

Abstract
Classic Galactosemia is an autosomal recessive disorder caused by the deficiency of galactose-1-phosphate uridylyltransferase (GALT), one of the key enzymes in the Leloir pathway of galactose metabolism. While the neonatal morbidity and mortality of the disease are now mostly prevented by newborn screening and galactose restriction, long-term outcome for older children and adults with this disorder remains unsatisfactory. The pathophysiology of Classic Galactosemia is complex, but there is convincing evidence that galactose-1-phosphate (gal-1P) accumulation is a major, if not the sole pathogenic factor. Galactokinase (GALK) inhibition will eliminate the accumulation of gal-1P from both dietary sources and endogenous production, and efforts towards identification of therapeutic small molecule GALK inhibitors are reviewed in detail. Experimental and computational high-throughput screenings of compound libraries to identify GALK inhibitors have been conducted, and subsequent studies aimed to characterize, prioritize, as well as to optimize the identified positives have been implemented to improve the potency of promising compounds. Although none of the identified GALK inhibitors inhibit glucokinase and hexokinase, some of them cross-inhibit other related enzymes in the GHMP small molecule kinase superfamily. While this finding may render the on-going hit-to-lead process more challenging, there is growing evidence that such cross-inhibition could also lead to advances in antimicrobial and anti-cancer therapies.
1. Introduction
Galactose is the C-4 epimer of glucose, with an identical molecular formula, but a distinct structural formula. Despite its strong structural similarity to glucose, the conversion from galactose into glucose requires a few evolutionarily-conserved enzymatic steps, all residing in the cytoplasm, known as the Leloir pathway of galactose metabolism [1]. The main source of galactose in humans is dietary, mainly dairy products containing lactose, but other non-dairy foodstuffs can also contain galactose moieties [2, 3]. In humans, galactose can also be produced endogenously, mostly through the enzymatic conversion between uridine diphosphate-glucose (UDP-glucose) and UDP-galactose, as well as the turnover of glycoprotein and glycolipids [4, 5]. Upon entry to the Leloir pathway, galactose is first phosphorylated by galactokinase (GALK) to form galactose-1-phosphate (gal-1P) [6]. Together with the second substrate UDP-glucose, gal-1P is converted by galactose-1-phosphate uridylyltransferase (GALT) to form UDP-galactose and glucose-1-phosphate [7]. The Leloir pathway is completed by reversibly forming UDP-glucose from UDP-galactose by UDP-galactose-4-epimerase (GALE) [8, 9] (see Fig. 1). Enzyme deficiencies in the Leloir pathway, caused by bi-allelic amorphic or hypomorphic mutations in any of the genes coding for the GAL enzymes have been described (see refs [10-14] for extensive reviews on this subject). Of these deficiencies, the most common disorder is Classic (Type I) Galactosemia, which is caused by bi-allelic amorphic mutations in the GALT gene, and is the main focus of this review. Infants born with Classic Galactosemia usually become ill within days after birth if exposed to breast milk or lactose-containing formula. Initially, the infant develops jaundice, and if lactose exposure continues, complications such as liver failure, Escherichia coli (E. coli) sepsis, coma, and death follow shortly after [13]. The main aspect of management is the replacement of lactose/galactose using soy-based formula, after which the infant usually recovers fairly quickly [13]. All 50 states in the U.S. and many developed countries have included Classic Galactosemia as one of the conditions screened for in the newborn period, ensuring that most infants survive without becoming ill [15].
Fig. 1. The metabolic pathway of galactose in humans [57].

Despite a galactose-restricted diet, most patients with Classic Galactosemia continue to accumulate significant amount of galactose, galactitol and gal-1P in their cells [13, 16-18]. Further, it has become clear that even with early detection and (early) dietary intervention, there is still a significant burden of this disease due to chronic complications that arise in childhood and adulthood. The most common complications are speech dyspraxia, ataxia, and premature ovarian insufficiency [19, 20]. To date, the pathophysiology of the acute toxicity syndrome and the chronic complications remains largely unknown, but it is reasonable to assume that any blockage in a metabolic pathway will lead to (i) accumulating precursor(s), (ii) alternate metabolites normally not encountered, or (iii) absent metabolites past the enzymatic block. Any, or a combination of these possibilities, could be responsible for the phenotypes associated with the enzymatic blockage. As to GALT-deficiency Classic Galactosemia, it is apparent that galactose and gal-1P accumulate in patients, with galactose being further metabolized through two alternative pathways to form galactitol and galactonate [17, 18, 21, 22]. Among all the metabolites formed, gal-1P and galactitol have received most attention. But what are the potential toxicity targets of these toxic metabolites, and between gal-1P and galactitol, which plays a more important role in the pathophysiology of Classic Galactosemia?
Various reports suggested that gal-1P competitively inhibited UDP-glucose pyrophosphorylase [23-25], inositol monophosphatase [25-28], phosphoglucomutase [29], glycogen phosphorylase [30], or even glucose-6-phosphatase [31], although none of these in vitro findings have been fully substantiated in human patients in vivo. Nevertheless, if inhibition of UDP-glucose pyrophosphorylase occurs in vivo, it could potentially reduce UDP-glucose/galactose formation in the cells, and could cause aberrant glycosylation of proteins and lipids. In fact, there are reports showing abnormal circulating proteins, including transferrin and follicle-stimulating hormone (FSH) in the blood of GALT-deficient patients with elevated erythrocyte gal-1P [32-34]. Because of these findings, Classic Galactosemia has been broadly regarded as a congenital disorder of glycosylation. Recently, there is a renewed interest in the potential link between inositol metabolism and galactose metabolism [35, 36]. If inhibition of inositol monophosphatases takes place in vivo as it was demonstrated in in vitro experiments [25-28], it could lead to reduced free inositol, and accumulation of inositol monophosphates in the cells, Indeed, decreased free and lipid-bound inositol in the tissues of both GALT-deficient patients and galactose-intoxicated rats has been reported [37, 38].
Regarding the toxicity targets of galactitol, Berry and coworkers hypothesized that galactitol may also inhibit Na+/myo-inositol transporter (SMIT1)-mediated myo-inositol transport in vivo by osmoregulatory control and jeopardize the availability of myo-inositol in the cellular level [35]. As excess galactose accumulates inside the GALT-deficient cells, it is reduced to galactitol by aldose reductase. Similar to sorbitol, the excess galactitol formed will cause osmotic imbalance inside the cells and through the action of TonEBP/OREBP and/or AP-1, lead to reduced transcription of the SLC5A3 gene, which encodes the SMIT1 transporter [39-43]. One of the important roles of myo-inositol is to serve as the precursor of phosphatidylinositol-4,5-biphosphate (PtdIns-4,5-P2), which is essential for numerous intracellular signal transduction pathways [44]. A severe deficiency of myo-inositol would therefore impair multiple signal transduction pathways such as the PI3K/AKT/mTOR cell growth pathway [45, 46] (Fig. 2). In fact, it has been shown that complete ablation of the myo-inositol transporter gene SLC5A3 is lethal in mice due to abnormal electrical discharge from the respiratory control center in the brain [47]. Administration of myo-inositol via the drinking water of the pregnant female carrier mouse can prevent the death of the knock-out mice [48], but as adults, they still have problems related to abnormal brain development [48, 49]. Similarly, deficiency of myo-inositol in patients with Classic Galactosemia caused by the inhibition of SMIT by galactitol, may also confer the central nervous system complications.
Fig. 2. Proposed effects of blockage in galactose metabolic pathway on the myo-inositol metabolic pathway and related signal transduction pathways.

Gal-1P (an intermediate) and galactitol (a by-product) of the blocked galactose metabolic pathway interfere inositol turnover and inositol entry by inhibiting IMPase and SMIT, respectively. This could potentially decrease cellular PIP2 content and adversely affect normal AKT function. Blockage of galactose metabolism has also been shown to up-regulate ARHI gene expression [132], which negatively regulate AKT activity in the cells. (Abbreviations: PIP2: phosphatidylinositol-4,5-biphosphate; IP3: inositol-1,4,5-triphosphate; IP2: inositol bisphosphate; IP1: inositol monophosphate; PIP3: phosphatidylinositol-3,4,5-triphosphate; SMIT1: Na+/myo-inositol transporter; IMPase: inositol monophosphatase; P13K: phosphatidylinositol 3-kinase; GPCR: G-protein coupled receptor. L: ligand for GPCR; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; PTEN: phosphatase and tensin homolog; ARHI: aplysia ras homolog I.)
