

Abstract
Objective
This prospective study aims to address mortality in the context of the early pulmonary immune response to burn and inhalation injury.
Methods
We collected bronchoalveolar lavage (BAL) fluid from 60 burn patients within 14 hours of their injury when smoke inhalation was suspected. Clinical and laboratory parameters and immune mediator profiles were compared to patient outcomes.
Results
Patients who succumbed to their injuries were older (p=0.005), had a larger % TBSA (total body surface area) burn (p<0.001), and required greater 24-hour resuscitative fluids (p=0.002). Non-survivors had lower BAL fluid concentrations of numerous immunomodulators, including C5a, IL-1β, IL-1RA, IL-8, IL-10, and IL-13 (p<0.05 for all). Comparing only those with the highest Baux scores to account for the effects of age and % TBSA burn on mortality, non-survivors also had reduced levels of IL-2, IL-4, G-CSF, IFN-γ, MIP-1β, and TNF-α (p<0.05 for all). The apparent pulmonary immune hyporesponsiveness in those who died was confirmed by in vitro culture, which revealed that pulmonary leukocytes from non-survivors had a blunted production of numerous immune mediators.
Conclusions
Our study demonstrates that the early pulmonary immune response to burn and smoke inhalation may be attenuated in patients who succumb to their injuries.
Keywords: Burn, Inhalation Injury, Cytokines, Chemokines, Mortality
Introduction
Enhanced resuscitative efforts, advancements in wound care and closure, improved prevention and treatment of infections, and more directed pulmonary management strategies have improved survival of burn-injured patients since the 1950’s.1 Yet, according to the National Burn Repository 2007 Report, there were only minimal changes in overall mortality during the previous decade, with the worst survival still in those with an associated inhalation injury.2 Meanwhile, and despite promising bench-level research, numerous trials designed to modulate the immune response to injury have fallen short of demonstrating a clear benefit to improving mortality.3–20 Indeed, this may be a function of the complex interplay between the severity of injury and inherent patient parameters such as age, gender, comorbidity, genetic polymorphisms, or even study design. As such, it appears that the immune response to injury remains incompletely understood and that additional effort is required to further improve survival of the burn-injured patient.
Our prospective study which targets the pulmonary immune response to burn and smoke inhalation injury demonstrates a pattern of early post-injury immune dysfunction that may be associated with burn patient mortality. These findings may explain failure of earlier trials designed to modulate the immune response to injury.
Methods
Patients and Parameters
From January 2007 to April 2010, bronchoalveolar lavage (BAL) fluid was collected from 60 patients admitted to the Burn Intensive Care Unit when inhalation injury was suspected by history, physical, and/or laboratory findings. Exclusion criteria were age less than 18 years, malignancy, use of immunosuppressive medications, or known autoimmune or chronic inflammatory diseases. Clinical variables and outcomes collected were: age, gender, race/ethnicity, presenting weight in kilograms (kg), presenting carboxyhemoglobin (COHb) level, % total body surface area (% TBSA) burn, presence of inhalation injury, inhalation injury grade, ratio of partial pressure of oxygen in arterial blood to fraction of inspired oxygen (PaO2/FiO2) at the time of bronchoscopy, BAL fluid cell count and differential, initial 24- and 72-hour fluid requirements (cc/kg), incidence of pneumonia, incidence of sepsis, incidence of tracheostomy, ICU and hospital length of stay, and mortality. Pneumonia and sepsis were defined according to American Burn Association Consensus Conference criteria.21 All BAL fluid samples were collected before any aerosolized pulmonary medications had been administered (β-agonists, heparin, etc.). This study was approved by the Institutional Review Board.
Bronchoalveolar Lavage
Following a standardized protocol, bronchoscopy and BAL were performed within 14 hours of injury in all subjects.22 In brief, the bronchoscope was directed into the left lower lobe, wedged, and 20 cc of saline instilled, aspirated, and discarded. After repositioning of the bronchoscope into a different subsegmental bronchus in the same lobe, another 20 cc of saline was instilled and then aspirated into a sterile Lukens’s fluid trap. Subsequently, up to four 20 cc aliquots were collected from the other four lobes as tolerated by the patient. A portion of the sample was sent for fluid cell count, differential, and culture. Any remaining BAL fluid not required for clinical analysis was then aliquoted for research purposes.
Grading of Inhalation Injury
Using a standardized bronchoscopic scoring system based on Abbreviated Injury Score Criteria the grade of inhalation injury was assessed, as previously described.23 Specifically, the severity of inhalation injury was categorized into one of five grades (0, 1, 2, 3, and 4) with 0 being the absence of visible injury (Figure 1).
Figure 1.
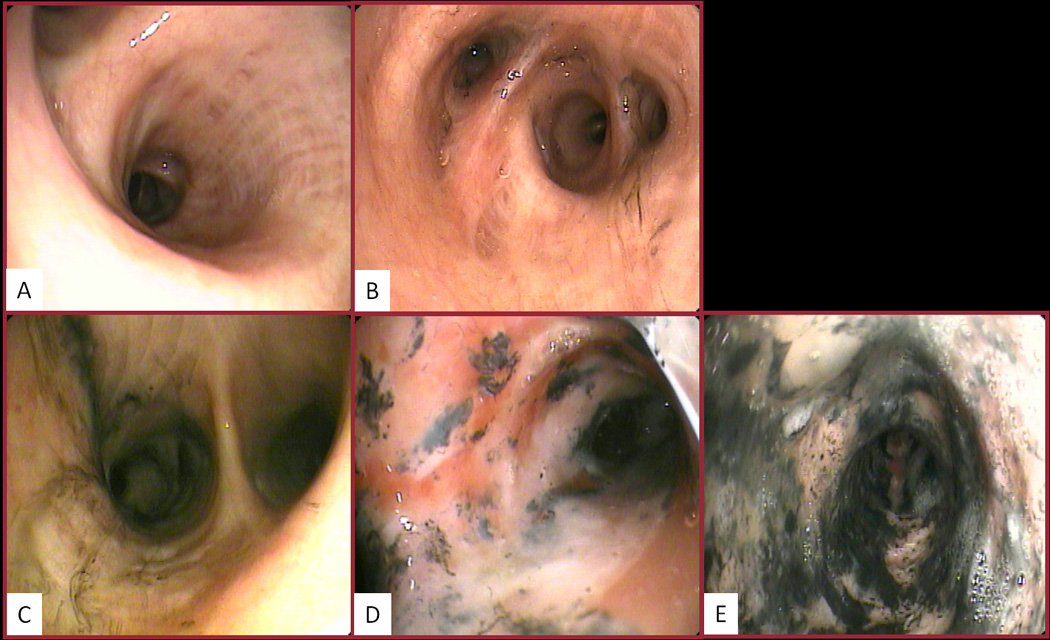
Bronchoscopic grading of inhalation injury: A. Grade 0 (no injury), absence of carbonaceous deposits, erythema, edema, bronchorrhea, or obstruction; B. Grade 1 (mild injury), minor or patchy areas of erythema, carbonaceous deposits in proximal or distal bronchi (any or combination); C. Grade 2 (moderate injury), moderate degree of erythema, carbonaceous deposits, bronchorrhea, with or without compromise of the bronchi (any or combination); D. Grade 3 (severe injury), severe inflammation with friability, copious carbonaceous deposits, bronchorrea, bronchial obstruction (any or combination); E. Grade 4 (massive injury), evidence of mucosal sloughing, necrosis, endoluminal obliteration (any or combination).
