 1988;85:3304–3308. doi: 10.1073/pnas.85.10.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buning H, Braun-Falco M, Hallek M. Progress in the use of adeno-associated viral vectors for gene therapy. Cells Tissues Organs. 2004;177(3):139–150. doi: 10.1159/000079988. [DOI] [PubMed] [Google Scholar]
- Buning H, Ried MU, Perabo L, Gerner FM, Huttner NA, Enssle J, Hallek M. Receptor targeting of adeno-associated virus vectors. Gene Ther. 2003;10(14):1142–51. doi: 10.1038/sj.gt.3301976. [DOI] [PubMed] [Google Scholar]
- Caspar DLD, Klug A. Physical principles in the constructuion of regular viruses. Cold Spring Harbor Symposium in Quantitative Biology. 1962;27:1–24. doi: 10.1101/sqb.1962.027.001.005. [DOI] [PubMed] [Google Scholar]
- Conrad HE. Heparin-Binding Proteins. 1st ed Academic Press; San Diego: 1998. [Google Scholar]
- Flotte TR. Gene therapy progress and prospects: Recombinant adeno-associated virus (rAAV) vectors. Gene Therapy. 2004;11(10):805–810. doi: 10.1038/sj.gt.3302233. [DOI] [PubMed] [Google Scholar]
- Glushakova LG, Lisankie MJ, Eruslanov EB, Ojano-Dirain C, Zolotukhin I, Liu C, Srivastava A, Stacpoole PW. AAV3-mediated transfer and expression of the pyruvate dehydrogenase E1 alpha subunit gene causes metabolic remodeling and apoptosis of human liver cancer cells. Mol Genet Metab. 2009 doi: 10.1016/j.ymgme.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini JA, Agbandje-McKenna M. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J Virol. 2006;80(23):11556–70. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Kern A, Pawlita M, Ferrari F, Samulski R, Kleinschmidt J. Titration of AAV-2 particles via a novel capsid ELISA: packaging of genomes can limit production of recombinant AAV-2. Gene Ther. 1999;6(7):1322–1330. doi: 10.1038/sj.gt.3300946. [DOI] [PubMed] [Google Scholar]
- Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, Kay MA. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. 2008;82(12):5887–911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa A, Muramatsu S, Qiu J, Mizukami H, Brown KE. Adeno-associated virus (AAV)-3-based vectors transduce haematopoietic cells not susceptible to transduction with AAV-2-based vectors. J Gen Virol. 2000;81(Pt 8):2077–2084. doi: 10.1099/0022-1317-81-8-2077. [DOI] [PubMed] [Google Scholar]
- Hildinger M, Auricchio A. Advances in AAV-mediated gene transfer for the treatment of inherited disorders. European Journal of Human Genetics. 2004;12(4):263–271. doi: 10.1038/sj.ejhg.5201153. [DOI] [PubMed] [Google Scholar]
- Innis CA, Hyvonen M. Crystal structures of the heparan sulfate-binding domain of follistatin. Insights into ligand binding. J Biol Chem. 2003;278(41):39969–77. doi: 10.1074/jbc.M211284200. [DOI] [PubMed] [Google Scholar]
- Kern A, Schmidt K, Leder C, Muller OJ, Wobus CE, Bettinger K, Von der Lieth CW, King JA, Kleinschmidt JA. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J Virol. 2003;77(20):11072–81. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Harris MR, Zou JY, Taylor TC, Wahlby A, Jones TA. The Uppsala Electron-Density Server. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2240–9. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- Lerch TF, Xie Q, Chapman MS. The structure of adeno-associated virus serotype 3B (AAV-3B): insights into receptor binding and immune evasion. Virology. 2010;403(1):26–36. doi: 10.1016/j.virol.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch TF, Xie Q, Ongley HM, Hare J, Chapman MS. Twinned crystals of adeno-associated virus serotype 3b prove suitable for structural studies. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2009;65(Pt 2):177–83. doi: 10.1107/S1744309109000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy HC, Bowman VD, Govindasamy L, McKenna R, Nash K, Warrington K, Chen W, Muzyczka N, Yan X, Baker TS, Agbandje-McKenna M. Heparin binding induces conformational changes in Adeno-associated virus serotype 2. J Struct Biol. 2009;165(3):146–56. doi: 10.1016/j.jsb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AM, Nicolson SC, Warischalk JK, Samulski RJ. AAV’s anatomy: roadmap for optimizing vectors for translational success. Curr Gene Ther. 2010;10(5):319–40. doi: 10.2174/156652310793180706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HJ, Lane MD, Padron E, Gurda B, McKenna R, Kohlbrenner E, Aslanidi G, Byrne B, Muzyczka N, Zolotukhin S, Agbandje-McKenna M. Structure of adeno-associated virus serotype 8, a gene therapy vector. J Virol. 