But between gal-1P and galactitol, which is a more important pathogenic factor for Classic Galactosemia? As GALK-deficient patients who accumulates galactitol but not gal-1P develop mostly cataracts [50, 51], rather than the long-term central nervous system (CNS) and ovarian complications seen in Classic Galactosemia [52, 53], it has been proposed that the accumulation of gal-1P, but not galactitol, is more crucial for the development of these complications. However, this does not necessarily preclude any potential synergistic effects of galactitol in the presence of elevated gal-1P.
Based on these important lines of evidence, which strongly suggested that gal-1P is the most important and determinant factor for developing long-term complications in Classic Galactosemia, we and others (other way around sounds awkward, even if polite) proposed that pharmacological inhibition GALK, in conjunction with dietary (ga-)lactose restriction, will provide a novel, improved treatment for this disorder by preventing gal-1P accumulation [54-57]. In the ensuing sections, we will evaluate the merits of pharmacological inhibition of GALK in the context of different therapeutic approaches to treat Inborn Errors of Metabolism (Section 2). In Section 3, we will explain why we prefer to inhibit human GALK enzymes using small molecule compounds among other approaches, and in Section 4, we will describe how we began to identify such inhibitors experimentally. However, identification of such first-generation small molecule inhibitors is only the first, albeit essential step of any drug-discovery process. In Sections 5 and 6, we will describe the required steps of characterization, prioritization and optimization that we have taken in order to yield selective (specific), safe and more potent derivatives. In Section 7, we will high-light some of the potential novel therapeutic uses of the GALK inhibitors in other common diseases. We will conclude, in Section 8, by discussing some of the challenges that we will face in bringing the GALK inhibitors that we discovered from bench to bedside.
2. Current Therapeutic Approaches to Inborn Errors of Metabolism
Inborn errors of metabolism (IEM) comprise a large group of disorders, commonly caused by inactivity of specific enzymatic steps, leading to compromised flux of molecules in evolutionarily conserved metabolic pathways. Traditionally, IEM are categorized as those involved in (a) synthesis or catabolism of complex molecules (e.g., congenital glycosylation disorders), (b) intermediate metabolism, (e.g., amino acidopathies, organic acidemias, urea cycle defects, and disorders of carbohydrate metabolism including Classic Galactosemia), and (c) energy metabolism, (e.g., disorders of gluconeogenesis, lactic acidemias, fatty acid oxidation defects, and Krebs cycle and mitochondrial respiratory chain disorders)[58].
Clearly, the symptomatology and approach to management of these disorders are diverse, and beyond the scope of this review. Here we will focus on highlighting the various approaches that have been attempted or could potentially be employed in the management of such disorders. In a way, one could categorize the various levels of management as those at organ/tissue level, those at the genome level, those at the RNA level, those at the protein/enzyme level, and those at the level of small molecules (Fig. 3) [58, 59]. We will briefly mention some of these (potential) approaches, with the realization that this overview cannot be exhaustive.
Fig. 3. Overview of current therapeutic treatment approaches for inborn error of metabolism at different levels of interaction [58, 59].

A. Approaches at the organ/tissue level
Transplantation
In some cases, the action of an enzymatic step may take place in a single organ, such as liver or in a single tissue, such as bone marrow; and by providing a tissue/organ with normal enzyme activity will benefit all other tissues/organs. Replacement of the affected organ/tissue with that from an unaffected donor may thus overcome the disorder, and restore normal metabolism. Examples of this approach are β-L-iduronidase deficiency (Hurler syndrome) [60, 61] and sickle cell disease [62, 63].
B. Approaches at the genome level
Gene therapy
This approach focuses on the insertion of a normal gene, often through the use of a (viral) vector, into cells of the recipient’s genome, with the aim that the gene contained within this vector will be translated, in order to overcome the metabolic block. Examples of this approach are gene therapies for adenosine deaminase (ADA) deficiency [64, 65], Leber’s congenital amaurosis [66], X-linked adrenoleukodystrophy (ALD) [67] and lysosomal storage diseases [68]. This approach is further strengthened by the recent advances in induced pluripotent stem (IPS) cells research. As IPS cells are actually induced from individual’s somatic cells [69-71], there is no immune incompatibility issue. Besides, these IPS cells can redifferentiate into many tissues, if the correct molecular signals and /or growth conditions were given [72, 73]. Introducing specific gene into IPS cells derived from patients and selecting proper colonies of IPS cells before re-introducing or redifferentiating and re-introducing back to patients, gene therapy can be performed in an easier, more efficient way nearly without concern of immune incompatibility. This approach has been achieved in metabolic disease animal models such as sickle cell anemia [74].
C. Approaches at the RNA level
Antisense-mediated exon skipping
This approach focuses on restoration of the open reading frame by selectively blocking one of more exons. While currently considered experimental, there are promising results in disorders as Duchenne muscular dystrophy [75].
Stop codon read-through
This approach focuses on restoration of protein translation in the presence of truncating mutations due to a premature stop codon. It is currently experimental, but has shown promise in disorders such as Duchenne muscular dystrophy [76], methylmalonic aciduria [77], Menkes disease [78], and β-L-iduronidase deficiency (Hurler syndrome) [79].
RNA interference (RNAi) therapy
This approach focuses on targeted gene silencing using small interfering RNA targeting mRNA for destruction. It is also currently experimental. Potential applications are in blocking flux in a metabolic pathway in patients affected by a more severe enzyme defect (and resulting accumulation of a toxic substrate) further downstream in the pathway [80].
D. Approaches at the protein/enzyme level
Enzyme replacement
This approach has been proven beneficial in many metabolic disorders, mostly storage disorders, as β-L-iduronidase deficiency (Hurler syndrome) [81, 82], Fabry disease [83, 84], Gaucher disease [85], but may also be beneficial in other disorders, such as phenylketonuria [86].
Chaperone therapy
This approach aims to prevent misfolding of a mutant protein. Potential applications explored thus far include cystic fibrosis [87] and β-1-antitrypsin deficiency [88].
Co-factor supplementation
This approach has been proven beneficial in many metabolic disorders, as homocystinuria [89] and phenylketonuria [90].
Small molecule inhibitors of an enzyme upstream of a blocked metabolic pathway
This approach has been proven beneficial in disorders as tyrosinemia type 1 (nitisinone as an inhibitor of 4-hydroxyphenylpyruvate dioxygenase) [91], and in gout (allopurinol as an inhibitor of xanthine oxidase) [92]
Small molecule analogs of missing substrate/activator
This approach has been proven beneficial in disorders as N-acetyl glutamate synthase (NAGS) deficiency (carglumic acid, a functional analogue of N-acetyl glutamate, restores the urea cycle by activating the carbamylphosphate synthetase) [93].
E. Approaches to control (small) molecule concentrations
Substrate reduction
This is a traditional approach in the management of many metabolic disorders. Through dietary manipulation such as the use of metabolic foods, the concentration of the accumulating toxic compound is reduced in conditions such as Classic Galactosemia as we discussed above.
Product supplementation
This is a traditional approach in the management of many metabolic disorders, often in combination with substrate reduction. It allows continued flux through vital metabolic pathways. An example is tyrosine supplementation in phenylketonuria [94].
Alternative elimination
This approach has been proven beneficial in the management of urea cycle disorders, where small molecules as sodium phenylacetate and sodium benzoate are provided to bind glutamine and glycine respectively [60, 95, 96].
3. Why is small molecule GALK inhibitor the preferred therapeutic approach for Classic Galactosemia?
In searching for an improved therapy for Classic Galactosemia, it is important to remember that this disorder is currently a chronic genetic disorder, associated with low mortality, but significant chronic morbidity which nonetheless does not appear to lead to shortened life expectancy. Hence, the treatment options considered for Classic Galactosemia should be relevant to the disorder and not by itself lead to more morbidity. As a result, liver transplantation and gene therapy are currently considered too high a risk to pursue in this disorder. Chaperone and co-factor therapy are unlikely to be beneficial as few if any GALT gene mutations are currently known to cause protein mis-folding, and as the GALT enzyme does not employ a co-factor. In terms of RNA-based therapies, antisense-mediated exon skipping and stop codon read-through could be attempted in some GALT gene mutations, but not in the most common mutations, including Q188R, K285N, L195P, S135L, 5kb. siRNA therapy, once it has become a mature approach, could be a successful therapeutic strategy, as down-regulation of GALK1 gene using this technology is expected to act similarly to a small molecule with GALK inhibitory properties. Substrate reduction is currently the mainstay of therapy, and will likely remain the cornerstone of therapy for years to come. If a GALK inhibitor were identified that is both safe and effective, it would still be prudent to continue lactose restriction to reduce galactitol production. Whether product supplementation is useful in Classic Galactosemia is in our opinion currently unclear, but could be considered adjunct therapy, if clinically possible. We have shown that there may be a partial UDP-galactose deficiency in the disease [24], which could be responsible for the glycosylation defects seen in Classic Galactosemia [24]. However, it is currently unclear if UDP-galactose supplementation will correct this deficiency, or even if UDP-galactose will reach the cytosolic compartment in the patient cells. Certainly, it remains controversial if uridine supplementation is beneficial [97]. While these questions remain without clear answers, it is our opinion and that of others that gal-1P toxicity will continue to be the most important target for novel therapeutic endeavors [54-57]. Moreover, with current technology, therapeutic endeavors using small molecule inhibitors are the best option. However, siRNA technology, to accomplish reduced GALK1 expression, may be a good future option.