Sample Processing, Pulmonary Leukocyte Culture, and Immunomodulator Detection
BAL samples were initially strained through sterile 100 micron nylon cell strainers (BD Biosciences, Bedford, MA) and the raw filtrate centrifuged at 1200 RPM for 5 minutes to separate the supernatant from the cellular component. The supernatant was then harvested, aliquoted, and frozen at −80° C. After re-suspending the cell pellet in culture medium (RPMI with 5% FBS and 1% PSG), leukocytes were assessed for viability with trypan blue dye exclusion, plated in duplicate at 105 cells/ml with or without LPS, and cultured at 37° C for 20 hours. Finally, the pulmonary culture supernatants were harvested, aliquoted, and frozen at −80° C.
The concentrations of 28 immunomodulating proteins were assayed in BAL fluid and culture supernatants via Bio-Rad Multiplex (Hercules, CA).24 In short, 50 µl of each sample was added to a 96 well filter-bottom plate containing 50 µl of antibody coated fluorescent beads. Following several incubation and washing steps, biotinylated detection and streptavidin-PE antibodies were added to the plate per manufacturer instructions. After re-suspension of the beads the plate was read on a Bio-Rad Multiplex reader. All samples were assayed in duplicate and the results analyzed using the Bio-Plex manager software, version 5.0. Enzyme linked immunosorbant assays (ELISA) were utilized to measure sample concentrations of complement component 5a (C5a), as it was not part of the multiplex assay, and interleukin (IL)-8, as concentrations were initially out of testing range requiring dilution and re-measurement. These assays were performed in duplicate according to manufacturer instructions (R&D Systems, Minneapolis, MN).
Statistical analysis
Patient demographics, outcomes, and immunomodulator concentrations were assessed for normality and parametric or non-parametric tests applied where appropriate. Continuous variables of parametric tests are reported as mean with standard deviation, and non-parametric tests reported as median with 25th and 75th percentiles. Otherwise dichotomous variables are reported as a number and percent. Logistic regression was performed based on log-transformed data in order to generate odds ratios and control for relevant confounders. Statistical analyses were calculated with SAS Version 9.1 (SAS Institute Inc., Cary, NC) and corresponding graphs created with GraphPad Prism 5 for Windows (GraphPad Software, La Jolla, CA). A difference between observed variables was considered significant when p < 0.05.
Results
Patient Demographics, Clinical Characteristics, and Outcomes
BAL fluid was collected from 63 patients admitted to the Burn Intensive Care Unit, of which 3 were excluded from study analysis for declining study participation. The demographics, clinical characteristics, and outcomes of the remaining 60 patients are shown in Table 1. As expected, age, % TBSA, Baux score (Age plus % TBSA), and fluid resuscitation were significantly associated with mortality.25 Further examination of our data with logistic regression upheld the association between mortality and age (OR 1.26; 95% CI 1.08:1.48) and % TBSA (OR 1.18; 95% CI 1.05:1.32). A subgroup analysis also revealed that mortality was 50% for those who received greater than 250 cc/kg resuscitation during the first 24 hr compared to just 21% of those who received less than 250 cc/kg, although this difference only trended toward statistical significance (p=0.098). Similarly, the percentage of those developing pneumonia or sepsis was not statistically different between those who succumbed to their injuries and those who did not (p=0.180 and 0.122, respectively). These differences may carry clinical relevance, however, in that two-thirds who died developed pneumonia compared to less than one-half those who survived, and the frequency of sepsis was twice that in the deceased compared to survivors. Finally, between survivors and non-survivors there were no differences in terms of inhalation injury frequency, inhalation injury severity, PaO2/FiO2, COHb levels, requirements for tracheostomy, and ventilator days.
Table 1.
