

Abstract
Suppression therapy utilizes compounds that suppress translation termination at in-frame premature termination codons (PTCs) to restore full-length, functional protein. This approach may provide a treatment for diseases caused by nonsense mutations such as mucopolysaccharidosis type I-Hurler (MPS I-H). MPS I-H is a lysosomal storage disease caused by severe α-L-iduronidase deficiency and subsequent lysosomal glycosaminoglycan (GAG) accumulation. MPS I-H represents a good target for suppression therapy because the majority of MPS I-H patients carry nonsense mutations, and restoration of even a small amount of functional α-L-iduronidase may attenuate the MPS I-H phenotype. In this study, we investigated the efficiency of suppression therapy agents to suppress the Idua-W392X nonsense mutation in an MPS I-H mouse model. The drugs tested included the conventional aminoglycosides gentamicin, G418, amikacin, and paromomycin. In addition, the designer aminoglycosides NB54 and NB84, two compounds previously designed to mediate efficient PTC suppression with reduced toxicity, were also examined. Overall, NB84 suppressed the Idua-W392X nonsense mutation much more efficiently than any of the other compounds tested. NB84 treatment restored enough functional α-L-iduronidase activity to partially reverse abnormal GAG accumulation and lysosomal abundance in mouse embryonic fibroblasts derived from the Idua-W392X mouse. Finally, in vivo administration of NB84 to Idua-W392X mice significantly reduced urine GAG excretion and tissue GAG storage. Together, these results suggest that NB84-mediated suppression therapy has the potential to attenuate the MPS I-H disease phenotype.
INTRODUCTION
Nonsense mutations are in-frame premature termination codons (PTCs) that convert a sense codon in the coding region of an mRNA to a stop codon (UGA, UAG, UAA). The introduction of a PTC frequently reduces the amount of functional protein so severely that a disease state results. In an effort to moderate the consequences of PTCs that cause genetic diseases, the concept of suppression therapy was developed. This is a therapeutic approach utilizing compounds that induce the translational machinery to recode an in-frame PTC into a sense codon. Suppression therapy increases the frequency that near-cognate aminoacyl-tRNAs bind at a PTC and subsequently transfer their amino acid to the nascent polypeptide. This “readthrough” mechanism allows the resumption of translation elongation in the correct reading frame to produce a full-length polypeptide.
Aminoglycoside antibiotics have been shown to suppress PTCs and restore functional protein for more than twenty genetic diseases [1–3]. However, the potential toxicity associated with aminoglycosides has limited their clinical application in suppression therapy. Strategies developed to overcome this issue include: discovery of non-aminoglycoside suppression agents such as PTC124 (Ataluren®) [4, 5]; combining aminoglycoside administration with poly-L-aspartate to reduce toxicity while also enhancing readthrough [6]; and using a medicinal chemistry approach to design new aminoglycoside derivatives capable of enhancing PTC suppression with reduced overall toxicity [7, 8].
Mucopolysaccharidosis type I (MPS I) is a lysosomal storage disorder caused by α-L-iduronidase deficiency. α-L-iduronidase (EC 3.2.1.76, encoded by the IDUA gene) catalyzes a step in the degradation of dermatan sulfate and heparan sulfate glycosaminoglycans (GAGs). A deficiency in this enzyme leads to abnormal lysosomal GAG accumulation. Multiple cellular dysfunctions downstream of GAG accumulation contribute to organ abnormalities and metabolic defects through poorly understood mechanisms [9–12]. MPS I disease severity and progression exhibit broad variations depending upon the amount of residual α-L-iduronidase activity. MPS I-Hurler (MPS I-H) is the most severe form of α-L-iduronidase deficiency and is characterized by severe skeletal and joint disease, short stature, coarse facial appearance, hepatosplenomegaly, corneal clouding, valvular heart disease, and neurological defects that result in severe mental disabilities. MPS I-H disease onset usually occurs in infancy followed by rapid disease progression leading to death within the first decade of life [11]. Nonsense mutations represent the majority of IDUA gene lesions found in MPS I-H patients and result in negligible α-L-iduronidase activity. In contrast, attenuated forms of α-L-iduronidase deficiency such as MPS I-Scheie (MPS I-S) or MPS I-Hurler/Scheie (MPS I-HS) retain residual α-L-iduronidase activity and are characterized by mild somatic symptoms that include an attenuated skeletal disease, but an absence of hepatosplenomegaly and no neurological involvement with normal intellect [11]. These patients experience a more delayed disease onset in early to late childhood, frequently survive into adulthood, and can have a normal lifespan. However, patients with attenuated MPS I may develop cardiac impairment and spinal cord compression later in life. Based on the generally strong genotype/phenotype correlation observed in MPS I patients, it has been proposed that restoration of as little as 0.1 – 0.3% of normal α-L-iduronidase activity in MPS I-H patients may lead to an improved phenotype [13, 14].
Using primary skin fibroblasts derived from an MPS I-H patient with PTCs, we previously found that suppression therapy using gentamicin restored enough α-L-iduronidase activity to normalize cellular GAG storage [15]. To further examine whether this approach can alleviate the MPS I-H disease in vivo, we generated a knock-in mouse model of MPS I-H that carries the Idua-W392X mutation [16]. Mice carrying this mutant allele, which corresponds to the IDUA-W402X mutation frequently found in MPS I-H patients, were found to exhibit a phenotype that closely resembles the human MPS I-H disease. In the current study, we investigated whether conventional and designer aminoglycosides can suppress the PTC in this MPS I-H mouse model. We found that the designer aminoglycoside NB84 most effectively suppressed the Idua-W392X mutation, resulting in a significant attenuation of the primary biochemical defects associated with MPS I-H both in vitro and in vivo.
MATERIALS AND METHODS
Cell Culture
Primary mouse embryonic fibroblasts (MEFs) were cultured from 13–14 day-old embryos derived by breeding homozygous Idua-W392X [16], Idua-knockout [17], and WT mice. All three lines were on the C57BL/6J background. Idua-W392X MEFs were immortalized by stable expression of the SV40 large T-antigen (Discovery BioMed, Inc.). MEFs and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, 100ug/ml streptomycin, and 1% v/v nonessential amino acids. MEFs were cultured in collagen-coated dishes.
Dual Luciferase Readthrough Assays
The p2luc dual luciferase readthrough constructs were a gift from Dr. John Atkins [18]. The p2luc construct was modified to express either the WT mouse Idua W392 codon (UGG) or the Idua W392X premature stop codon (UAG) along with three codons of upstream and downstream mouse Idua sequence. Complementary oligonucleotides for the Idua-W392X construct: [5′-TCGACG GAACAA CTCTAG GCAGAG GTCG-3′; 5′-GATCCG ACCTCT GCCTAG AGTTGT TCCG-3′], and the Idua-WT construct: [5′-TCGACG GAACAA CTCTGG GCAGAG GTCG-3′; 5′-GATCCG ACCTCT GCCCAG AGTTGT TCCG-3′] were annealed to generate double-stranded DNA fragments that were ligated into the SalI and BamHI restriction sites of the p2luc vector, yielding the Idua-W392X (pDB1134) and Idua-WT (pDB1133) constructs. HEK293T cells were transfected with each reporter construct using Lipofectamine (Invitrogen). Cells were grown in the presence of drugs at the indicated concentrations for 24hrs. Luciferase assays were performed with the Dual Luciferase Assay System (Promega) using a Berthold Lumat LB9507 luminometer. The percent readthrough was calculated as the ratio of firefly/Renilla luciferase units expressed from the W392X construct relative to the ratio of the firefly/Renilla luciferase units produced from the WT construct × 100.