4. How can we identify therapeutic small molecule GALK inhibitors?
It should be noted that prior to our efforts of identifying small molecule GALK inhibitors with diverse chemical scaffolds, there was no known effort to systematically search for such molecules. Our first attempt was to identify derivatives of galactose which can compete with the binding of this hexose towards GALK. One of the derivatives we selected was 2-deoxygalactose. Although 2-deoxygalactose is recognized and phosphorylated by GALK, it did not result in GALK inhibition (unpublished data). Also for other hexoses, as glucose and fructose, there was no inhibition on GALK at low millimolar (mM) range (unpublished data). We therefore proceeded to develop a new way to identify GALK inhibitors by adopting the experimental high-throughput screening (HTS) strategy employed by many successful drug development programs (Fig. 4). The key step for experimental HTS is developing a miniaturized, robust assay with a high signal-to-noise ratio resulting in good readout and consistency. Using the Kinase-Glo™ reagent (Promega), we developed a miniaturized, two-step HTS assay for GALK. This primary assay measures GALK activity indirectly by determining the amount of ATP remains after completion of the GALK-mediated reaction (step 1): Galactose + ATP → gal-1P + ADP. If there is ample GALK activity, most ATP will be used up in this step and little will be left for the luciferase reaction (step 2): ATP + Luciferin & Luciferase (Kinase Glo™) → oxyluciferin + light. Therefore, the luminescent signal recorded as relative luminescence units (RLU) at the end of step 2 is inversely proportional to the amount of kinase activity in step 1. Since this two-step assay involves luciferase, we were aware of that compounds competing with ATP for luciferase will not be identified and therefore, result in false negatives. Although these false-negatives might result in an ‘undesirable outcome’, one must realize that the compounds that are missed in such case are likely to be the less selective inhibitors because they are likely to interact with the ATP-binding sites of enzymes. As a result, this assay might, in the long run, reduce the number of non-selective hits for GALK. With this assay setup, we have successfully screened over 350,000 compounds experimentally from two different HTS facilities: (a) Broad Institute at Harvard University and MIT and (b) National Chemical Genomics Center (NCGC) at the National Institutes of Health (Fig. 4). In this review, we will focus on the first screen, through which we identified about 150 chemicals with inhibition at 30μM; we then selected 34 compounds for further study [57].
Fig. 4. Two high-throughput screening strategies employed to identify GALK inhibitors.

Experimental and computational approaches were used in the high-throughput screenings of human GALK inhibitors. The identified inhibitors will be characterized by a series of tertiary assays to determine the efficacy, toxicity, selectivity. Lead compounds with the optimal profile will be selected for further optimization to improve their drug-like properties.
In addition to experimental HTS mentioned above, we also employed high-precision molecular docking programs in computational “virtual” high-throughput screening of chemical compounds libraries to identify novel GALK inhibitors (Fig. 4). Due to advances in computational sciences, virtual screening has emerged as an effective paradigm for discovering enzyme inhibitors in recent years [98]. Compared to experimental HTS, neither expensive robotic instruments nor large collection of compounds libraries are needed for virtual screening. The technique employed is based on the prediction of binding modes and binding affinities of each compound in the dataset against an X-ray crystallographic structure of the target.
For our endeavor, 160,000 compounds were chosen randomly from ZINC compound database (http://zinc.docking.org) against the active site of GALK [99]. The top 200 hits were re-evaluated and six hits were purchased, four of them were shown to be active to GALK, with IC50’s ranging from 70μM to 400μM (Table 1). Although the data suggested that these compounds are not necessarily ideal lead compounds to pursue, these results provided evidence that virtual screening can be used to identify GALK inhibitors from large chemical compound or fragment libraries which would be helpful in our continuing effort in GALK inhibitors identification and optimization.
Table 1. Tested top-scoring compounds from Virtual Screening of Zinc “clean-druglike” library.
| Structure | XP score | IC50(μM) |
|---|---|---|
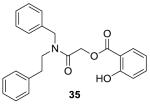
|
−9.16 | Not active |
|
−8.5 | Not active |
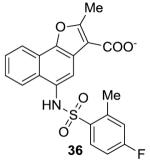
|
−8.8 | 70 |
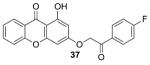
|
−8.4 | 200 |
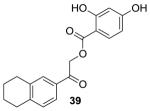
|
−8.07 | 250 |
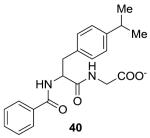
|
−8.42 | 400 |
Six out of 20 top scoring compounds, based on their XP score, were purchased and tested for efficacy to inhibit GALK in vitro.
5. How can we prioritize identified small molecule GALK inhibitors?
In order to prioritize the identified positives from the experimental HTS, we subjected the selected compounds to further characterization.
(a) IC50 and kinase selectivity determination
The IC50 of the 34 compounds identified from the first HTS ranged from 0.7μM to 33μM as shown in Table 2. While it is important to focus on more potent compounds, other properties must be considered for the long-term success of the project. Human GALK belongs to the GHMP small molecule kinase superfamily, which was named after the four characteristic family members: galactokinase (GALK), homoserine kinase (HSK), mevalonate kinase (MVK), and phosphomevalonate kinase (PMVK) [100]. Members of this kinase superfamily share highly conserved substrates-binding motifs and therefore, it is important to test if the GALK inhibitors identified are selective (i.e., specific) to avoid potential adverse interactions between the chosen compounds and other GHMP kinases. We tested all 34 GALK inhibitors from experimental HTS against several typical GHMP kinases as well as other sugar kinases, glucokinase and hexokinase. As shown in Table 2, some of GALK inhibitors did interact with other GHMP kinases but not the other two sugar kinases, which again proved members of GHMP kinase family shared similar structure folding [101]. Yet, six compounds (1, 4, 6, 23, 24, and 32) had no detectable inhibition against any of the tested GHMP kinases at a concentration up to 60μM, thus indicating that the selective GALK inhibitors do exist [101].
Table 2.
Efficacy of selected GALK inhibitors against selected GHMP kinases and sugar kinases.
| GALK (μM) |
MVK (μM) |
HSK (μM) |
CDP-ME kinase(μM) |
Glucokinase /Hexokinase (μM) |
GALK (μM) |
MVK (μM) |
HSK (μM) |
CDP-ME kinase(μM) |
Glucokinase/ Hexokinase (μM) |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.7 | >60 | >60 | >60 | >60 | 18 | 12.5 | 23 | >60 | >60 | >60 |
| 3 | 10.5 | 24.5 | >60 | >60 | >60 | 20 | 25 | 13 | >60 | 5.5 | >60 |
| 4 | 1.5 | >60 | >60 | >60 | >60 | 21 | 18.5 | 15 | >60 | 5.5 | >60 |
| 5 | 12 | 20 | >60 | 18 | >60 | 22 | 30 | 21.3 | >60 | >60 | >60 |
| 6 | 16.5 | >60 | >60 | >60 | >60 | 23 | 11.5 | >60 | >60 | >60 | >60 |
| 8 | 24 | 25.2 | >60 | 5 | >60 | 24 | 6.3 | >60 | >60 | >60 | >60 |
| 9 | 5.2 | 8.2 | >60 | 11 | >60 | 26 | 19.3 | 11.5 | >60 | >60 | >60 |
| 10 | 6.5 | 10.3 | 15.8 | >60 | >60 | 27 | 32 | 30 | >60 | >60 | >60 |
| 11 | 33.3 | 40 | >60 | >60 | >60 | 28 | 20 | 21 | >60 | >60 | >60 |
| 13 | 33.3 | 20 | >60 | 17 | >60 | 30 | 21.1 | 14.5 | >60 | >60 | Weak inhibition for Glucokinase |
| 14 | 27.1 | 16 | >60 | >60 | >60 | 31 | 21 | 18.5 | >60 | >60 | >60 |
| 16 | 30 | 40 | >60 | Not tested | >60 | 32 | 20.3 | >60 | >60 | >60 | >60 |
| 17 | 12.1 | 15.2 | >60 | 28 | >60 |
Note for the Table: Compounds were tested against mevalonate kinase (MVK), homoserine kinase (HSK), CDP-ME kinase, glucokinase and hexokinase, and IC50’s are shown. IC50’s for GALK are provided for comparison.