Demographics, Clinical Characteristics, Outcomes and Mortality
| Survivors n=45 |
Deceased n=15 |
p Value | |
|---|---|---|---|
| Age | 49 (33–61) | 67 (47–80) | 0.005 |
| Gender | 0.646 | ||
| Male | 27 (60) | 10 (67) | |
| Female | 18 (40) | 5 (33) | |
| Race/Ethnicity | 0.769 | ||
| Caucasian | 26 (58) | 10 (67) | |
| African American | 12 (27) | 2 (13) | |
| Hispanic | 4 (9) | 1 (6) | |
| Asian | 1 (2) | 1 (6) | |
| Other/Unknown | 2 (4) | 1 (6) | |
| TBSA (%) | 9 (1–21) | 42 (15–71) | <0.001 |
| Baux Score (Age + TBSA) | 62.4 (± 3.1) | 108.5 (±5.9) | <0.0001 |
| Initial 24 hr fluid resuscitation (cc/kg) |
73.3 (40.3–151.4) | 202.5 (142.9–305.2) | 0.002 |
| Initial 72 hr fluid resuscitation (cc/kg) |
185.6 (131.3–329.1) | 365.0 (285.6–472.2) | <0.001 |
| Inhalation Injury | 37 (82) | 14 (93) | 0.427 |
| Inhalation Injury Grade | 0.210 | ||
| Grade 0 | 8 (18) | 1 (6) | |
| Grade 1 | 8 (18) | 7 (47) | |
| Grade 2 | 13 (29) | 2 (13) | |
| Grade 3 | 14 (31) | 4 (27) | |
| Grade 4 | 2 (4) | 1 (6) | |
| COHb (%) | 7.3 (3.4–12.5) | 2.2 (1.0–10.8) | 0.074 |
| PaO2/FiO2 | 340.6 (±18.0) | 344.3 (±30.0) | 0.917 |
| Pneumonia | 21 (47) | 10 (67) | 0.180 |
| Tracheostomy | 16 (36) | 5 (33) | 0.876 |
| Ventilator days | 10 (3–27) | 13 (6–34) | 0.255 |
| Sepsis | 6 (13) | 4 (27) | 0.122 |
Data presented as n (%), mean (±SD), or median (interquartile range) where appropriate. COHb, Carboxyhemoglobin; ICU, Intensive Care Unit; LOS, Length of Stay; PaO2/FiO2, partial pressure of oxygen / fraction of inspired oxygen at the time of bronchoscopy; TBSA, Total Body Surface Area skin burn.
BAL Fluid and Pulmonary Leukocyte Culture Findings
The primary focus of our study was the relationship between the pulmonary immune response to burn and inhalation injury and the ultimate outcome, that being mortality. The results of this analysis are shown in Table 2. Fibroblast growth factor was undetectable in 93% of samples and was excluded from statistical interpretation. When we compared the BAL fluid concentrations of the remaining immunomodulators in the entire study cohort of survivors and non-survivors we found that the concentrations of complement component 5a (C5a), interleukin (IL)-1β, IL-1 receptor antagonist (RA), IL-8, IL-10, and IL-13 were significantly lower in the deceased when normalized to volume of BAL fluid (p≤0.05 for all); likewise, the BAL fluid concentrations of IL-1RA, IL-4, IL-10, and granulocyte-macrophage colony-stimulating factor (GM-CSF) were also lower in the deceased when normalized to total protein concentration (p<0.05 for all). In addition to these results and those shown in Table 1, survivors and non-survivors were no different in terms of overall BAL fluid protein concentration, white blood cell (WBC) count, or WBC differential. Understanding that our results might be explained by differences in age and % TBSA, we compared only survivors and non-survivors with a Baux score above the median. In this fashion, the groups did not differ in terms of age, gender, % TBSA, inhalation injury frequency, inhalation injury severity, COHb level, resuscitation requirements, BAL fluid WBC differential, and BAL fluid protein concentration; the sole variable that varied between these groups was the total WBC count, which was lower in those who had died (median 141 per µL, vs. 1019 per µL from survivors; p=0.036). Interestingly, even after comparing only those with Baux scores above the median, the BAL fluid concentration (normalized to volume) of many immune mediators was still lower in the deceased, including IL-1β, IL-1RA, IL-2, IL-4, IL-8, IL-10, granulocyte-colony stimulating factor (G-CSF), interferon (IFN)-γ, macrophage inflammatory protein (MIP)-1β, and tumor necrosis factor (TNF)-α (p<0.05 for all) (Table 3). Finally, these overall results persisted even after normalizing the BAL fluid immune mediators to total protein concentration (Table 3).
Table 2.
BAL Fluid Cytokine, Chemokine, and Growth Factors as Related to Mortality, Entire Cohort
| Concentration (pg/ml fluid) | Concentration (pg/mg protein) | |||||
|---|---|---|---|---|---|---|
| Protein | Survivors n=45 |
Deceased n=15 |
p Value | Survivors n=45 |
Deceased n=15 |
p Value |
| C5a | 405.5 (222.6–1086) | 192.9 (89.5–448.5) | 0.025 | 369.1 (209.4–931.1) | 292.9(184.7–405.5) | 0.199 |
| IL-1β | 33.4 (10.4–83.2) | 3.1 (1.4–21.3) | 0.045 | 46.2 (11.8–83.6) | 8.3 (1.6–84.2) | 0.078 |
| IL-1RA | 316.1 (174.7–888.6) | 184.9 (43.8–328.5) | 0.020 | 440.9 (175.1–729.1) | 163.0 (97.6–589.7) | 0.048 |
| IL-2 | 1.4 (0–4.7) | 0.3 (0–2.0) | 0.256 | 0.9 (0–4.8) | 0.9 (0–2.2) | 0.357 |
| IL-4 | 1.2 (0–3.4) | 0 (0–2.3) | 0.138 | 318.6 (0–992.3) | 0 (0–222.2) | 0.015 |
| IL-5 | 0.2 (0–0.5) | 0 (0–0.4) | 0.277 | 0.2 (0–0.5) | 0.1 (0–0.6) | 0.566 |
| IL-6 | 179.5 (52.2–519.5) | 139.7 (62.7–246.2) | 0.445 | 195.8 (82.7–516.5) | 152.6 (62.7–441.8) | 0.567 |