Iduronidase Activity Assays
Primary WT, Idua knockout, and immortalized Idua-W392X MEFs were cultured +/− drug for 48 hrs. The cells were lysed in Pierce Mammalian Protein Extraction Reagent containing a protease inhibitor cocktail (Roche) and the total protein concentration was determined using the Bio-Rad Protein Assay. Cell lysates containing 50–80 ug of total protein were incubated with 0.12 mM 4-methyl-umbelliferyl-α-L-iduronide (Gold Biotech) and 0.42 mg/ml of D-saccharic acid 1,4-lactone monohydrate (a β-glucuronidase inhibitor) (Sigma) in 130 mM sodium formate buffer, pH 3.5. After 48 hrs of incubation at 37°C, the reaction was quenched with glycine buffer, pH 10.8 and samples were aliquoted into methacrylate cuvettes. Fluorescence was measured using a Cary Eclipse fluorometer at excitation= 365 nm and emission= 450 nm. Specific activity was calculated as picomoles of FMU released per milligram of total protein per hour. Alpha-L-iduronidase activity remained linear over the 48 hr incubation time. Inhibitory factors in lysates from mutant mouse tissues precluded α-L-iduronidase activity measurements.
MEF Sulfated GAG Assay
Primary WT and immortalized Idua-W392X MEFs were grown in 100mm dishes +/− aminoglycosides for 48 hrs. The cells were lysed in Pierce Mammalian Protein Extraction Reagent and the total protein concentration was determined using the Bio-Rad Protein Assay. Sulfated GAG quantitation was performed using a Blyscan kit (Biocolor Ltd. UK). GAG levels were calculated as the nanograms of GAGs per milligrams total protein.
Confocal Fluorescence Microscopy
Primary MEFs were grown on coverslips +/− 2 mg/ml of NB84 for 48 hrs. 200 nM LysoTracker Red DND-99 (Invitrogen) was added to the culture medium. After incubation at 37°C with 5% CO2 for 1 hr, coverslips were immersed in PBS containing calcium and magnesium, and 2.5 μg/ml CellMask Deep Red plasma membrane stain (Invitrogen) at room temperature for 2 minutes. Cells were subsequently fixed with 4% formaldehyde at room temperature and washed with PBS four times before mounted onto a No.1 thickness coverslip in VECTASHIELD mounting medium with DAPI nuclear stain. Confocal fluorescence microscopy was performed using a Zeiss LSM-710 confocal microscope. Z-stack images and pixel intensities were analyzed using Zen software. LysoTracker signal abundance was calculated as the ratio of the total number of red pixels (LysoTracker) to the total number of blue pixels (DAPI). LysoTracker signal intensity was calculated as the ratio of the total red pixel intensities (LysoTracker) to the total number of blue pixels (DAPI). At least 100 cells from 8 to 11 fields were analyzed for each condition.
Readthrough Drugs and Animal Treatment
Paromomycin (cat #P5057) and G418 (cat #G8168) were purchased from Sigma-Aldrich. Amikacin (Bedford Labs) and gentamicin (Hospira, Inc.) were purchased from UAB University Hospital. The Baasov lab synthesized NB54 and NB84 [7, 8]. Readthrough compounds were dissolved in sterilized PBS and administered to mice that were 9–11 weeks old once daily via subcutaneous injections for 14 days. All animal protocols used in this study were reviewed and approved by UAB IACUC.
Urine GAG Assays
Three mice from each treatment group were housed together in a diuresis cage (Nalgene) for 2–4 hrs to collect urine. Collected urine was filtered through a sterile 0.45 μM filter (Whatman) and stored at 4°C until assay. The amount of GAGs excreted in urine was quantitated using a 1, 9-dimethylmethylene blue chloride (Sigma) assay and normalized to creatinine as previously described [16].
Tissue GAG Assays
Mice were perfused with 25 mls of ice-cold PBS and organs were dissected and stored at −80°C until assay. Tissue was delipidated by homogenization in chloroform: methanol (2:1) using a rotor-stator homogenizer (Biospec) and then dried in a SpeedVac concentrator (Savant). GAGs were released by overnight papain digestion at 60°C. The level of sulfated GAGs was determined using a Blyscan kit (Biocolor Ltd. UK) and the data was calculated as the micrograms of GAGs per milligram of defatted, dried tissue.
Analysis of Glycosphingolipids
Each lyophilized brain sample was extracted twice with 700 μl of chloroform:methanol (2:1) in a 1.5 ml Teflon Eppendorf tube using a BioSpect Tissue Tearor with a 4.5 mm probe and centrifuged at 10,000 rpm for 20 minutes using a Sorvall Micro 21R centrifuge. The two extracts were pooled, evaporated to dryness, dissolved in 150 μl of chloroform:methanol (2:1) and 5 μl-aliquot was applied onto a Silica Gel 60 High Performance Thin Layer Chromatography (HPTLC) plate (Merk Darmstadt, Germany). The plate was developed twice with a freshly prepared solvent mixture of chloroform:methanol:12 mM MgCl2 in water (60:35:8). Between the two developments, the plate was thoroughly air-dried. The plate was sprayed with diphenylamine-aniline-phosphoric acid reagent [19] and heated at 110° C for 15 minutes to reveal glycoconjugates. GM3, GM2 and the standard ganglioside mixture were purchased from Matreya (Pleasant Gap, PA). N-glycolylneuraminic acid-containing GM2 (Gc-GM2) and N-glycolylneuraminic acid-containing GM3 (Gc-GM3) were gifts of Prof. Tamio Yamakawa (University of Tokyo).
Statistics
Statistical analysis was performed using the GraphPad Instat 3.0 software (GraphPad Software, Inc.). All p values were calculated using the unpaired two-tail Student’s t-test.
RESULTS
Investigating the ability of aminoglycosides to suppress the Idua-W392X nonsense mutation using in vitro reporter assays
The designer aminoglycosides NB54 and NB84 contain structural components predicted to better suppress PTCs with less toxicity than conventional aminoglycosides [7, 8]. NB54 was derived from components of paromomycin and amikacin, while NB84 was composed of structural groups of paromomycin, amikacin, and G418 (Figure 1). We tested NB54, NB84, and the conventional aminoglycosides gentamicin, G418, amikacin, and paromomycin for their ability to suppress the mouse Idua-W392X PTC using dual luciferase readthrough reporters in HEK293T cells.
Figure 1. Chemical structures of readthrough compounds.