2007;81(22):12260–71. doi: 10.1128/JVI.01304-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng R, Govindasamy L, Gurda BL, McKenna R, Kozyreva OG, Samulski RJ, Parent KN, Baker TS, Agbandje-McKenna M. Structural characterization of the dual glycan binding adeno-associated virus serotype 6. J Virol. 2010;84(24):12945–57. doi: 10.1128/JVI.01235-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell J, Taylor KA, Chapman MS. Adeno-associated virus-2 and its primary cellular receptor--Cryo-EM structure of a heparin complex. Virology. 2009;385(2):434–43. doi: 10.1016/j.virol.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opie SR, Warrington KH, Jr, Agbandje-McKenna M, Zolotukhin S, Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J Virol. 2003;77(12):6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. Jr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Perabo L, Goldnau D, White K, Endell J, Boucas J, Humme S, Work LM, Janicki H, Hallek M, Baker AH, Buning H. Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J Virol. 2006;80(14):7265–9. doi: 10.1128/JVI.00076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, Samulski RJ. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76(2):791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RJ. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr A. 1986;42:140–149. [Google Scholar]
- Rossmann MG. The Canyon Hypothesis. Journal of Biological Chemistry. 1989;264:14587–90. [PubMed] [Google Scholar]
- Rutledge E, Halbert C, Russell D. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. Journal of Virology. 1998;72(1):309–319. doi: 10.1128/jvi.72.1.309-319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi WF, Bartlett JS. RGD inclusion in VP3 provides adeno-associated virus type 2 (AAV2)-based vectors with a heparan sulfate-independent cell entry mechanism. Molecular Therapy. 2003;7(4):515–525. doi: 10.1016/s1525-0016(03)00042-x. [DOI] [PubMed] [Google Scholar]
- Shi X, Fang G, Shi W, Bartlett JS. Insertional mutagenesis at positions 520 and 584 of adeno-associated virus type 2 (AAV2) capsid gene and generation of AAV2 vectors with eliminated heparin- binding ability and introduced novel tropism. Hum Gene Ther. 2006;17(3):353–61. doi: 10.1089/hum.2006.17.353. [DOI] [PubMed] [Google Scholar]
- Thompson LD, Pantoliano MW, Springer BA. Energetic characterization of the basic fibroblast growth factor-heparin interaction: identification of the heparin binding domain. Biochemistry. 1994;33(13):3831–40. doi: 10.1021/bi00179a006. [DOI] [PubMed] [Google Scholar]
- Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002;13(16):1935–43. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- Van Vliet KM, Blouin V, Brument N, Agbandje-McKenna M, Snyder RO. The role of the adeno-associated virus capsid in gene transfer. Methods Mol Biol. 2008;437:51–91. doi: 10.1007/978-1-59745-210-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus CE, Hugle-Dorr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74(19):9281–93. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A, Chapman MS. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U S A. 2002;99(16):10405–10. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Hare J, Turnigan J, Chapman MS. Large-scale production, purification and crystallization of wild-type adeno-associated virus-2. Journal of Virological Methods. 2004;122(1):17–27. doi: 10.1016/j.jviromet.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Zaiss AK, Muruve DA. Immune responses to adeno-associated virus vectors. Curr Gene Ther. 2005;5(3):323–31. doi: 10.2174/1566523054065039. [DOI] [PubMed] [Google Scholar]