| IL-7 | 5.8 (2.0–8.4) | 3.3 (0.9–6.1) | 0.277 | 4.4 (1.6–11.3) | 3.3 (1.2–9.6) | 0.611 |
| IL-8 | 4845 (2191–16563) | 1345 (573.3–3668) | 0.005 | 5401 (2555–17240) | 2346 (1211–5604) | 0.082 |
| IL-9 | 6.3 (0–12.9) | 7.2 (4.0–14.3) | 0.556 | 4.8 (0–15.0) | 5.9 (2.6–18.6) | 0.536 |
| IL-10 | 7.3 (3.7–13.9) | 2.5 (1.7–7.4) | 0.008 | 6.8 (4.1–22.0) | 3.9 (2.2–6.3) | 0.038 |
| IL-12 | 3.4 (0–11.2) | 0 (0–5.7) | 0.250 | 1.9 (0–14.4) | 0 (0–3.5) | 0.157 |
| IL-13 | 3.5 (1.6–5.4) | 2.0 (0.7–3.7) | 0.050 | 3.6 (1.6–5.6) | 1.7 (0.9–3.9) | 0.065 |
| IL-15 | 9.1 (1.2–9.1) | 4.7 (0.6–14.4) | 0.545 | 3.3 (0.2–8.1) | 4.2 (1.5–9.0) | 0.626 |
| IL-17 | 0 (0–2.9) | 0 (0–0) | 0.255 | 0 (0–2.1) | 0 (0–0) | 0.408 |
| Eotaxin | 50.2 (12.6–80.5) | 16.1 (0–128.1) | 0.332 | 37.9 (9.0–87.9) | 25.5 (0–12.8) | 0.501 |
| G-CSF | 104.8 (43.1–259.7) | 68.2 (40.6–118.8) | 0.250 | 96.7 (45.3–272.6) | 151.4 (30.9–173.2) | 0.738 |
| GM-CSF | 2.5 (0–7.6) | 0 (0–5.1) | 0.177 | 2.7 (0–7.2) | 0 (0–2.2) | 0.027 |
| IFN-γ | 59.7 (22.1–147) | 23.7 (8.1–85.5) | 0.131 | 72.0 (18.7–154.5) | 25.1 (9.3–99.8) | 0.183 |
| IP-10 | 1042 (388.7–2576) | 640.6 (151.5–2786) | 0.325 | 958.4 (324.8–3742) | 584.3 (278.3–5557) | 0.901 |
| MCP-1 | 75.7 (35.3–255.8) | 123.4 (40.2–639.2) | 0.431 | 122.2 (35.0–291.9) | 285.6 (53.7–480.8) | 0.336 |
| MIP-1α | 0 (0–20.7) | 0 (0–5.4) | 0.360 | 0 (0–14.2) | 0 (0–6.0) | 0.510 |
| MIP-1β | 63.6 (25.8–168.5) | 28.1 (7.8–63.9) | 0.067 | 63.5 (26.5–171.4) | 56.3 (20.5–95.8) | 0.365 |
| PDGF | 26.3 (8.4–41.9) | 9.6 (0–34.9) | 0.207 | 20.7 (8.3–65.0) | 10.3 (0–24.7) | 0.285 |
| RANTES | 48.8 (21.2–85.4) | 20.5 (9.8–64.3) | 0.101 | 65.0 (25.5–119.1) | 31.0 (18.3–73.0) | 0.245 |
| TNF-α | 29.5 (0–63.2) | 4.8 (0–60.9) | 0.201 | 18.8 (0–79.5) | 0 (0–33.0) | 0.090 |
| VEGF | 131.4 (54.1–261.5) | 68.0 (25.9–164.8) | 0.106 | 100.3 (45.4–348.5) | 76.3 (24.9–373.3) | 0.443 |
Data represented as median (interquartile range).
Table 3.
BAL Fluid Cytokine, Chemokine, and Growth Factors as Related to Mortality, Patients Matched According to Baux Score > Median (69.5)
| Concentration (pg/ml fluid) | Concentration (pg/mg protein) | |||||
|---|---|---|---|---|---|---|
| Protein | Survivors n=15 |
Deceased n=15 |
p Value | Survivors n=15 |
Deceased n=15 |
p Value |
| C5a | 382.1 (233.2–843.7) | 192.9 (89.5–448.5) | 0.063 | 503.1 (212.0–956.8) | 292.9 (184.7–405.5) | 0.141 |
| IL-1β | 47.1 (15.4–72.2) | 3.1 (1.4–21.3) | 0.042 | 51.9 (33.1–123.7) | 8.3 (1.6–84.2) | 0.036 |
| IL-1RA | 342.8 (219.3–940.7) | 184.9 (43.8–328.5) | 0.023 | 354.4 (295.3–730.6) | 163.0 (97.6–589.7) | 0.027 |
| IL-2 | 2.7 (0.5–6.9) | 0.3 (0–2.0) | 0.032 | 3.8 (0.9–6.8) | 0.9 (0–2.2) | 0.026 |
| IL-4 | 1.8 (0.7–6.9) | 0 (0–2.3) | 0.021 | 690.3 (95.1–947.8) | 0 (0–222.2) | 0.006 |
| IL-5 | 0.2 (0–0.5) | 0 (0–.04) | 0.279 | 0.3 (0.1–0.9) | 0.1 (0–0.6) | 0.281 |
| IL-6 | 171.4 (51.4–831.8) | 139.7 (62.7–246.2) | 0.562 | 245.4 (90.6–658.9) | 152.6 (62.7–441.8) | 0.383 |
| IL-7 | 5.4 (1.3–8.4) | 3.3 (0.9–6.1) | 0.506 | 4.5 (2.0–16.5) | 3.3 (1.2–9.6) | 0.383 |
| IL-8 | 5377 (2810–29399) | 1345 (573.3–3668) | 0.002 | 6363 (3843–32780) | 2346 (1211–5604) | 0.011 |
| IL-9 | 11.2 (6.8–22.5) | 7.2 (4.0–14.3) | 0.177 | 11.3 (7.5–28.6) | 5.9 (2.6–18.6) | 0.158 |
| IL-10 | 7.5 (5.0–26.6) | 2.5 (1.7–7.4) | 0.003 | 12.3 (4.9–29.9) | 3.9 (2.2–6.3) | 0.004 |
| IL-12 | 4.4 (0–14.0) | 0 (0–5.7) | 0.189 | 5.9 (0–14.7) | 0 (0–3.5) | 0.089 |
| IL-13 | 3.0 (1.6–7.4) | 2.0 (0.7–3.7) | 0.165 | 3.9 (2.3–6.0) | 1.7 (0.9–3.9) | 0.081 |
| IL-15 | 3.4 (1.1–24.9) | 4.7 (0.6–14.4) | 0.431 | 4.0 (1.3–16.5) | 4.2 (1.5–9.0) | 0.878 |
| IL-17 | 0 (0–15.2) | 0 (0–0) | 0.162 | 0 (0–7.1) | 0 (0–0) | 0.385 |
| Eotaxin | 50.2 (0.8–93.4) | 16.1 (0–128.1) | 0.490 | 58.2 (0.8–108.6) | 25.5 (0–12.8) | 0.621 |
| G-CSF | 166.5 (71.1–734.7) | 68.2 (40.6–118.8) | 0.016 | 272.5 (81.6–651.6) | 151.4 (30.9–173.2) | 0.081 |
| GM-CSF | 4.2 (0–11.9) | 0 (0–5.1) | 0.052 | 4.5 (0–10.7 | 0 (0–2.2) | 0.016 |
| IFN-γ | 81.0 (42.7–159.3) | 23.7 (8.1–85.5) | 0.025 | 108.5 (72.8–193.7) | 25.1 (9.3–99.8) | 0.012 |
| IP-10 | 884.2 (257.7–2018) | 640.6 (151.5–2786) | 0.616 | 774.1 (274.2–1510) | 584.3(278.3–5557) | 0.849 |
| MCP-1 | 190.0 (102.2–386.5) | 123.4 (40.2–639.2) | 0.499 | 267.6 (145.8–515.9) | 285.6 (53.7–480.8) | 0.568 |
| MIP-1α | 0 (0–50.4) | 0 (0–5.4) | 0.245 | 0 (0–26.0) | 0 (0–6.0) | 0.469 |
| MIP-1β | 106.8 (31.0–256.9) | 28.1 (7.8–63.9) | 0.028 | 171.4 (49.1–264.5) | 56.3 (20.5–95.8) | 0.024 |
| PDGF | 28.7 (9.0–62.6) | 9.6 (0–34.9) | 0.224 | 31.0 (7.9–66.9) | 10.3 (0–24.7) | 0.270 |
| RANTES | 37.6 (21.2–68.2) | 20.5 (9.8–64.3) | 0.145 | 65.0 (38.1–76.1) | 31.0 (18.3–73.0) | 0.200 |
| TNF-α | 36.3 (7.4–205.4) | 4.8 (0–60.9) | 0.044 | 25.3 (9.6–109.0) | 0 (0–33.0) | 0.053 |
| VEGF | 135.8 (58.3–516.5) | 68.0 (25.9–164.8) | 0.089 | 222.2 (68.2–571.7) | 76.3 (24.9–373.3) | 0.166 |
Data represented as median (interquartile range).