(A) Conventional aminoglycosides evaluated for the ability to suppress the Idua-W392X nonsense mutation. (B). Designer aminoglycosides evaluated for the ability to suppress the Idua-W392X nonsense mutation. The chemical groups shaded in paromomycin, amikacin and G418 were used to generate the designer aminoglycosides NB54 and NB84.
The identity of a PTC and its surrounding mRNA sequence context can strongly influence the ability of drugs to suppress PTCs [20–23]. Accordingly, we monitored the readthrough of the mouse Idua-W392X PTC in its natural mRNA sequence context using a dual luciferase readthrough reporter that contained three codons of both upstream and downstream mouse Idua sequence context surrounding the PTC (or the corresponding sense codon control) (Figure 2A). A concentration range was tested for each compound using the reporters to find the optimal dose that produced the maximum W392X readthrough without cell toxicity. The percent of Idua-W392X readthrough obtained using the optimal concentration(s) for each suppression agent is shown (Figure 2B & 2C).
Figure 2. Idua-W392X readthrough reporter assays.

(A) A schematic of the dual luciferase readthrough reporters. The sequences of the Idua-WT (UGG) and Idua-W392X (UAG) readthrough cassettes are shown. (B) The percent readthrough values of the Idua-W392X nonsense mutation +/− various readthrough agents are shown. Also shown is the percent readthrough values obtained using a reporter that expresses a UGAC termination signal within a context that is particularly susceptible to suppression. The fold-increase signifies the percent readthrough obtained with treatment relative to the percent readthrough obtained without treatment for the UGAC and the Idua-W392X contexts, respectively. Gent: gentamicin, Paro: paromomycin, Amik: amikacin. (C) The percent readthrough values of the Idua-W392X nonsense construct +/− varying concentrations of NB54 or NB84 are shown. The fold-increase signifies the percent readthrough obtained with treatment relative to the percent readthrough obtained without treatment for the Idua-W392X context. The data shown is expressed as the mean +/− SD of at least six replicates.
In contrast to a control readthrough reporter (UGAC) that contained a UGA codon in a context that is susceptible to suppression [6, 24, 25], the mouse Idua-W392X mutation in its natural sequence context was quite resistant to suppression (Figure 2B). The basal readthrough (without drug) of the Idua-W392X mutation was 50% lower than that of the UGAC control. We found that gentamicin induced a 25-fold increase in readthrough of the UGAC control, but stimulated readthrough by only 1.7-fold in the Idua-W392X context (Figure 2B). Similar results were observed for paromomycin and amikacin. G418 treatment stimulated a 2.9-fold increase in W392X readthrough (Figure 2B), while NB54 and NB84 induced readthrough by 4.8-fold and 11.3-fold, respectively (Figure 2C). These results indicate that NB54 and NB84 suppressed the Idua-W392X mutation more efficiently than the conventional aminoglycosides tested.
Investigating the ability of aminoglycosides to restore functional α-L-iduronidase and reduce GAG accumulation in Idua-W392X MEFs
We next performed functional assays using mouse embryonic fibroblasts (MEFs) derived from homozygous Idua-W392X and WT mice to determine whether conventional and designer aminoglycosides can suppress the endogenous Idua-W392X mutation and restore some level of α-L-iduronidase activity. We utilized an α-L-iduronidase enzymatic assay to monitor the level of functional enzyme restored after suppression therapy. We found α-L-iduronidase specific activity in lysates from untreated Idua-W392X MEFs was negligible (Figure 3A). However, treatment with conventional or designer aminoglycosides significantly increased α-L-iduronidase activity in Idua-W392X MEFs. The conventional aminoglycosides gentamicin, amikacin, and G418 restored α-L-iduronidase activity at levels that maximally ranged from 0.02% to 0.08% of WT α-L-iduronidase activity (Figure 3A). The designer aminoglycosides NB54 and NB84 induced substantially more α-L-iduronidase activity than the conventional aminoglycosides. NB54 induced levels of enzymatic activity that were 9.5-fold, 6.1-fold, and 2.5-fold greater than gentamicin, amikacin, or G418, respectively. NB84 restored 15.5-fold, 10-fold, 4.1-fold, and 1.6-fold greater α-L-iduronidase activity than gentamicin, amikacin, G418, or NB54, respectively. NB54 treatment restored up to 0.2% of WT α-L-iduronidase activity, while NB84 treatment restored up to 0.32% of WT α-L-iduronidase activity (Figure 3A). In order to verify that the drugs tested did not stimulate α-L-iduronidase activity by a mechanism other than suppression of the W392X PTC, we utilized MEFs derived from Idua knockout mice that carried a targeted Idua disruption that would not be susceptible to readthrough [17]. When Idua knockout MEFs were cultured with NB84 under the same conditions used for the Idua-W392X MEFs, no increase in α-L-iduronidase activity was detected. These results confirmed that NB84 restored α-L-iduronidase activity in Idua-W392X MEFs as a result of PTC suppression.
Figure 3. Evaluation of Idua-W392X suppression using MEFs.

(A) α-L-iduronidase specific activity in Idua-W392X MEFs cultured +/− varying concentrations of conventional and designer aminoglycosides. The dashed line indicates the α-L-iduronidase specific activity value that is 0.2% of WT as reference. The data shown is expressed as the mean +/− SD of at least six replicates. (B) Sulfated GAG quantitation in wild-type MEFs (WT) and Idua-W392X MEFs (W392X) cultured in the absence or presence of G418 or NB84. The dashed line represents the WT GAG level as a reference to compare with the GAG levels in the Idua-W392X MEFs. The data shown is representative data expressed as the mean +/− SD of at least three replicates.
Since loss of α-L-iduronidase results in lysosomal GAG accumulation, we next determined whether the amount of α-L-iduronidase activity restored in MEFs by PTC suppression was sufficient to reduce GAG storage. In untreated Idua-W392X MEFs, GAGs were elevated 4-fold above normal (Figure 3B). In Idua-W392X MEFs treated with G418, excess GAG storage was decreased 49%. In Idua-W392X MEFs treated with NB84, excess GAG levels decreased 80% (Figure 3B). Together, these experiments showed that suppression therapy restored sufficient α-L-iduronidase activity to significantly reduce GAG accumulation in Idua-W392X MEFs.