(b) Molecular interactions with GALK
In order to improve the binding affinity of selected inhibitors, good understandings of how these inhibitors bind to the GALK enzyme at the structural and mechanistic levels are required. Pioneer efforts by laboratories of Holden and Timson [99, 102-105] have helped facilitate this objective through their intensive structural studies of the GALK enzyme. To date, GALK structures from four different species have been solved, two from bacteria [102, 106], one from yeast [102], and one from humans [99], together with different ligands - galactose, ADP, as well as the non-hydrolysable ATP analog, AMPPNP. The human GALK was co-crystallized with galactose and AMPPNP and the structure was determined with a resolution of 2.5Å [99] (Fig. 5a). Not surprisingly, all conserved GHMP kinase motifs in GALK are involved in substrate binding of galactose and AMPPNP. The availability of ligand-bound human GALK crystal structure allow us to perform high-precision docking experiments to predict the molecular interactions identified small molecule GALK inhibitors with high degree of confidence. The docking poses resulted will not only help confirm the binding modes of the inhibitors determined through kinetic analyses, but also aid the future optimization schemes of these first-generation compounds. To validate the results of the docking experiments, we have performed site-directed mutagenesis studies and showed that among the six compounds which selectively inhibit GALK, two of them actually interacted with one of the non-conserved amino acid residue, Ser140, in the conserved Motif II of the GHMP kinases [101]. Changing serine to glycine, by site-directed mutagenesis weakened the binding of two of the six compounds to GALK. Docking these two compounds to GALK active site showed that these compounds formed hydrogen bond with Ser140, and the change of serine to glycine abolished such hydrogen bond interaction which resulted in unfavorable change in binding energy [101] (Fig. 5b). More interestingly, most of GHMP kinases have a glycine moiety at this position [107], which may explain the selectivity of these two compounds to GALK among GHMP family. These results further confirmed that we could identify selective compounds inhibiting GALK with our HTS strategy.
Fig. 5. Molecular structure of ligands binding pocket of human GALK (1wuu)[102].


(a) Amino acid residues of human GALK located within 3.5Å of the ligands were represented here. Molecules in green carbon atoms are galactose and AMPPNP. Amino acid residues are depicted in gray. MI: motif I; MII: motif II; MIII: motif III. (b) Docking pose of compound 4 to GALK crystal structure. Using high-precision docking software GLIDE, compound 4 was docked to the GALK structure. Molecule in green and ball-and-stick conformation is compound 4. Side chain of Ser140 of GALK is in dark blue, and the side-chains of other amino acid residues selected for the site-directed mutagenesis studies are colored in brown. Side-chains of other amino acid residues surrounding the compound 4 are colored in gray. Yellow dotted lines are hydrogen-bonds formed between compound 4 and the side-chain of Ser140.
(c) Toxicity to human cells in culture
To date, many of the highly selective GALK inhibitors we identified were well-tolerated as no signs of gross cell death were observed when added to human fibroblasts at concentrations up to 100μM (data not shown). Moreover, the incubation of skin fibroblasts with some of the inhibitors did not significantly alter the intracellular concentrations of over 60 metabolites (including both abundant and rare metabolites) (toxicometabolomic data were obtained at the Metabolomics Core of the University of Utah, not shown). These preliminary tests, although not definitive in terms of safety, provided some evidence that the selected compounds are not highly toxic and also some indications on whether they are metabolized within cells. This sets the stage for more rigorous techniques for assaying cytotoxicity such as toxicogenomics and Ames test for the promising compounds.
(d) Effect of selective compounds on intracellular gal-1P accumulation and gal-1P-induced stress response in patient cells
Since some of the selected compounds were well-tolerated by cultured cells, we tested whether they were effective in preventing gal-1P accumulation in primary fibroblasts from Classic Galactosemia patients upon challenge with galactose. In the example shown in Fig. 6, compound BI-01 caused a dose-dependent decrease of gal-1P accumulation in patient cells. The reduced gal-1P accumulation also prevented the elevated expression of the BiP (GRP78) stress response protein, previously shown to be up-regulated in GALT-deficient cells under galactose challenge [28] (data not shown). These “proof-of-concept” results are highly significant because, for the first time, we demonstrated that inhibition of GALK in Classic Galactosemia patient cells cannot only lower gal-1P accumulation, but we also established the cause-and-effect relationship between gal-1P and cellular stress; which was significantly reduced by the novel, selective and non-toxic GALK inhibitors identified.
Fig. 6. GALK inhibitor prevents gal-1P accumulation in GALT-deficient fibroblasts [101].

Gal-1P level was measured after SV-40 transformed GALT-deficient fibroblasts were treated with compound BI-01 prior to 0.05% galactose challenge.
6. How can we optimize lead GALK inhibitors?
While the IC50 values of the identified GALK inhibitors shown in Table 2 are promising, they are far from practical in terms of therapeutic dosage. As a result, we have adopted structure-based optimization strategies to improve the potency of a few selected positives [108]. In this review, we will use one of the hit compounds, namely compound 5 (Fig. 7) as an example to illustrate the optimization process. Compound 5, was selected for optimization because it shares a similar aromatic core and functionality array with many of the positive hits from the first high-throughput screening [57]. This compound has two aromatic rings A and C, and a central thiazinone core B (Fig. 7). Through vigorous interrogation of similar compounds obtained from commercial vendors (compounds 41-49, Fig. 7), we identified from our structure-activity relationship (SAR) analyses that the prominent areas in the right (A) and left (C) sides of this molecule are indeed relevant for potent inhibition as well. In the end, compound 41 with the lowest IC50 value was used as a template to synthesize a new set of compounds in our laboratory for further SAR analyses
Fig. 7. Compounds from commercial sources selected for SAR studies [108].

Nine compounds (41-49) were selected from commercial sources based on their structural similarity to compound 5. These compounds were tested for their in vitro GALK inhibitory properties.
Through computational docking of compound 41 to the active site of GALK, we realized that three critical pieces of information on the A ring are needed for a rational design of an improved GALK inhibitor. The calculated pKa of the para hydroxyl group is 12.79, providing clear evidence that the para hydroxyl group is playing two important roles: (1) it is involved in hydrogen bonding with Ser141, and (2) it is in a close proximity to Mg2+ for chelation with the metal ion needed for enzymatic activity at the active site of GALK. Apparently, the adjacent hydroxyl group with an estimated pKa of 9.5 has the least interaction with amino acid residues forming the active site. Consequently, it would be necessary to investigate their roles in the binding to the GALK active site and the chelation to Mg2+ ion through medicinal chemistry approach (compounds 50, 51, 54, 55, 56, Fig. 8). We also attempted to mask the hydrophilic hydroxyl functionalities with the methoxyl or benzo[d][1,3]dioxole group functional groups in order to understand the absolute requirements of the hydroxyl groups in GALK inhibition (compounds 52, 53, Fig. 8) [108].
Fig. 8. Synthesized compounds selected for SAR studies [108].

Seven compounds (50-56) were synthesized to test the importance of substitutions at the A-ring in compound 41.
B ring of compound 41 was not investigated because computational analyses suggested that optimization of A- and C-rings would render more impact in improving the potency and pharmacokinetic properties of this compound. On the other hand, computational modeling studies suggested that the meta position to the thiobenzyl group of the C ring could be modified without causing loss of potency to the inhibitor because it extends out into the hydrophobic region of the adenine binding pocket. Any chemical group like morpholine or piperazyl group can theoretically improve the potency of the inhibitor through hydrophobic interaction with the adenine binding area (Fig. 5a). As a result, compounds 57-59 (Fig. 9) with a morpholine group at the meta position of the thiobenzyl group of the C ring were synthesized and tested. To date, the best optimized compound derived from compound 41 is morpholine derivative 57 because it has IC50 value of 1.2μM, compared to 12μM of compound 5 from which all these analogs derived [108]. Future optimization of this compound will focus on improving its in vivo potency.
Fig. 9. GALK inhibitors modified with morpholine moiety [108].

Morpholino substitutions at the C-ring were synthesized and tested for potential improvement in binding to the adenine binding pocket.
7. Other potential novel therapeutic uses of GALK inhibitors
(a) Antimicrobials
In all living organisms, isoprenoid biosynthesis is an important key step to survival because isoprenoids play important roles in biological functions, including electron transport in respiration and photosynthesis, hormone-based signaling, apoptosis, meiosis, protein cleavage and degradation [109-111]. While most eukaryotes use the mevalonate pathway in their isoprenoid biosynthesis [112-114], Gram-negative bacteria (e.g., E. coli), some Gram-positive bacteria (e.g., Mycobacterium tuberculosis), Chlamydia, a few apicocomplexan protozoa (e.g., Plasmodium sp.), and all plants use the non-mevalonate pathway [115-119]. Many of the serious human diseases such as leprosy, malaria, bacterial meningitis, various gastrointestinal and sexually transmitted infections, tuberculosis, and certain types of pneumonia emanate from human pathogens that use the non-mevalonate pathway [120].