Without any obvious confounder to explain the lower concentrations of pulmonary immunomodulators in the deceased, and because those who died from their injuries tended to have fewer WBCs in the airway, we suspected that a global immune hyporesponsiveness, if not paralysis, was responsible for our findings. To investigate these suspicions, we assessed the immonomodulator production of pulmonary leukocytes in culture with and without LPS. The immune mediator production by leukocytes derived from patients that would succumb to their injuries was blunted as compared to survivors, as depicted in Figure 2. This was particularly evident after in vitro stimulation with LPS. Indeed, this pattern persisted for all immune mediators measured from the culture supernatants except IL-1β, IL-4, IL-7, and IL-12. Statistical significance may not have been reached for IL-8 given low sample size as result of needing to repeat measurements. Nonetheless, the obvious pattern of blunted pulmonary leukocyte responsiveness remained.
Figure 2.

Immune mediator production by pulmonary leukocytes after culture for 20 hours in medium alone or in medium plus LPS. Comparisons are between survivors and deceased with medium alone, and survivors and deceased with medium plus LPS. *p< 0.05, vs. survivor with medium plus LPS.
Finally, in an effort to direct future research, we incorporated a logistic regression analysis comparing BAL fluid immunomodulator concentrations between survivors and deceased which controlled for the effects of age and % TBSA. Shown in Table 4, these results demonstrate that with increasing BAL fluid concentrations of numerous immunomodulators there is a concurrent reduction in the odds of death. For the crude analysis, this reduction in the odds of death with increasing immunomodulator concentrations was statistically significant for C5a, IL-1RA, IL-8, IL-10, IL-13, and MIP-1β (p<0.05 for all). After controlling for the effects of age and % TBSA, the adjusted odds for death were reduced with increasing BAL fluid concentrations of IL-8, IL-10, and MIP-1β with near significance (p<0.06 for all).
Table 4.
Crude and adjusted odds ratios for death associated with BAL fluid cytokine, chemokine, and growth factor concentrations
| Protein | Crude OR | 95% CI | p value | Adjusted OR* |
Adjusted 95% CI* |
Adjusted p value* |
|---|---|---|---|---|---|---|
| C5a | 0.56 | 0.32–0.99 | 0.046 | 0.52 | 0.21–1.27 | 0.150 |
| IL-1β | 0.72 | 0.51–1.02 | 0.065 | 0.74 | 0.41–1.33 | 0.319 |
| IL-1RA | 0.53 | 0.30–0.91 | 0.021 | 0.50 | 0.17–1.46 | 0.205 |
| IL-2 | 0.58 | 0.27–1.2 | 0.157 | 0.08 | 0.01–1.16 | 0.064 |
| IL-4 | 0.54 | 0.25–1.18 | 0.123 | 0.30 | 0.05–1.70 | 0.173 |
| IL-5 | 1.12 | 0.29–4.31 | 0.867 | 1.74 | 0.29–10.58 | 0.550 |
| IL-6 | 0.88 | 0.61–1.27 | 0.502 | 0.85 | 0.40–1.82 | 0.674 |
| IL-7 | 0.76 | 0.41–1.41 | 0.375 | 0.72 | 0.20–2.53 | 0.606 |
| IL-8 | 0.46 | 0.26–0.83 | 0.009 | 0.01 | 0.001–1.08 | 0.058 |
| IL-9 | 1.24 | 0.78–1.96 | 0.360 | 0.65 | 0.26–1.62 | 0.355 |
| IL-10 | 0.40 | 0.19–0.87 | 0.021 | 0.08 | 0.01–1.06 | 0.056 |
| IL-12 | 0.67 | 0.40–1.13 | 0.132 | 0.46 | 0.16–1.27 | 0.133 |
| IL-13 | 0.34 | 0.12–0.95 | 0.040 | 0.50 | 0.07–3.50 | 0.484 |
| IL-15 | 1.12 | 0.68–1.85 | 0.667 | 1.42 | 0.57–3.59 | 0.454 |
| IL-17 | 0.70 | 0.39–1.27 | 0.241 | 0.61 | 0.19–1.95 | 0.408 |
| Eotaxin | 0.81 | 0.59–1.11 | 0.188 | 1.36 | 0.73–2.51 | 0.333 |
| G-CSF | 0.71 | 0.43–1.20 | 0.201 | 0.15 | 0.01–1.90 | 0.143 |
| GM-CSF | 0.67 | 0.37–1.19 | 0.167 | 0.94 | 0.37–2.36 | 0.893 |
| IFN-γ | 0.77 | 0.55–1.08 | 0.125 | 0.41 | 0.16–1.05 | 0.063 |
| IP-10 | 0.83 | 0.55–1.27 | 0.393 | 1.07 | 0.60–2.03 | 0.849 |
| MCP-1 | 1.17 | 0.79–1.73 | 0.422 | 0.83 | 0.36–1.94 | 0.665 |
| MIP-1α | 0.81 | 0.54–1.20 | 0.291 | 0.76 | 0.38–1.53 | 0.445 |
| MIP-1β | 0.62 | 0.39–0.98 | 0.041 | 0.24 | 0.06–1.01 | 0.052 |
| PDGF | 0.78 | 0.52–1.15 | 0.211 | 1.07 | 0.49–2.34 | 0.861 |
| RANTES | 0.76 | 0.48–1.21 | 0.248 | 0.66 | 0.32–1.36 | 0.258 |
| TNF-α | 0.80 | 0.59–1.08 | 0.141 | 0.90 | 0.54–1.52 | 0.701 |
| VEGF | 0.66 | 0.40–1.09 | 0.104 | 0.53 | 0.19–1.53 | 0.241 |
BAL fluid cytokine, chemokine, and growth factor concentrations and their associated crude and adjusted odds of death.
Adjusted for Age and % TBSA burn.
Discussion
As expected, we found that increased age and % TBSA, particularly as manifested together in the Baux score, were associated with mortality in burn-injured patients. Most importantly, our results are suggestive that in some patients mortality may be associated with an immune hyporesponsiveness demonstrable soon after burn and inhalation injury.