NB84 alleviated elevated lysosome abundance in Idua-W392X MEFs
We previously reported that primary skin fibroblasts derived from an MPS I-H patient affected by IDUA nonsense mutations (Q70X/W402X) exhibited increased lysosomal abundance, and that treatment with gentamicin restored sufficient α-L-iduronidase activity to largely reverse that mutant phenotype [15]. To examine lysosomal abundance in Idua-W392X MEFs, we used the lysosome-specific dye LysoTracker Red (Figure 4). Untreated Idua-W392X MEFs showed a 2.4-fold elevation in lysosomal signal abundance (defined as the total LysoTracker-positive pixels normalized to total DAPI-positive pixels) compared to WT MEFs (Figure 4G). Similarly, we observed a 3.5-fold increase in lysosomal signal intensity (defined as the total LysoTracker-positive pixel intensity normalized to total DAPI-positive pixels) compared to WT MEFs (Figure 4H). These results are consistent with an increase in lysosomal abundance in Idua-W392X MEFs. Since NB84 treatment restored enough α-L-iduronidase activity to decrease GAG storage in Idua-W392X MEFs (Figure 3B), we next asked whether abnormal lysosomal abundance was also reduced. NB84-treated Idua-W392X MEFs showed a 69% decrease in lysosome signal abundance, resulting in a level within 1.4-fold of WT (Figure 4G); and a 60% decrease in lysosome signal intensity, reducing intensity to within 2-fold of WT (Figure 4H). This experiment indicated that in addition to partially correcting two primary MPS I-H biochemical defects (enzymatic activity and GAG accumulation), NB84 also partially alleviated the secondary phenotype of abnormal lysosome abundance.
Figure 4. Lysosome abundance in MEFs.

(A–F) Representative confocal fluorescence microscopy images of wild-type MEFs (WT), Idua-W392X MEFs (W392X), and Idua-W392X MEFs treated with 2 mg/ml NB84 (W392X+NB84) for 48 hours. (A, C, E) LysoTracker Red stain and DAPI stain are pseudo-colored as red and blue, respectively. (B, D, F) LysoTracker Red stain, DAPI stain and CellMask Deep Red plasma membrane stain are pseudo-colored as red, blue, and green, respectively. Scale bar = 50 microns. (G) Quantitation of LysoTracker signal abundance normalized to DAPI in WT, W392X, and W392X + NB84 MEF cell cultures. (H) LysoTracker signal intensity normalized to DAPI in WT, W392X, and W392X+NB84 MEF cell cultures. The data is expressed as the mean +/− SD for approximately ≥100 cells for each condition. ** p <0.01, * p < 0.05. Image capture and quantitation were performed under identical microscopy settings. For the purpose of better visualization of images in Panels A–F, the contrast of the LysoTracker signal was increased by 50%. Additional quantitation information is found in the Materials and Methods.
NB84 treatment significantly decreased urine GAG excretion and tissue GAG accumulation in Idua-W392X mice
The Idua-W392X mouse model is characterized by elevated urinary GAG excretion and tissue GAG accumulation that are typical clinical manifestations found in MPS I-H patients [16]. Using this mouse model, we evaluated the ability of aminoglycosides to suppress the Idua-W392X mutation and alleviate the MPS I-H phenotype in vivo. To determine if these drugs could suppress the Idua-W392X PTC in vivo, we treated groups of Idua-W392X mice with escalating doses of gentamicin, NB54, or NB84 using once daily subcutaneous injections for 14 days. The effectiveness of treatment with each drug was monitored by assaying urine GAG excretion and tissue GAG storage in the spleen, heart, and brain of treated Idua-W392X mice compared to untreated mutant and WT controls.
Idua-W392X mice treated once daily with 30 or 60 mg/kg gentamicin did not show a significant decrease in urine GAG excretion or tissue GAG accumulation (data not shown). In contrast, mutant mice treated once daily with 30 mg/kg NB54 showed significant GAG reduction in the spleen, heart, and brain (Figure 5). Spleen GAG accumulation decreased 16%, heart GAG storage decreased 20%, and brain GAG storage decreased 21% (Figure 5). GAG reduction was not observed at higher doses (60 or 120 mg/kg) of NB54 (data not shown). In addition, no reduction in urine GAG excretion was observed in NB54-treated mice at any dose (data not shown). When taken together, these results demonstrate that NB54 can induce a small but significant decrease in GAG accumulation in multiple tissues, but this effect is only observed within a narrow dose range.
Figure 5. In vivo treatment with NB54.
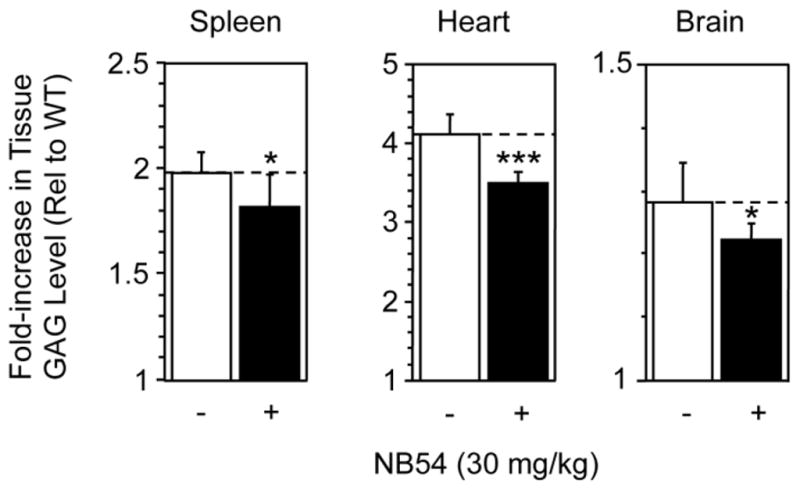
The level of GAGs was assayed in spleen, heart, and brain mouse tissues collected from Idua-W392X mice treated with 30 mg/kg NB54 and compared to the GAG levels in the same tissues from untreated Idua-W392X mice and WT controls. The data shown is the fold-increase in tissue GAG levels in untreated and treated Idua-W392X mice relative to WT controls. The dashed lines provide a reference for the GAG levels in untreated mutant mice. The data is expressed as the mean +/− SD of 9 assays from samples collected from 3 mice. *** p < 0.001, * p < 0.05.
We next examined the ability of NB84 to reduce GAG accumulation in Idua-W392X mice. NB84 administered once daily at 15 or 30 mg/kg reduced urine GAG levels 17% and 25%, respectively, compared to untreated mutant mice (Figure 6A). Urine GAG levels were not significantly reduced in Idua-W392X mice treated with 60 mg/kg NB84 once daily. Mutant mice treated with NB84 doses ranging from 15 to 60 mg/kg showed dose-dependent decreases in excess GAGs ranging from 30–65% in the spleen (Figure 6B). GAG storage was unchanged in the heart of Idua-W392X mice treated with 15 mg/kg NB84, but significant reductions in excess GAGs ranging from 13–23% were observed at higher doses. Finally, NB84 resulted in significant reductions in excess brain GAG storage that ranged from 27–40% when administered at doses of 30 and 60 mg/kg, respectively. When taken together, these results demonstrate that NB84 induced significant decreases in GAG accumulation in multiple tissues of Idua-W392X mice.
Figure 6. In vivo treatment with NB84.