One of the key enzymes of the non-mevalonate pathway is 4-Diphosphocytidyl-2C-methyl-d-erythritol (CDP-ME) kinase, which belongs to the GHMP kinase family [100, 103]. Because of its absence in humans, this enzyme is an attractive target for the development of novel antimicrobials and herbicides. Since GALK and CDP-ME kinases are from the same GHMP kinase family, they share conserved amino acid sequences that are involved in the formation of the active sites [100, 103]. A less selective GALK inhibitor may therefore cross-inhibit against CDP-ME kinase. In fact, close scrutiny of previous high-throughput screening for GALK inhibitors revealed that some of the GALK inhibitors could indeed inhibit CDP-ME kinase from E. coli (Tables 2) [121]. Therefore, the on-going development of GALK inhibitors provides new chemical templates for CDP-ME kinase inhibitors as novel antimicrobial agents.
(b) Anti-cancer therapeutics
AKT is a serine/threonine kinase and has normal cellular functions in cell proliferation, survival, angiogenesis, differentiation through phosphorylation and/or interaction with a number of molecules [45]. It is also the downstream target of phosphatidyl-inositol 3-kinase (P13K) and phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase (PTEN) [122]. Dysregulation of the PI3K/PTEN/AKT pathway can lead to the abnormal activation of AKT, triggering excessive cell proliferation through a chain of signaling events culminating in the immortalization of cancer cells.
Recently, it has been reported that expression of the GALK1 gene are significantly up-regulated in some tumors (colon, head/neck, lung, ovary, pancreas, skin and stomach), and this phenomenon has been attributed to malfunctioning of the PI3K/PTEN/AKT pathway in these tumors [123]. To further support this notion, investigators have found that when the over-expressed GALK1 gene is knocked down by GALK1-specific siRNA, the PI3K/PTEN/AKT pathway is restored back to normal. For example, in cell-based assays, AKT activity in the human tumor cell lines A549; MDA-MB231-T and PC-3 cells was reduced by at least 20% when these cells were treated with GALK1 siRNA. Simultaneously, increased caspase cleavage activity and phosphorylation of histone H2B in apoptosis assays have been observed in these treated tumor cell lines as well [123]. While these findings with GALK1 siRNA are exciting, there is reason to believe that similar results can be obtained by treating the cancer cells with GALK inhibitors, the same inhibitors that were intended for the reduction cellular accumulation of toxic gal-1P in patients with Classic Galactosemia.
8. Looking to the future
Despite decades of intensive research, the pathophysiology of Classic Galactosemia remains largely unknown, with limited and partly ineffective therapeutic options. In the past few years alone, at least six groups of investigators reported that the health-related quality of life consequences of galactosemic patients and their parents were worse than generally thought [124-129]. Such outcry of concerns for a relatively rare disease suggested that the stressful conditions suffered by the patients and their families have been overlooked for too long, and swift actions are required to improve the current treatments. In this review, we evaluated the different approaches for treating inborn errors of metabolism and concluded that pharmacological inhibition of the human galactokinase (GALK), in combination with a galactose-restricted diet, appears to be the best option for novel therapy. We have conducted both experimental and virtual screenings for potent, selective and non-toxic GALK inhibitors. During these activities, we also learned that such compounds could potentially be used as novel antimicrobial and anti-cancer agents. However, significant challenges remain ahead in this process of drug discovery. For instance, there will be a need for a mammalian GALT-deficient model to assess the in vivo safety and efficacy of these newly identified compounds. Although the GALT-knockout (KO) mice models constructed by Leslie and co-workers did not manifest any disease phenotypes of the patients [130], they nonetheless exhibited significant accumulation of gal-1P. Thus, if nothing else, this model is ideal to assess GALK inhibitors with regards to efficacy and safety in reducing the accumulation of gal-1P in vivo. Recently, we have identified a human gene called aplysia ras homolog I (ARHI) that is absent in rodents due to an ancestral recombination event [131]. Interestingly, this tumor suppressor gene was found to be abnormally up-regulated in patients with Classic Galactosemia [132]. We have proposed that the lack of ARHI gene in rodents may explain the absence of human disease phenotypes in the original GALT-KO mice. Perhaps we could re-introduce this gene in the GALT-KO mice and then examine this model for manifestations of human disease phenotypes. If our hypothesis is correct, the ARHI+ GALT-KO mice will be a useful model to ascertain the efficacy of our identified GALK inhibitors for the prevention of the chronic complications of Classic Galactosemia.
Highlights of this mini-review.
-
➢
The unsatisfactory outcome of patients with Classic Galactosemia calls for a more effective therapy.
-
➢
Evaluation of current therapeutic approaches to IEMs and existing information on the pathophysiology of the disease led us to conclude that pharmacological inhibition human galactokinase (GALK) can result in an improved therapy for Classic Galactosemia.
-
➢
Experimental and computational screenings for therapeutic GALK inhibitors were performed against chemical libraries of over 500,000 small molecule chemical compounds.
-
➢
Subsequent characterization and optimization of selected inhibitors have identified a few lead compounds that are potent and effective in lowering galactose-1-phosphate accumulation in patient cells in culture.
-
➢
GALK inhibitors can be further developed into novel antimicrobials and anti-cancer therapies.
Acknowledgement
Grant support to Kent Lai include U.S. National Institutes of Health Grants 5R01HD054744-05 & 3R01HD054744-04S1.
Footnotes
Authors contributed equally to this work.
References
- 1.Leloir LF. The enzymatic transformation of uridine diphosphate glucose into a galactose derivative. Arch Biochem. 1951;33(2):186–90. doi: 10.1016/0003-9861(51)90096-3. [DOI] [PubMed] [Google Scholar]
- 2.Acosta PB, Gross KC. Hidden sources of galactose in the environment. Eur J Pediatr. 1995;154(7 Suppl 2):S87–92. doi: 10.1007/BF02143811. [DOI] [PubMed] [Google Scholar]
- 3.Berry GT, et al. The effect of dietary fruits and vegetables on urinary galactitol excretion in galactose-1-phosphate uridyltransferase deficiency. J Inherit Metab Dis. 1993;16(1):91–100. doi: 10.1007/BF00711320. [DOI] [PubMed] [Google Scholar]
- 4.Berry GT, et al. Endogenous synthesis of galactose in normal men and patients with hereditary galactosaemia. Lancet. 1995;346(8982):1073–4. doi: 10.1016/s0140-6736(95)91745-4. [DOI] [PubMed] [Google Scholar]
- 5.Berry GT, et al. The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol Genet Metab. 2004;81(1):22–30. doi: 10.1016/j.ymgme.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 6.Cardini CE, Leloir LF. Enzymic phosphorylation of galactosamine and galactose. Arch Biochem Biophys. 1953;45(1):55–64. doi: 10.1016/0003-9861(53)90404-4. [DOI] [PubMed] [Google Scholar]
- 7.Kalckar HM, Braganca B, Munch-Petersen HM. Uridyl transferases and the formation of uridine diphosphogalactose. Nature. 1953;172(4388):1038. [PubMed] [Google Scholar]
- 8.Leloir LF. Enzymic isomerization and related processes. Adv Enzymol Relat Subj Biochem. 1953;14:193–218. doi: 10.1002/9780470122594.ch6. [DOI] [PubMed] [Google Scholar]
- 9.Darrow RA, Rodstrom R. Purification and properties of uridine diphosphate galactose 4-epimerase from yeast. Biochemistry. 1968;7(5):1645–54. doi: 10.1021/bi00845a005. [DOI] [PubMed] [Google Scholar]
- 10.Berry GT, Elsas LJ. Introduction to the Maastricht workshop: lessons from the past and new directions in galactosemia. J Inherit Metab Dis. 2010 doi: 10.1007/s10545-010-9232-1. [DOI] [PubMed] [Google Scholar]
- 11.Fridovich-Keil J, Walter J. In: Chapter 72: Galactosemia in Online Metabolic and Molecular Bases of Inherited Diseases-OMMBID. Valle D, et al., editors. McGraw-Hill; New York: 2008. www.ommbid.com [Google Scholar]