The epidemiological importance of smoke inhalation in burn care cannot be overstated, as up to one-third of those presenting with major burns have a concurrent inhalation injury.26, 27 Additionally, the pathophysiologic effects of inhalation injury are profound, as both animal experiments and human observations have shown that smoke inhalation may decrease PaO2/FiO2 while increasing the following: lung edema, pulmonary artery shunting, fluid resuscitation requirements, COHb levels, duration of ventilator support, incidence of pneumonia and sepsis, and length of hospital stay.27–35 Further yet, prior studies have demonstrated that smoke inhalation evokes an enhanced immune response, with greater production of pulmonary inflammatory mediators in parallel to activation of neutrophils and macrophages.36–38 Indeed, our own earlier work showed that BAL fluid concentrations of numerous pulmonary inflammatory mediators are increased in response to not only the presence of smoke inhalation, but in particular to worse severities of inhalation injury.39
It has long been held that increasing age, % TBSA, and inhalation injury are amongst the most important predictors of mortality in burn-injured patients, of which supporting evidence is found in even the most recent appraisals.2, 40 The Baux score (defined as Age + % TBSA) has persisted for fifty years as one of the most consistent prognosticators of death from burn injury and has even been molded into a predictive scale that includes inhalation injury.2, 25, 41, 42 Likewise, numerous studies indicate that inhalation injury severity, fluid resuscitation, COHb, PaO2/FiO2, pneumonia, and sepsis have utility in predicting death after burn injury.2, 23, 43, 44 Specific to our study population of burn-injured patients with suspected inhalation injury we found that age, % TBSA, Baux score, and fluid resuscitation had significant associations with mortality. Moreover, the frequency of pneumonia and sepsis were considerably higher in non-survivors compared to survivors (67% vs 47% and 27% vs 13%, respectively). We would argue that the lack of statistical significance for the difference in survival between groups for these latter parameters, as well as inhalation injury, inhalation injury severity, COHb level, tracheostomy, ventilator days, and PaO2/FiO2 are likely the result of patient selection bias (85% of the study population had a visible inhalation injury upon bronchoscopy), study sample size, and/or length of survival following injury. Nonetheless, both the differences and similarities we noted between survivors and non-survivors were essential to investigating the unexpected and profound differences in the immune response to injury when comparing these two groups.
The most important finding of our study was an apparent pulmonary immune hyporesponsiveness pervasive to those who did not survive their injuries. For instance, non-survivors had lower BAL fluid concentrations of the majority of immune mediators, differences of which remained even after comparing only those with the highest Baux scores in order to account for the most likely confounders as well as after normalization of the BAL fluid results to total protein concentration. Furthermore, these findings were substantiated by our in vitro culture data, which showed that pulmonary leukocytes from those who perished had less production of numerous immune mediators, especially in response to culture with LPS. This finding was particularly intriguing given that non-survivors had fewer total WBC’s in their BAL fluid than survivors, which is suggestive of impaired leukocyte migration in the potential setting of immunoparalysis.
The results of our study only allow us to speculate as to the cause of early pulmonary immune hyporesponsiveness to burn and inhalation injury that were observed in non-survivors. Review of the literature suggests that the most likely explanations for this finding are: 1) impaired chemotaxis, 2) genetic permutations, and/or 3) an imbalance of pro- and anti-inflammatory signaling. First, our study results support the potential for impaired leukocyte migration and activation as the result of deficient chemotaxis in non-survivors. For instance, the BAL fluid total WBC count was lower in the deceased, as were the concentrations of MIP-1β and IL-8. Moreover, with increasing concentrations of these chemokines there was a nearly significant decrease in the odds of death, even after controlling for age and % TBSA. These findings do, however, appear to conflict with the literature to date, which indicates that there is a detrimental effect of increased pulmonary IL-8 signaling. For instance, in a study of 88 patients with burn injury, Rodriguez et al found that higher levels of pulmonary IL-8 were linked to early pulmonary physiologic dysfunction (p=0.006) and lung infection (p=0.040).45 Similarly, in a recent study of sheep with burn and smoke inhalation, blockade of IL-8 signaling attenuated lung permeability; yet, the authors of this study found no improvements in oxygenation with inhibition of IL-8 signaling.46 Thus, it appears that the relevance of chemotaxis to the burn-injured patient may require further study.