(A) Urine GAG levels from untreated wild-type mice (WT) and Idua-W392X mice (W392X), or W392X mice after treatment with 15, 30 and 60 mg/kg of NB84. The data is expressed as the mean +/− SD of 8 assays collected from two groups of mice (3 mice/group). The levels of GAGs stored in (B) spleen, (C) heart, and (D) brain mouse tissues were assayed in Idua-W392X mice treated with 15, 30, and 60 mg/kg NB84 and compared to the GAG levels in untreated Idua-W392X mice and WT controls. The data shown is the fold-increase in tissue GAG levels in untreated and treated Idua-W392X mice relative to WT controls. The dashed lines provide a reference for the GAG levels in untreated mutant mice. The data is expressed as the mean +/− SD for 18 assays from 6 mice. *** p < 0.001, ** p < 0.01, ns = not significant. (E) Glycosphingolipid analysis of brain lysates from Idua-W392X mice treated with 30 mg/kg NB84 for two weeks (lane 6) compared to untreated WT (lane 4) and mutant (lane 5) controls. Glycosphingolipid standards are shown in lanes 1–3 and 7–8, where Ac-GM2 and Ac-GM3 respectively stand for N-acetylneuraminic acid-containing GM2 and N-acetylneuraminic acid-containing GM3. Likewise Gc-GM2 and Gc-GM3 respectively stand for N-glycolylneuraminic acid-containing GM2 and N-glycolylneuraminic acid-containing GM3. Asterisks (*) indicate the location of the Gc-GM2 and Gc-GM3 species found to be elevated in Idua-W392X mice.
We further evaluated the effect of NB84 treatment on an additional central nervous system (CNS) marker for MPS I-H. GM2 and GM3 gangliosides have been found to accumulate in the brain tissue of MPS I-H patients and animal models secondary to GAG accumulation [26, 27]. GM2 and GM3 gangliosides that appear to be N-glycolylneuraminic acid species [28, 29] were also found to be elevated in the brain of Idua-W392X mice compared to wild-type controls (Figure 6E). In Idua-W392X mice treated with 30 mg/kg NB84 for two weeks, we found that the GM2 and GM3 ganglioside levels remained elevated (Figure 6E). This indicated that while the experimental NB84 treatment conditions used restored enough α-L-iduronidase activity to significantly decrease GAG storage in the brain of Idua-W392X mice, they were not sufficient to reduce secondary ganglioside storage.
DISCUSSION
Aminoglycosides bind to the decoding center, a region of the ribosomal RNA conserved in prokaryotes and eukaryotes. The decoding center monitors base pairing between codons and anticodons to ensure the correct amino acid is incorporated into the nascent polypeptide chain [30, 31]. Aminoglycosides bind strongly to the prokaryotic decoding center. This association induces high levels of translational misreading at low drug concentrations, and completely blocks protein synthesis at higher concentrations. Previous studies have shown that two critical nucleotide differences in the eukaryotic decoding center significantly weaken its affinity for aminoglycoside binding [32–34]. These differences in binding affinity allow aminoglycosides to be used successfully as antibiotics. However, it has been shown that clinically relevant doses of certain aminoglycosides such as gentamicin can suppress translation termination at PTCs in mammalian cells [1–3]. Based on these findings, a subset of aminoglycosides has been repurposed as suppression agents to treat genetic diseases caused by nonsense mutations.
A major limitation of this approach is the observation that aminoglycosides are frequently nephrotoxic and/or ototoxic, which prohibits their long-term clinical use. Three distinct mechanisms have been proposed to underlie aminoglycoside toxicity. First, aminoglycosides become positively charged in the low pH of the lysosome and bind to negatively charged phospholipids in the lysosomal membrane [35, 36]. This interaction is thought to inhibit various membrane-associated biochemical pathways such as phospholipase signaling, which results in significant cellular pathology. Second, the charged nature of aminoglycosides induces formation of reactive oxygen species and oxidative damage [37, 38]. Third, the decoding center of mitochondrial ribosomes is similar to its bacterial counterpart, and some aminoglycosides inhibit mitochondrial translation [39].
Since aminoglycoside toxicity is not caused by stop codon suppression during cytoplasmic translation, it is feasible that structural elements within aminoglycosides that induce toxicity can be separated from those that induce PTC suppression. Refinement of ribosomal structures has provided better visualization of the differences in aminoglycoside binding to the eukaryotic and prokaryotic decoding centers. Using systematic structure-function analyses, structural elements within conventional aminoglycosides predicted to increase binding to the eukaryotic ribosome and reduce binding to the bacterial (or mitochondrial) ribosome were identified, including a paromamine backbone in paromomycin, the (S)-4-amino-hydroxybutanoic acid (AHB) group in amikacin, and the (R)-6′-methyl group in G418 (Figure 1). This strategy resulted in the generation of novel drug candidates such as NB54 and NB84 that are predicted to have enhanced efficacy for suppression therapy while also having significantly reduced toxicity [7, 8, 40, 41].
In this study, we evaluated the ability of the designer aminoglycosides NB54 and NB84 as well as several conventional aminoglycosides to suppress the mouse Idua-W392X mutation that causes MPS I-H. Readthrough reporter data indicated that the Idua-W392X sequence context is particularly resistant to suppression compared to other disease-causing PTCs (Figure 2) [7, 8, 42]. NB54 and NB84 suppressed the Idua-W392X PTC much more effectively than the conventional aminoglycosides gentamicin, amikacin, and G418, which are considered to be potent PTC suppressors. Using MEFs derived from the Idua-W392X mouse, we found NB84 restored a higher level of α-L-iduronidase than any of the other drugs tested, which corresponded to 0.32% of wild-type α-L-iduronidase activity (Figure 3A). A similar level of α-L-iduronidase activity was observed in skin fibroblasts from patients with milder forms of MPS I, including MPS I-Scheie and MPS I-Hurler/Scheie [13, 14]. In agreement with these previous observations, we found that this level of α-L-iduronidase activity restored by suppression therapy was sufficient to significantly reduce GAG levels (Figure 3B) and lysosomal abundance (Figure 4) in Idua-W392X MEFs.
Consistent with our in vitro data, NB54 and NB84 were found to be superior to gentamicin in alleviating the MPS I-H phenotype in vivo. Gentamicin administered at 30 or 60 mg/kg did not significantly reduce excess GAGs in the urine or tissues of Idua-W392X mice (data not shown). However, NB54 administered at 30 mg/kg reduced excess GAG storage ≈20% in multiple organs (Figure 5), but urine GAGs remained unchanged (data not shown). Idua-W392X mice administered NB84 at 30 mg/kg showed a 25% reduction in urine GAGs and a 23–55% reduction in excess tissue GAGs, suggesting that NB84 was superior to both NB54 and gentamicin in alleviating the phenotype in the Idua-W392X mouse. Together these data support the hypothesis that more efficacious suppression agents with decreased toxicity can be generated by rational drug design. This study represents the first in vivo demonstration of the effectiveness of NB84 as a suppression drug. In addition, this is the first in vivo demonstration that suppression therapy can attenuate the primary biochemical defect associated with the lysosomal storage disease MPS I-H.