- 12.Lai K, Elsas LJ, Wierenga KJ. Galactose toxicity in animals. IUBMB Life. 2009;61(11):1063–74. doi: 10.1002/iub.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segal S, Berry GT. Disorders of galactose metabolism. In: Scriver CR, et al., editors. The Metabolic Basis of Inherited Diseases. McGraw-Hill; New York: 1995. pp. 967–1000. [Google Scholar]
- 14.McCorvie TJ, Timson DJ. The structural and molecular biology of type I galactosemia: Enzymology of galactose 1-phosphate uridylyltransferase. IUBMB Life. 2011 doi: 10.1002/iub.511. [DOI] [PubMed] [Google Scholar]
- 15.Kaye CI, et al. Newborn screening fact sheets. Pediatrics. 2006;118(3):e934–63. doi: 10.1542/peds.2006-1783. [DOI] [PubMed] [Google Scholar]
- 16.Donnell GN, et al. Galactose-1-phosphate in galactosemia. Pediatrics. 1963;31:802–10. [PubMed] [Google Scholar]
- 17.Gitzelmann R. Galactose-1-phosphate in the pathophysiology of galactosemia. Eur J Pediatr. 1995;154(7 Suppl 2):S45–9. doi: 10.1007/BF02143803. [DOI] [PubMed] [Google Scholar]
- 18.Abukawa D, Ohura T, Tazawa Y. Disorders of fructose and galactose metabolism. Ryoikibetsu Shokogun Shirizu. 1995;(8):371–4. [PubMed] [Google Scholar]
- 19.Waisbren SE, et al. The adult galactosemic phenotype. J Inherit Metab Dis. 2011 doi: 10.1007/s10545-011-9372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waggoner D. Buist, NRM, Long-term complications in treated galactosemia - 175 U.S. cases. International Pediatrics. 1993;8:97–100. [Google Scholar]
- 21.Gitzelmann R, Wells HJ, Segal S. Galactose metabolism in a patient with hereditary galactokinase deficiency. Eur J Clin Invest. 1974;4(2):79–84. doi: 10.1111/j.1365-2362.1974.tb00376.x. [DOI] [PubMed] [Google Scholar]
- 22.Gitzelmann R. Formation of galactose-1-phosphate from uridine diphosphate galactose in erythrocytes from patients with galactosemia. Pediatr Res. 1969;3(4):279–86. doi: 10.1203/00006450-196907000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Lai K, Elsas LJ. Overexpression of human UDP-glucose pyrophosphorylase rescues galactose-1-phosphate uridyltransferase-deficient yeast. Biochem Biophys Res Commun. 2000;271(2):392–400. doi: 10.1006/bbrc.2000.2629. [DOI] [PubMed] [Google Scholar]
- 24.Lai K, et al. GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology. 2003;13(4):285–94. doi: 10.1093/glycob/cwg033. [DOI] [PubMed] [Google Scholar]
- 25.Mehta DV, Kabir A, Bhat PJ. Expression of human inositol monophosphatase suppresses galactose toxicity in Saccharomyces cerevisiae: possible implications in galactosemia. Biochim Biophys Acta. 1999;1454(3):217–26. doi: 10.1016/s0925-4439(99)00037-x. [DOI] [PubMed] [Google Scholar]
- 26.Bhat PJ. Galactose-1-phosphate is a regulator of inositol monophosphatase: a fact or a fiction? Med Hypotheses. 2003;60(1):123–8. doi: 10.1016/s0306-9877(02)00347-x. [DOI] [PubMed] [Google Scholar]
- 27.Parthasarathy R, Parthasarathy L, Vadnal R. Brain inositol monophosphatase identified as a galactose 1-phosphatase. Brain Res. 1997;778(1):99–106. doi: 10.1016/s0006-8993(97)01042-1. [DOI] [PubMed] [Google Scholar]
- 28.Slepak TI, et al. Involvement of endoplasmic reticulum stress in a novel Classic Galactosemia model. Mol Genet Metab. 2007;92(1-2):78–87. doi: 10.1016/j.ymgme.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stempfel RS, Jr., Sidbury JB, Jr., Migeon CJ. beta-Glucuronidase hydrolysis of urinary corticosteroid conjugates: the effect of salicylate glucuronoside as a competing substrate and the effect of enzyme inactivation. J Clin Endocrinol Metab. 1960;20:814–24. doi: 10.1210/jcem-20-6-814. [DOI] [PubMed] [Google Scholar]
- 30.Maddaiah VT, Madsen NB. Kinetics of purified liver phosphorylase. J Biol Chem. 1966;241(17):3873–81. [PubMed] [Google Scholar]
- 31.Kalckar HM, Maxwell ES. Biosynthesis and metabolic function of uridine diphosphoglucose in mammalian organisms and its relevance to certain inborn errors. Physiol Rev. 1958;38(1):77–90. doi: 10.1152/physrev.1958.38.1.77. [DOI] [PubMed] [Google Scholar]
- 32.Charlwood J, et al. Defective galactosylation of serum transferrin in galactosemia. Glycobiology. 1998;8(4):351–7. doi: 10.1093/glycob/8.4.351. [DOI] [PubMed] [Google Scholar]
- 33.Jaeken J, Kint J, Spaapen L. Serum lysosomal enzyme abnormalities in galactosaemia. Lancet. 1992;340(8833):1472–3. doi: 10.1016/0140-6736(92)92664-2. [DOI] [PubMed] [Google Scholar]
- 34.Prestoz LL, et al. Altered follicle stimulating hormone isoforms in female galactosaemia patients. Eur J Pediatr. 1997;156(2):116–20. doi: 10.1007/s004310050568. [DOI] [PubMed] [Google Scholar]
- 35.Berry GT. The unexplored potential of the pentose phosphate pathway in health and disease. J Inherit Metab Dis. 2008;31(6):661. doi: 10.1007/s10545-008-9971-4. [DOI] [PubMed] [Google Scholar]
- 36.Berry GT. Is prenatal myo-inositol deficiency a mechanism of CNS injury in galactosemia? J Inherit Metab Dis. 2011;34(2):345–55. doi: 10.1007/s10545-010-9260-x. [DOI] [PubMed] [Google Scholar]
- 37.Wells WW, et al. The Isolation And Identification Of Galactitol From The Brains Of Galactosemia Patients. J Biol Chem. 1965;240:1002–4. [PubMed] [Google Scholar]
- 38.Quan-Ma R, Wells WW. The distribution of galactitol in tissues of rats fed galactose. Biochem Biophys Res Commun. 1965;20(4):486–90. doi: 10.1016/0006-291x(65)90605-4. [DOI] [PubMed] [Google Scholar]
- 39.Dahl SC, Handler JS, Kwon HM. Hypertonicity-induced phosphorylation and nuclear localization of the transcription factor TonEBP. Am J Physiol Cell Physiol. 2001;280(2):C248–53. doi: 10.1152/ajpcell.2001.280.2.C248. [DOI] [PubMed] [Google Scholar]
- 40.Jeon US, et al. How tonicity regulates genes: story of TonEBP transcriptional activator. Acta Physiol (Oxf) 2006;187(1-2):241–7. doi: 10.1111/j.1748-1716.2006.01551.x. [DOI] [PubMed] [Google Scholar]
- 41.Miyakawa H, et al. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc Natl Acad Sci U S A. 1999;96(5):2538–42. doi: 10.1073/pnas.96.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irarrazabal CE, et al. Activator protein-1 contributes to high NaCl-induced increase in tonicity-responsive enhancer/osmotic response element-binding protein transactivating activity. J Biol Chem. 2008;283(5):2554–63. doi: 10.1074/jbc.M703490200. [DOI] [PubMed] [Google Scholar]
- 43.Berry GT, et al. The human osmoregulatory Na+/myo-inositol cotransporter gene (SLC5A3): molecular cloning and localization to chromosome 21. Genomics. 1995;25(2):507–13. doi: 10.1016/0888-7543(95)80052-n. [DOI] [PubMed] [Google Scholar]
- 44.Tsui MM, York JD. Roles of inositol phosphates and inositol pyrophosphates in development, cell signaling and nuclear processes. Adv Enzyme Regul. 50(1):324–37. doi: 10.1016/j.advenzreg.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osaki M, and M. Oshimura,, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9(6):667–76. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 47.Berry GT, et al. Loss of murine Na+/myo-inositol cotransporter leads to brain myo-inositol depletion and central apnea. J Biol Chem. 2003;278(20):18297–302. doi: 10.1074/jbc.M213176200. [DOI] [PubMed] [Google Scholar]
- 48.Berry GT. Galactosemia and amenorrhea in the adolescent. Ann N Y Acad Sci. 2008;1135:112–7. doi: 10.1196/annals.1429.038. [DOI] [PubMed] [Google Scholar]
- 49.Agam G, et al. Knockout mice in understanding the mechanism of action of lithium. Biochem Soc Trans. 2009;37(Pt 5):1121–5. doi: 10.1042/BST0371121. [DOI] [PubMed] [Google Scholar]
- 50.Murata M, et al. The role of aldose reductase in sugar cataract formation: aldose reductase plays a key role in lens epithelial cell death (apoptosis) Chem Biol Interact. 2001;130(1-3):617–25. doi: 10.1016/s0009-2797(00)00289-1. [DOI] [PubMed] [Google Scholar]
- 51.Kubo E, et al. Cataract formation through the polyol pathway is associated with free radical production. Exp Eye Res. 1999;68(4):457–64. doi: 10.1006/exer.1998.0624. [DOI] [PubMed] [Google Scholar]
- 52.Gitzelmann R. Deficiency of erythrocyte galactokinase in a patient with galactose diabetes. Lancet. 1965;2(7414):670–1. doi: 10.1016/s0140-6736(65)90400-9. [DOI] [PubMed] [Google Scholar]
- 53.Gitzelmann R. Letter: Additional findings in galactokinase deficiency. J Pediatr. 1975;87(6 Pt 1):1007–8. doi: 10.1016/s0022-3476(75)80937-1. [DOI] [PubMed] [Google Scholar]
- 54.Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29(4):516–25. doi: 10.1007/s10545-006-0382-0. [DOI] [PubMed] [Google Scholar]
- 55.Fridovich-Keil J. Toward Improved Intervention for Galactosemia. 2007 Available from: http://www.galactosemia.org/PGC_awards.asp.