Several recent reports have investigated the role of genetic subtypes to outcomes following trauma and burns. For example, McDaniel et al found that genetic polymorphisms of IL-10 and IFN-γ varied significantly between patients that developed sepsis and those that did not.7 Similarly, Huebinger et al identified a reduction in the risk of death after burn injury in carriers of particular alleles of the IL-10 promotor.47 Though our study was not designed to assess the impact of genetic subtypes on outcomes after burn and inhalation injury such an investigation might prove useful.
Finally, earlier studies have suggested that some patients have a blunted immune response to injury. For instance, Mokart et al found that early elevated levels of the anti-inflammatory cytokine IL-1RA were associated with patients who developed septic shock soon after major surgical trauma.48 Along these lines, the importance of a balanced inflammatory response to injury was highlighted by an editorial describing the successes and failures of IFN-γ treatment after injury.49 In this editorial the authors suggested that an inadequate inflammatory response (or “immunoparalysis”) may be just as detrimental to outcome as too much inflammation. The argument of these authors was supported by the study of Nakos et al, who found that immunoparalyzed patients (defined as a decreased level of human leukocyte antigen-DR expression by alveolar macrophages) that were given IFN-γ had a reduction in the incidence of ventilator-associated pneumonia.50 Indeed, the results of our own study may lend credence to these earlier suggestions, and reason for a more targeted approach to immunomodulation in future trials.
We acknowledge several weaknesses of our study. First, our results are subject to considerable selection bias, as we could only perform the invasiveness of bronchoscopy on those suspected of inhalation injury; therefore, many patients with isolated burn injury were excluded. Investigation of how the immune system responds to isolated burn or inhalation injury may eliminate potential confounding effects. Second, our report lacks data on the systemic immune response profile as may be derived from analysis of plasma immunomodulator concentrations and plasma leukocyte functionality. Third, our data are limited to the first 14 hours following injury, and analysis of later time points may prove useful in determining which factors may contribute or change in response to events, be they pneumonia, sepsis, or multiple organ dysfunction. Undoubtedly, such comparisons will be the target of future research, especially with a greater number of study patients as may be afforded by collaboration with other centers.
In conclusion, our study has shown that: 1. Some burn and smoke-injured patients have a pulmonary immune hyporesponsiveness that is evident within 14 hours of injury; and 2. Immune hyporesponsiveness may be a contributing factor in the subsequent odds of mortality in these patients. Perhaps a better understanding of this early pulmonary immune dysfunction will allow for therapies that further improve outcomes in burn care.
Acknowledgements
We would especially like to thank the nurses, respiratory therapists, and staff in the burn ICU for their dedicated assistance in study sample handling, as well as the burn research nurses for their management of this prospectively designed study. Finally, the graciousness, friendship, and contributions of Dr. Young-Ku Choi will forever by remembered.
Funded by the National Institutes of Health (T32 GM008750, T32 AA013527, R01 AA012034, P30 AA019373), the Department of Defense (W81XWH-09-1-0619), the International Association of Fire Fighters, and the Dr. Ralph and Marian C. Falk Medical Research Trust
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Christopher S. Davis, Burn and Shock Trauma Institute, Department of Surgery, Loyola University Medical Center, Maywood, IL.
Joslyn M. Albright, Burn and Shock Trauma Institute, Department of Surgery, Loyola University Medical Center, Maywood, IL.
Stewart R. Carter, Burn and Shock Trauma Institute, Department of Surgery, Loyola University Medical Center, Maywood, IL.
Luis Ramirez, Burn and Shock Trauma Institute, Department of Surgery, Loyola University Medical Center, Maywood, IL.
Hajwa Kim, Center for Clinical and Translational Science, University of Illinois at Chicago, Chicago, IL.
Richard L. Gamelli, Burn and Shock Trauma Institute, Department of Surgery, Loyola University Medical Center, Maywood, IL.
Elizabeth J. Kovacs, Burn and Shock Trauma Institute, Department of Surgery, Loyola University Medical Center, Maywood, IL.