This initial evaluation of drug efficacy for the suppression of the Idua-W392X nonsense mutation, restoration of functional α-L-iduronidase, and reduction in excess GAG storage was conducted in 8–11 week old mice for a two-week period to determine which drugs might prove to be effective in treating MPS I-H. Many MPS I-H phenotypes including bone and heart abnormalities as well as other tissue and organ aberrations associated with MPS I-H do not occur in Idua-W392X mice until they are approximately 20–40 weeks old [16]. Longer term studies will be required to determine whether the level of GAG reduction observed in NB84-treated mice is sufficient to prevent the onset of these defects. It was previously shown that the level of residual α-L-iduronidase activity in MPS I patient skin fibroblasts can roughly be correlated with the severity of the clinical phenotype [13, 14]. In general, patients with the severe Hurler form of MPS I had less than 0.1% of normal α-L-iduronidase activity. Patients with the attenuated Hurler-Scheie and Scheie forms of MPS I had higher levels of residual α-L-iduronidase activity that generally ranged from 0.1% – 7% of normal. We were unable to determine the level of α-L-iduronidase activity restored in vivo due to inhibitory factors in the mouse tissue lysates that greatly reduced our assay sensitivity. However, we did find that NB84 restored 0.32% of normal α-L-iduronidase activity in MEFs derived from the Idua-W392X mouse, which significantly alleviated both biochemical and morphological abnormalities. The correlation between the level of tissue GAG storage and the severity of the MPS I phenotype has not been as clearly defined. However, one study found that skin fibroblasts from patients with mild forms of MPS I retained 20–70% fewer GAGs than cells from patients with the severe form of MPS I [14]. Based on these values, the 23% – 65% reduction in excess tissue GAGs observed in Idua-W392X mice treated with NB84 may be sufficient to attenuate the MPS I-H phenotype in at least some tissues.
Three therapeutic approaches are currently available for MPS I-H patients: hematopoietic stem cell transplantation (HSCT), enzyme replacement therapy (ERT) and substrate reduction therapy (SRT). Each effectively treats many, but not all, aspects of MPS I-H disease. HSCT is unable to correct skeletal and heart defects. In order to prevent the onset of neurological dysfunction, HSCT must be performed at an early age and patients are subjected to a high risk of morbidity and mortality due to engraftment complications [43, 44]. In contrast, ERT is unable to improve neurological dysfunction in MPS I-H patients because the recombinant enzyme cannot cross the blood-brain barrier [45]. SRT using the isoflavone genistein was found to inhibit GAG synthesis and partially reduce excess GAG storage [46]. Preliminary clinical trials demonstrated that SRT using genistein improved cognitive function in patients with MPS III (Sanfillipo) disease, suggesting that genistein reduces GAG storage in the CNS and may be applied as a treatment for other MPS diseases [47]. Suppression therapy may be a suitable supplement to these current MPS I-H treatments because: 1) nonsense mutations represent the majority of lesions in many MPS I-H populations [48]; 2) MPS I-H has a low threshold for phenotypic improvement, since only 0.1–0.3% of normal enzyme levels can reduce disease severity [13, 14]; and 3) aminoglycosides such as gentamicin can cross the blood-brain barrier to some extent, potentially enabling α-L-iduronidase function to be partially restored in the CNS [49, 50]. Importantly, after two weeks of NB84 treatment, GAGs in the Idua-W392X mouse were reduced to levels that were equal to or greater than the levels of GAG reduction observed after ten weeks of genistein treatment in an MPS II mouse model [51]. This further suggests that suppression therapy may provide a viable way to reduce excess GAG accumulation associated with lysosomal storage diseases that are caused by nonsense mutations.
Aminoglycosides are primarily endocytosed into mammalian cells via the megalin receptor. Megalin is expressed in epithelial cells, including the choroid plexus modified ependymal cells that line the cerebral ventricles, which directs a portion of circulating aminoglycosides to the cerebrospinal fluid (CSF) [52]. In healthy animal models, aminoglycosides accumulate in the CSF at levels that are approximately 20% of serum aminoglycoside levels [53, 54]. Based on those results, we expected at least a portion of the aminoglycosides that we tested to cross the blood-brain barrier. Significantly, we consistently observed a 20–40% decrease in excess GAG storage in the brain tissues of NB54 and NB84 treated Idua-W392X mice compared to untreated controls (Figures 5 & 6D). Although an assay to directly measure NB54 or NB84 concentrations in brain lysates is not yet available, these results indicate that a functionally significant amount of NB54 and NB84 can enter the CSF and suppress the Idua-W392X mutation within the CNS tissues. The observation that NB54 and NB84 can at least partially alleviate one aspect of MPS I-H CNS dysfunction suggests that other PTC-induced disorders that exhibit CNS dysfunction such as Rett syndrome may also benefit from these designer drugs [55].
Since the Idua-W392X PTC is particularly resistant to readthrough, it is extremely promising that a significant decrease in GAG accumulation could be observed using this mouse model. In this regard, NB54 and NB84 provide promising scaffolds for the development of additional aminoglycoside derivatives with even greater PTC suppression efficacy for MPS I-H as well as other diseases caused by PTCs. Additionally, the co-administration of poly-L-aspartate may provide a way to enhance the effects of NB54 and NB84. This polymer has not only been shown to alleviate aminoglycoside-induced nephrotoxicity [56], but to also enhance the efficiency of in vivo gentamicin-mediated PTC suppression by increasing the cytoplasmic aminoglycoside concentration [6]. Finally, combining suppression therapy with other current MPS I-H treatments such as HSCT, ERT, or SRT [43, 44, 57] may enhance the therapeutic benefits in organs that are resistant to current treatments, such as the brain, heart, or bone. A combinatorial approach may not only prove to be valuable for the treatment of MPS I-H, but to a wide variety of genetic diseases caused by nonsense mutations.
Highlights.
Various aminoglycosides that suppress nonsense mutations with were tested.
NB54 and NB84 suppressed a nonsense mutation in an MPS I-H mouse.
NB84 significantly reduced GAG levels in the Idua-W392X mouse.
Acknowledgments
This research was supported by grants from the NIH/NINDS (1 R01 NS057412-04 for DMB & KMK), the University of Pennsylvania Improved Therapies for MPS I Grant Program (MPS I-11-001-01 for DMB & KMK), the NIH/NIGMS (1 R01 GM094792-01A1 for TB & DMB), and the US-Israel Binational Science Foundation (2006/301 for TB). We wish to thank Shawn Williams with the UAB High Resolution Imaging Facility for technical advice and Prof. Tamio Yamakawa (University of Tokyo) for providing N-glycolyl-GM2 and N-glycolyl-GM3 standards. We also wish to thank Yanying Dai for her assistance in mouse colony maintenance. VB acknowledges financial support by the Center of Absorption in Science, the Ministry of Immigration Absorption, and the Ministry of Science and Technology, Israel (Kamea Program). JK acknowledges the Schulich Postdoctoral Fellowship.