- 56.Timson DJ. GHMP Kinases - Structures, Mechanisms and Potential for Therapeutically Relevant Inhibition. Current Enzyme Inhibition. 2007;(3):77–94. [Google Scholar]
- 57.Wierenga KJ, et al. High-throughput screening for human galactokinase inhibitors. J Biomol Screen. 2008;13(5):415–23. doi: 10.1177/1087057108318331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walter J, Wraith JE. Chapter 5: Treatment: present status and new trends in Metabolic Diseases-Diagnosis and Treatment. 4th ed Springer Medizin Verlag; 2006. [Google Scholar]
- 59.Saudubray JM, et al. Inborn Metabolic Diseases - Diagnosis and Treatment. 4th ed Springer Medizin Verlag; 2006. Chapter 1: A clinical approach to inherited metabolic diseases. [Google Scholar]
- 60.Souillet G, et al. Outcome of 27 patients with Hurler’s syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003;31(12):1105–17. doi: 10.1038/sj.bmt.1704105. [DOI] [PubMed] [Google Scholar]
- 61.Staba SL, et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N Engl J Med. 2004;350(19):1960–9. doi: 10.1056/NEJMoa032613. [DOI] [PubMed] [Google Scholar]
- 62.Hsieh MM, Fitzhugh CD, Tisdale JF. Allogeneic hematopoietic stem cell transplant for sickle cell disease: the time is now. Blood. 2011 doi: 10.1182/blood-2011-01-332510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson FL, et al. Bone-marrow transplantation in a patient with sickle-cell anemia. N Engl J Med. 1984;311(12):780–3. doi: 10.1056/NEJM198409203111207. [DOI] [PubMed] [Google Scholar]
- 64.Ariga T. Gene therapy for adenosine deaminase (ADA) deficiency: review of the past, the present and the future. Nihon Rinsho. 2001;59(1):72–5. [PubMed] [Google Scholar]
- 65.Blaese RM, et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science. 1995;270(5235):475–80. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 66.Maguire AM, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374(9701):1597–605. doi: 10.1016/S0140-6736(09)61836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cartier N, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326(5954):818–23. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 68.Seregin SS, Amalfitano A. Gene Therapy for Lysosomal Storage Diseases: Progress, Challenges and Future Prospects. Curr Pharm Des. 2011 doi: 10.2174/138161211797247578. [DOI] [PubMed] [Google Scholar]
- 69.Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451(7175):141–6. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 70.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 71.Kim JB, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008;454(7204):646–50. doi: 10.1038/nature07061. [DOI] [PubMed] [Google Scholar]
- 72.Mohamadnejad M, Swenson ES. Induced pluripotent cells mimicking human embryonic stem cells. Arch Iran Med. 2008;11(1):125–8. [PubMed] [Google Scholar]
- 73.Kishore R, Krishnamurthy P, Losordo DW. Career moves: induced pluripotent cells from human aortic smooth muscle cells can efficiently redifferentiate into parental phenotype. Circ Res. 2010;106(1):7–9. doi: 10.1161/CIRCRESAHA.109.210351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanna J, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318(5858):1920–3. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 75.Goemans NM, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 364(16):1513–22. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 76.Howard MT, et al. Sequence specificity of aminoglycoside-induced stop condon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol. 2000;48(2):164–9. [PubMed] [Google Scholar]
- 77.Buck NE, et al. Stop codon read-through of a methylmalonic aciduria mutation. Mol Genet Metab. 2009;97(4):244–9. doi: 10.1016/j.ymgme.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 78.Kaler SG, et al. Translational read-through of a nonsense mutation in ATP7A impacts treatment outcome in Menkes disease. Ann Neurol. 2009;65(1):108–13. doi: 10.1002/ana.21576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hein LK, et al. alpha-L-iduronidase premature stop codons and potential read-through in mucopolysaccharidosis type I patients. J Mol Biol. 2004;338(3):453–62. doi: 10.1016/j.jmb.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 80.Sato A, et al. Small interfering RNA delivery to the liver by intravenous administration of galactosylated cationic liposomes in mice. Biomaterials. 2007;28(7):1434–42. doi: 10.1016/j.biomaterials.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 81.Kakkis ED, et al. Long-term and high-dose trials of enzyme replacement therapy in the canine model of mucopolysaccharidosis I. Biochem Mol Med. 1996;58(2):156–67. doi: 10.1006/bmme.1996.0044. [DOI] [PubMed] [Google Scholar]
- 82.Germain DP, et al. Enzyme replacement therapy of lysosomal storage diseases. Rev Med Interne. 2010;31(Suppl 2):S279–91. doi: 10.1016/S0248-8663(10)70028-X. [DOI] [PubMed] [Google Scholar]
- 83.Schiffmann R, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743–9. doi: 10.1001/jama.285.21.2743. [DOI] [PubMed] [Google Scholar]
- 84.Eng CM, et al. A phase 1/2 clinical trial of enzyme replacement in fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet. 2001;68(3):711–22. doi: 10.1086/318809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Arikan-Ayyildiz Z, et al. Immunoglobulin abnormalities and effects of enzyme replacement therapy in children with Gaucher disease. Pediatr Blood Cancer. 2010 doi: 10.1002/pbc.22863. [DOI] [PubMed] [Google Scholar]
- 86.Kang TS, et al. Converting an injectable protein therapeutic into an oral form: phenylalanine ammonia lyase for phenylketonuria. Mol Genet Metab. 2010;99(1):4–9. doi: 10.1016/j.ymgme.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sampson HM, et al. Identification of a NBD1-binding pharmacological chaperone that corrects the trafficking defect of F508del-CFTR. Chem Biol. 2011;18(2):231–42. doi: 10.1016/j.chembiol.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 88.Kuznetsov G, Nigam SK. Folding of secretory and membrane proteins. N Engl J Med. 1998;339(23):1688–95. doi: 10.1056/NEJM199812033392307. [DOI] [PubMed] [Google Scholar]
- 89.McCully KS. Homocysteine, vitamins, and vascular disease prevention. Am J Clin Nutr. 2007;86(5):1563S–8S. doi: 10.1093/ajcn/86.5.1563S. [DOI] [PubMed] [Google Scholar]
- 90.Shintaku H, et al. Long-term treatment and diagnosis of tetrahydrobiopterin-responsive hyperphenylalaninemia with a mutant phenylalanine hydroxylase gene. Pediatr Res. 2004;55(3):425–30. doi: 10.1203/01.PDR.0000111283.91564.7E. [DOI] [PubMed] [Google Scholar]
- 91.Lindstedt S, et al. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340(8823):813–7. doi: 10.1016/0140-6736(92)92685-9. [DOI] [PubMed] [Google Scholar]
- 92.Yue TF, Gutman AB. Effect Of Allopurinol (4-Hydroxypyrazolo-(3,4-D)Pyrimidine) On Serum And Urinary Uric Acid In Primary And Secondary Gout. Am J Med. 1964;37:885–98. doi: 10.1016/0002-9343(64)90131-7. [DOI] [PubMed] [Google Scholar]
- 93.Daniotti M, et al. New developments in the treatment of hyperammonemia: emerging use of carglumic acid. Int J Gen Med. 2011;4:21–8. doi: 10.2147/IJGM.S10490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Spronsen FJ, et al. Phenylketonuria: tyrosine supplementation in phenylalanine-restricted diets. Am J Clin Nutr. 2001;73(2):153–7. doi: 10.1093/ajcn/73.2.153. [DOI] [PubMed] [Google Scholar]
- 95.Batshaw ML, Brusilow SW. Evidence of lack of toxicity of sodium phenylacetate and sodium benzoate in treating urea cycle enzymopathies. J Inherit Metab Dis. 1981;4(4):231. doi: 10.1007/BF02263659. [DOI] [PubMed] [Google Scholar]