Footnotes
DISCLOSURES
DMB and KMK have proprietary and financial interests in the treatment of lysosomal storage diseases with aminoglycosides (Patent # 7,749,971). TB has proprietary and financial interests in the treatment of genetic diseases with novel aminoglycosides (PCT application # WO 2007113841 A2 20071011). DMB is a paid consultant for PTC Therapeutics, Incorporated.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Keeling KM, Bedwell DM. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. WIREs RNA. 2011 doi: 10.1002/wrna.95. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keeling KM, Bedwell DM. Recoding therapies for genetic diseases. In: Atkins JF, Gesteland RF, editors. Recoding: Expansion of Decoding Rules Enriches Gene Expression. Springer Publishing; New York: 2010. [Google Scholar]
- 3.Linde L, Kerem B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24:552–563. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Du L, Damoiseaux R, Nahas S, Gao K, Hu H, Pollard JM, Goldstine J, Jung ME, Henning SM, Bertoni C, Gatti RA. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J Exp Med. 2009;206:2285–2297. doi: 10.1084/jem.20081940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 6.Du M, Keeling KM, Fan L, Liu X, Bedwell DM. Poly-L-aspartic Acid Enhances and Prolongs Gentamicin-mediated Suppression of the CFTR-G542X Mutation in a Cystic Fibrosis Mouse Model. J Biol Chem. 2009;284:6885–6892. doi: 10.1074/jbc.M806728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nudelman I, Glikin D, Smolkin B, Hainrichson M, Belakhov V, Baasov T. Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg Med Chem. 2010;18:3735–3746. doi: 10.1016/j.bmc.2010.03.060. [DOI] [PubMed] [Google Scholar]
- 8.Nudelman I, Rebibo-Sabbah A, Cherniavsky M, Belakhov V, Hainrichson M, Chen F, Schacht J, Pilch DS, Ben-Yosef T, Baasov T. Development of Novel Aminoglycoside (NB54) with Reduced Toxicity and Enhanced Suppression of Disease-Causing Premature Stop Mutations. J Med Chem. 2009;52:2836–2845. doi: 10.1021/jm801640k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793:684–696. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–565. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 11.Neufeld EF, Muenzer J. The Mucopolysaccharidoses, The Online Metabolic & Molecular Bases of Inherited Disease. The McGraw-Hill Companies; 2001. pp. 3421–3452. ( www.ommbid.com) [Google Scholar]
- 12.Walkley SU. Pathogenic cascades in lysosomal disease-Why so complex? J Inherit Metab Dis. 2009;32:181–189. doi: 10.1007/s10545-008-1040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashton LJ, Brooks DA, McCourt PA, Muller VJ, Clements PR, Hopwood JJ. Immunoquantification and enzyme kinetics of alpha-L-iduronidase in cultured fibroblasts from normal controls and mucopolysaccharidosis type I patients. Am J Hum Genet. 1992;50:787–794. [PMC free article] [PubMed] [Google Scholar]
- 14.Bunge S, Clements PR, Byers S, Kleijer WJ, Brooks DA, Hopwood JJ. Genotype-phenotype correlations in mucopolysaccharidosis type I using enzyme kinetics, immunoquantification and in vitro turnover studies. Biochim Biophys Acta. 1998;1407:249–256. doi: 10.1016/s0925-4439(98)00046-5. [DOI] [PubMed] [Google Scholar]
- 15.Keeling KM, Brooks DA, Hopwood JJ, Li P, Thompson JN, Bedwell DM. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum Mol Genet. 2001;10:291–299. doi: 10.1093/hmg/10.3.291. [DOI] [PubMed] [Google Scholar]
- 16.Wang D, Shukla C, Liu X, Schoeb TR, Clarke LA, Bedwell DM, Keeling KM. Characterization of an MPS I-H knock-in mouse that carries a nonsense mutation analogous to the human IDUA-W402X mutation. Mol Genet Metab. 2010;99:62–71. doi: 10.1016/j.ymgme.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke LA, Russell CS, Pownall S, Warrington CL, Borowski A, Dimmick JE, Toone J, Jirik FR. Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum Mol Genet. 1997;6:503–511. doi: 10.1093/hmg/6.4.503. [DOI] [PubMed] [Google Scholar]
- 18.Grentzmann G, Ingram JA, Kelly PJ, Gesteland RF, Atkins JF. A dual-luciferase reporter system for studying recoding signals. RNA. 1998;4:479–486. [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson K, Li SC, Li YT. Diphenylamine-aniline-phosphoric acid reagent, a versatile spray reagent for revealing glycoconjugates on thin-layer chromatography plates. Anal Biochem. 2000;287:337–339. doi: 10.1006/abio.2000.4829. [DOI] [PubMed] [Google Scholar]
- 20.Martin R. On the relationship between preferred termination codon contexts and nonsense suppression in human cells. Nucleic Acids Res. 1994;22:15–19. doi: 10.1093/nar/22.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips-Jones MK, Hill LS, Atkinson J, Martin R. Context effects on misreading and suppression at UAG codons in human cells. Mol Cell Biol. 1995;15:6593–6600. doi: 10.1128/mcb.15.12.6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–1055. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cassan M, Rousset JP. UAG readthrough in mammalian cells: effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol Biol. 2001;2:3. doi: 10.1186/1471-2199-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howard MT, Anderson CB, Fass U, Khatri S, Gesteland RF, Atkins JF, Flanigan KM. Readthrough of dystrophin stop codon mutations induced by aminoglycosides. Ann Neurol. 2004;55:422–426. doi: 10.1002/ana.20052. [DOI] [PubMed] [Google Scholar]
- 25.Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Sequence specificity of aminoglycoside-induced stop codon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol. 2000;48:164–169. [PubMed] [Google Scholar]
- 26.Walkley SU. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin Cell Dev Biol. 2004;15:433–444. doi: 10.1016/j.semcdb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Russell C, Hendson G, Jevon G, Matlock T, Yu J, Aklujkar M, Ng KY, Clarke LA. Murine MPS I: insights into the pathogenesis of Hurler syndrome. Clin Genet. 1998;53:349–361. doi: 10.1111/j.1399-0004.1998.tb02745.x. [DOI] [PubMed] [Google Scholar]
- 28.Hashimoto Y, Otsuka H, Yamakawa T. The occurrence of GM4 and GM2 in erythrocytes from inbred strains of mice. J Biochem. 1982;91:1039–1046. doi: 10.1093/oxfordjournals.jbchem.a133753. [DOI] [PubMed] [Google Scholar]
- 29.Yamakawa T, Suzuki S. The Chemistry of the Lipids of Posthemolytic Residue or Stroma of Erythrocytes II. On the Structure of Hemataminic. Acid J Biochem. 1951;39:175–184. [Google Scholar]
- 30.Recht MI, Fourmy D, Blanchard SC, Dahlquist KD, Puglisi JD. RNA sequence determinants for aminoglycoside binding to an A-site rRNA model oligonucleotide. J Mol Biol. 1996;262:421–436. doi: 10.1006/jmbi.1996.0526. [DOI] [PubMed] [Google Scholar]