- 96.Batshaw ML, et al. Therapy of urea cycle enzymopathies: three case studies. Johns Hopkins Med J. 1981;148(1):34–40. [PubMed] [Google Scholar]
- 97.Holton JB, de la Cruz F, Levy HL. Galactosemia: the uridine diphosphate galactose deficiency-uridine treatment controversy. J Pediatr. 1993;123(6):1009–14. doi: 10.1016/s0022-3476(05)80404-4. [DOI] [PubMed] [Google Scholar]
- 98.Sudha KN, et al. Virtual screening for novel COX-2 inhibitors using the ZINC database. Bioinformation. 2008;2(8):325–9. doi: 10.6026/97320630002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thoden JB, et al. Molecular structure of human galactokinase: implications for type II galactosemia. J Biol Chem. 2005;280(10):9662–70. doi: 10.1074/jbc.M412916200. [DOI] [PubMed] [Google Scholar]
- 100.Bork P, Sander C, Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993;2(1):31–40. doi: 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tang M, et al. Molecular and biochemical characterization of human galactokinase and its small molecule inhibitors. Chem Biol Interact. 2010;188(3):376–385. doi: 10.1016/j.cbi.2010.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thoden JB, Holden HM. Molecular structure of galactokinase. J Biol Chem. 2003;278(35):33305–11. doi: 10.1074/jbc.M304789200. [DOI] [PubMed] [Google Scholar]
- 103.Timson DJ. GHMP Kinases - Structures, Mechanisms and Potential for Therapeutically Relevant Inhibition. Current Enzyme Inhibition. 2007;3(1):77–94. [Google Scholar]
- 104.Timson DJ, Reece RJ. Functional analysis of disease-causing mutations in human galactokinase. Eur J Biochem. 2003;270(8):1767–74. doi: 10.1046/j.1432-1033.2003.03538.x. [DOI] [PubMed] [Google Scholar]
- 105.Timson DJ, Reece RJ. Sugar recognition by human galactokinase. BMC Biochem. 2003;4:16. doi: 10.1186/1471-2091-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Debouck C, et al. Structure of the galactokinase gene of Escherichia coli, the last (?) gene of the gal operon. Nucleic Acids Res. 1985;13(6):1841–53. doi: 10.1093/nar/13.6.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Andreassi JL, 2nd, et al. Crystal structure of the Streptococcus pneumoniae mevalonate kinase in complex with diphosphomevalonate. Protein Sci. 2007;16(5):983–9. doi: 10.1110/ps.072755707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Odejinmi S, et al. Structure-activity analysis and cell-based optimization of human galactokinase inhibitors. ACS Medicinal Chemistry Letters. 2011 doi: 10.1021/ml200131j. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Edwards PA, Ericsson J. Signaling molecules derived from the cholesterol biosynthetic pathway: mechanisms of action and possible roles in human disease. Curr Opin Lipidol. 1998;9(5):433–40. doi: 10.1097/00041433-199810000-00007. [DOI] [PubMed] [Google Scholar]
- 110.Miallau L, et al. Biosynthesis of isoprenoids: crystal structure of 4-diphosphocytidyl-2C-methyl-D-erythritol kinase. Proc Natl Acad Sci U S A. 2003;100(16):9173–8. doi: 10.1073/pnas.1533425100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sacchettini JC, Poulter CD. Creating isoprenoid diversity. Science. 1997;277(5333):1788–9. doi: 10.1126/science.277.5333.1788. [DOI] [PubMed] [Google Scholar]
- 112.Balibar CJ, Shen X, Tao J. The mevalonate pathway of Staphylococcus aureus. J Bacteriol. 2009;191(3):851–61. doi: 10.1128/JB.01357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 114.Smit A, Mushegian A. Biosynthesis of isoprenoids via mevalonate in Archaea: the lost pathway. Genome Res. 2000;10(10):1468–84. doi: 10.1101/gr.145600. [DOI] [PubMed] [Google Scholar]
- 115.Dubey VS, Bhalla R, Luthra R. An overview of the non-mevalonate pathway for terpenoid biosynthesis in plants. J Biosci. 2003;28(5):637–46. doi: 10.1007/BF02703339. [DOI] [PubMed] [Google Scholar]
- 116.Eisenreich W, et al. Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell Mol Life Sci. 2004;61(12):1401–26. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hunter WN. The non-mevalonate pathway of isoprenoid precursor biosynthesis. J Biol Chem. 2007;282(30):21573–7. doi: 10.1074/jbc.R700005200. [DOI] [PubMed] [Google Scholar]
- 118.Lichtenthaler HK, et al. The non-mevalonate isoprenoid biosynthesis of plants as a test system for new herbicides and drugs against pathogenic bacteria and the malaria parasite. Z Naturforsch C. 2000;55(5-6):305–13. doi: 10.1515/znc-2000-5-601. [DOI] [PubMed] [Google Scholar]
- 119.Eoh H, Brennan PJ, Crick DC. The Mycobacterium tuberculosis MEP (2C-methyl-d-erythritol 4-phosphate) pathway as a new drug target. Tuberculosis (Edinb) 2009;89(1):1–11. doi: 10.1016/j.tube.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Boucher Y, Doolittle WF. The role of lateral gene transfer in the evolution of isoprenoid biosynthesis pathways. Mol Microbiol. 2000;37(4):703–16. doi: 10.1046/j.1365-2958.2000.02004.x. [DOI] [PubMed] [Google Scholar]
- 121.Tang M, et al. Identification and Identification of novel small molecule inhibitors of 4-disphosphocytidyl-2-C-methyl D-erythritol (CDP-ME) kinase of Gram-negative bacteria. Bioorganic and Medicinal Chemistry. 2011 doi: 10.1016/j.bmc.2011.08.012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cheng GZ, et al. Advances of AKT pathway in human oncogenesis and as a target for anti-cancer drug discovery. Curr Cancer Drug Targets. 2008;8(1):2–6. [PubMed] [Google Scholar]
- 123.Brace A, et al. US Patent Application Publication - Application#11/993,191. USA: Jul 22, 2010. GALK1s as modifiers of the PTEN/AKT Pathway and methods of use. [Google Scholar]
- 124.Antshel KM, Epstein IO, Waisbren SE. Cognitive strengths and weaknesses in children and adolescents homozygous for the galactosemia Q188R mutation: a descriptive study. Neuropsychology. 2004;18(4):658–64. doi: 10.1037/0894-4105.18.4.658. [DOI] [PubMed] [Google Scholar]
- 125.Bosch AM, et al. Living with classical galactosemia: health-related quality of life consequences. Pediatrics. 2004;113(5):e423–8. doi: 10.1542/peds.113.5.e423. [DOI] [PubMed] [Google Scholar]
- 126.Lambert C, Boneh A. The impact of galactosaemia on quality of life--a pilot study. J Inherit Metab Dis. 2004;27(5):601–8. doi: 10.1023/b:boli.0000042957.98782.e4. [DOI] [PubMed] [Google Scholar]
- 127.Ridel KR, Leslie ND, Gilbert DL. An updated review of the long-term neurological effects of galactosemia. Pediatr Neurol. 2005;33(3):153–61. doi: 10.1016/j.pediatrneurol.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 128.Waisbren SE, et al. Effect of expanded newborn screening for biochemical genetic disorders on child outcomes and parental stress. Jama. 2003;290(19):2564–72. doi: 10.1001/jama.290.19.2564. [DOI] [PubMed] [Google Scholar]
- 129.Waisbren SE, et al. Brief report: Predictors of parenting stress among parents of children with biochemical genetic disorders. J Pediatr Psychol. 2004;29(7):565–70. doi: 10.1093/jpepsy/jsh058. [DOI] [PubMed] [Google Scholar]
- 130.Leslie ND, et al. A mouse model of galactose-1-phosphate uridyl transferase deficiency. Biochem Mol Med. 1996;59(1):7–12. doi: 10.1006/bmme.1996.0057. [DOI] [PubMed] [Google Scholar]
- 131.Fitzgerald J, Bateman JF. Why mice have lost genes for COL21A1, STK17A, GPR145 and AHRI: evidence for gene deletion at evolutionary breakpoints in the rodent lineage. Trends Genet. 2004;20(9):408–12. doi: 10.1016/j.tig.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 132.Lai K, et al. ARHI: A new target of galactose toxicity in Classic Galactosemia. Biosci Hypotheses. 2008;1(5):263–271. doi: 10.1016/j.bihy.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]