- 31.Blanchard SC, Fourmy D, Eason RG, Puglisi JD. rRNA chemical groups required for aminoglycoside binding. Biochemistry (Mosc) 1998;37:7716–7724. doi: 10.1021/bi973125y. [DOI] [PubMed] [Google Scholar]
- 32.Lynch SR, Puglisi JD. Structural origins of aminoglycoside specificity for prokaryotic ribosomes. J Mol Biol. 2001;306:1037–1058. doi: 10.1006/jmbi.2000.4420. [DOI] [PubMed] [Google Scholar]
- 33.Lynch SR, Puglisi JD. Structure of a eukaryotic decoding region A-site RNA. J Mol Biol. 2001;306:1023–1035. doi: 10.1006/jmbi.2000.4419. [DOI] [PubMed] [Google Scholar]
- 34.Fan-Minogue H, Bedwell DM. Eukaryotic ribosomal RNA determinants of aminoglycoside resistance and their role in translational fidelity. RNA. 2007;14:148–157. doi: 10.1261/rna.805208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagai J, Takano M. Molecular aspects of renal handling of aminoglycosides and strategies for preventing the nephrotoxicity. Drug Metab Pharmacokinet. 2004;19:159–170. doi: 10.2133/dmpk.19.159. [DOI] [PubMed] [Google Scholar]
- 36.Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 1999;43:1003–1012. doi: 10.1128/aac.43.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawamoto K, Sha SH, Minoda R, Izumikawa M, Kuriyama H, Schacht J, Raphael Y. Antioxidant gene therapy can protect hearing and hair cells from ototoxicity. Mol Ther. 2004;9:173–181. doi: 10.1016/j.ymthe.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 38.Sha SH, Schacht J. Antioxidants attenuate gentamicin-induced free radical formation in vitro and ototoxicity in vivo: D-methionine is a potential protectant. Hear Res. 2000;142:34–40. doi: 10.1016/s0378-5955(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 39.Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Bottger EC. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Natl Acad Sci U S A. 2008;105:20888–20893. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pokrovskaya V, Nudelman I, Kandasamy J, Baasov T. Aminoglycosides redesign strategies for improved antibiotics and compounds for treatment of human genetic diseases. Methods Enzymol. 2010;478:437–462. doi: 10.1016/S0076-6879(10)78021-6. [DOI] [PubMed] [Google Scholar]
- 41.Nudelman I, Rebibo-Sabbah A, Shallom-Shezifi D, Hainrichson M, Stahl I, Ben-Yosef T, Baasov T. Redesign of aminoglycosides for treatment of human genetic diseases caused by premature stop mutations. Bioorg Med Chem Lett. 2006;16:6310–6315. doi: 10.1016/j.bmcl.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 42.Keeling KM, Bedwell DM. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J Mol Med. 2002;80:367–376. doi: 10.1007/s00109-001-0317-z. [DOI] [PubMed] [Google Scholar]
- 43.Aldenhoven M, Boelens JJ, de Koning TJ. The clinical outcome of Hurler syndrome after stem cell transplantation. Biol Blood Marrow Transplant. 2008;14:485–498. doi: 10.1016/j.bbmt.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Krivit W, Aubourg P, Shapiro E, Peters C. Bone marrow transplantation for globoid cell leukodystrophy, adrenoleukodystrophy, metachromatic leukodystrophy, and Hurler syndrome. Curr Opin Hematol. 1999;6:377–382. doi: 10.1097/00062752-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 45.Wraith EJ, Hopwood JJ, Fuller M, Meikle PJ, Brooks DA. Laronidase treatment of mucopolysaccharidosis I. BioDrugs. 2005;19:1–7. doi: 10.2165/00063030-200519010-00001. [DOI] [PubMed] [Google Scholar]
- 46.Wegrzyn G, Jakobkiewicz-Banecka J, Gabig-Ciminska M, Piotrowska E, Narajczyk M, Kloska A, Malinowska M, Dziedzic D, Golebiewska I, Moskot M, Wegrzyn A. Genistein: a natural isoflavone with a potential for treatment of genetic diseases. Biochem Soc Trans. 2010;38:695–701. doi: 10.1042/BST0380695. [DOI] [PubMed] [Google Scholar]
- 47.Piotrowska E, Jakobkiewicz-Banecka J, Maryniak A, Tylki-Szymanska A, Puk E, Liberek A, Wegrzyn A, Czartoryska B, Slominska-Wojewodzka M, Wegrzyn G. Two-year follow-up of Sanfilippo Disease patients treated with a genistein-rich isoflavone extract: Assessment of effects on cognitive functions and general status of patients. Med Sci Monit. 2011;17:CR196–202. doi: 10.12659/MSM.881715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bunge S, Kleijer WJ, Steglich C, Beck M, Zuther C, Morris CP, Schwinger E, Hopwood JJ, Scott HS, Gal A. Mucopolysaccharidosis type I: identification of 8 novel mutations and determination of the frequency of the two common alpha-L-iduronidase mutations (W402X and Q70X) among European patients. Hum Mol Genet. 1994;3:861–866. doi: 10.1093/hmg/3.6.861. [DOI] [PubMed] [Google Scholar]
- 49.McCracken GH, Jr, Chrane DF, Thomas ML. Pharmacologic evaluation of gentamicin in newborn infants. J Infect Dis. 1971;124:Suppl 124, 214. doi: 10.1093/infdis/124.supplement_1.s214. [DOI] [PubMed] [Google Scholar]
- 50.Newman RL, Holt RJ. Intrathecal gentamicin in treatment of ventriculitis in children. Br Med J. 1967;2:539–542. [PMC free article] [PubMed] [Google Scholar]
- 51.Friso A, Tomanin R, Salvalaio M, Scarpa M. Genistein reduces glycosaminoglycan levels in a mouse model of mucopolysaccharidosis type II. Br J Pharmacol. 2010;159:1082–1091. doi: 10.1111/j.1476-5381.2009.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002;3:256–266. doi: 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- 53.Strausbaugh LJ, Brinker GS. Effect of osmotic blood-brain barrier disruption on gentamicin penetration into the cerebrospinal fluid and brains of normal rabbits. Antimicrob Agents Chemother. 1983;24:147–150. doi: 10.1128/aac.24.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith AL, Daum RS, Siber GR, Scheifele DW, Syriopoulou VP. Gentamicin penetration into cerebrospinal fluid in experimental Haemophilus influenzae meningitis. Antimicrob Agents Chemother. 1988;32:1034–1039. doi: 10.1128/aac.32.7.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brendel C, Belakhov V, Werner H, Wegener E, Gartner J, Nudelman I, Baasov T, Huppke P. Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J Mol Med. 2010;89:389–398. doi: 10.1007/s00109-010-0704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swan SK, Kohlhepp SJ, Kohnen PW, Gilbert DN, Bennett WM. Long-term protection of polyaspartic acid in experimental gentamicin nephrotoxicity. Antimicrob Agents Chemother. 1991;35:2591–2595. doi: 10.1128/aac.35.12.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hawkins-Salsbury JA, Reddy AS, Sands MS. Combination therapies for lysosomal storage disease: is the whole greater than the sum of its parts? Hum Mol Genet. 2011;20:R54–60. doi: 10.1093/hmg/ddr112. [DOI] [PMC free article] [PubMed] [Google Scholar]