References
- 1.Pruitt BA, Wolf SE, Mason AD. Total Burn Care. 3rd Edition. Philadelphia: Elsevier; 2007. Epidemiological, demographic, and outcome characteristics of burn injury; pp. 14–32. [Google Scholar]
- 2.Miller SF, Bessey P, Lentz CW, et al. ABA NBR Committee. National burn repository 2007 report: a synopsis of the 2007 call for data. J Burn Care Res. 2008;29(6):862–870. doi: 10.1097/BCR.0b013e31818cb046. discussion 871. [DOI] [PubMed] [Google Scholar]
- 3.Schröder O, Laun RA, Held B, et al. Association of interleukin-10 promoter polymorphism with the incidence of multiple organ dysfunction following major trauma: results of a prospective pilot study. Shock. 2004;21(4):306–310. doi: 10.1097/00024382-200404000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Schroeder O, Schulte KM, Schroeder J, et al. The -1082 interleukin-10 polymorphism is associated with acute respiratory failure after major trauma: a prospective cohort study. Surgery. 2008;143(2):233–242. doi: 10.1016/j.surg.2007.07.040. [DOI] [PubMed] [Google Scholar]
- 5.Huebinger RM, Rivera-Chavez F, Chang LY, et al. IL-10 polymorphism associated with decreased risk for mortality after burn injury. J Surg Res. 2010;164(1):e141–e145. doi: 10.1016/j.jss.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu W, Zeng L, Zhou J, et al. Clinical relevance of 13 cytokine gene polymorphisms in Chinese major trauma patients. Intensive Care Med. 2010;36(7):1261–1265. doi: 10.1007/s00134-010-1797-5. [DOI] [PubMed] [Google Scholar]
- 7.McDaniel DO, Hamilton J, Brock M, et al. Molecular analysis of inflammatory markers in trauma patients at risk of postinjury complications. J Trauma. 2007;63(1):147–157. doi: 10.1097/TA.0b013e31806bf0ab. discussion 157–8. [DOI] [PubMed] [Google Scholar]
- 8.Ami K, Kinoshita M, Yamauchi A, et al. IFN-gamma production from liver mononuclear cells of mice in burn injury as well as in postburn bacterial infection models and the therapeutic effect of IL-18. J Immunol. 2002;169(8):4437–4442. doi: 10.4049/jimmunol.169.8.4437. [DOI] [PubMed] [Google Scholar]
- 9.Göebel A, Kavanagh E, Lyons A, et al. Injury induces deficient interleukin-12 production, but interleukin-12 therapy after injury restores resistance to infection. Ann Surg. 2000;231(2):253–261. doi: 10.1097/00000658-200002000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Suilleabhain C, O'Sullivan ST, Kelly JL, et al. Interleukin-12 treatment restores normal resistance to bacterial challenge after burn injury. Surgery. 1996;120(2):290–296. doi: 10.1016/s0039-6060(96)80300-x. [DOI] [PubMed] [Google Scholar]
- 11.Lyons A, Goebel A, Mannick JA, et al. Protective effects of early interleukin 10 antagonism on injury-induced immune dysfunction. Arch Surg. 1999;134(12):1317–1323. doi: 10.1001/archsurg.134.12.1317. discussion 1324. [DOI] [PubMed] [Google Scholar]
- 12.O'Riordain MG, O'Riordain DS, Molloy RG, et al. Dosage and timing of anti-TNF-alpha antibody treatment determine its effect of resistance to sepsis after injury. J Surg Res. 1996;64(1):95–101. doi: 10.1006/jsre.1996.0312. [DOI] [PubMed] [Google Scholar]
- 13.Gamelli RL, He LK, Liu H. Recombinant human granulocyte colony-stimulating factor treatment improves macrophage suppression of granulocyte and macrophage growth after burn and burn wound infection. J Trauma. 1995;39(6):1141–1146. doi: 10.1097/00005373-199512000-00023. discussion 1146–7. [DOI] [PubMed] [Google Scholar]
- 14.Molloy RG, Holzheimer R, Nestor M, et al. Granulocyte-macrophage colony-stimulating factor modulates immune function and improves survival after experimental thermal injury. Br J Surg. 1995;82(6):770–776. doi: 10.1002/bjs.1800820618. [DOI] [PubMed] [Google Scholar]
- 15.Laffon M, Pittet JF, Modelska K, et al. Interleukin-8 mediates injury from smoke inhalation to both the lung endothelial and the alveolar epithelial barriers in rabbits. Am J Respir Crit Care Med. 1999;160(5 Pt 1):1443–1449. doi: 10.1164/ajrccm.160.5.9901097. [DOI] [PubMed] [Google Scholar]
- 16.Sakurai H, Soejima K, Schmalstieg FC, et al. Inhibition of lung permeability changes after burn and smoke inhalation by an anti-interleukin-8 antibody in sheep. Surg Today. 2009;39(5):399–406. doi: 10.1007/s00595-008-3879-3. [DOI] [PubMed] [Google Scholar]
- 17.Polk HC, Jr, Cheadle WG, Livingston DH, et al. A randomized prospective clinical trial to determine the efficacy of interferon-gamma in severely injured patients. Am J Surg. 1992;163(2):191–196. doi: 10.1016/0002-9610(92)90099-d. [DOI] [PubMed] [Google Scholar]
- 18.Dries DJ. Interferon gamma in trauma-related infections. Intensive Care Med. 1996;22(Suppl 4):S462–S467. doi: 10.1007/BF01743725. [DOI] [PubMed] [Google Scholar]
- 19.Wasserman D, Ioannovich JD, Hinzmann RD, et al. Interferon-gamma in the prevention of severe burn-related infections: a European phase III multicenter trialThe Severe Burns Study Group. Crit Care Med. 1998;26(3):434–439. doi: 10.1097/00003246-199803000-00010. [DOI] [PubMed] [Google Scholar]
- 20.Park KH, Lee KH, Kim H, et al. The anti-inflammatory effects of ulinastatin in trauma patients with hemorrhagic shock. J Korean Med Sci. 2010;25(1):128–134. doi: 10.3346/jkms.2010.25.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenhalgh DG, Saffle JR, Holmes JHt, et al. American Burn Association consensus conference to define sepsis and infection in burns. J Burn Care Res. 2007;28(6):776–790. doi: 10.1097/BCR.0b013e3181599bc9. [DOI] [PubMed] [Google Scholar]
- 22.Wahl WL, Ahrns KS, Brandt MM, et al. Bronchoalveolar lavage in diagnosis of ventilator-associated pneumonia in patients with burns. J Burn Care Rehabil. 2005;26(1):57–61. doi: 10.1097/01.bcr.0000150305.25484.1a. [DOI] [PubMed] [Google Scholar]
- 23.Endorf FW, Gamelli RL. Inhalation injury, pulmonary perturbations, and fluid resuscitation. J Burn Care Res. 2007;28(1):80–83. doi: 10.1097/BCR.0B013E31802C889F. [DOI] [PubMed] [Google Scholar]
- 24.Bird MD, Zahs A, Deburghgraeve C, et al. Decreased pulmonary inflammation following ethanol and burn injury in mice deficient in TLR4 but not TLR2 signaling. Alcohol Clin Exp Res. 2010;34(10):1733–1741. doi: 10.1111/j.1530-0277.2010.01260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baux S. Contribution à l’Etude du traitement local des brûlures thermiques étendues. Paris: Thèse; 1961. [Google Scholar]
- 26.Herndon DN, Curreri PW, Abston S, et al. Treatment of Burns. Curr Probl Surg. 1987;24(6):341–397. doi: 10.1016/0011-3840(87)90010-4. [DOI] [PubMed] [Google Scholar]
- 27.Shirani KZ, Pruitt BA, Jr, Mason AD., Jr The influence of inhalation injury and pneumonia on burn mortality. Ann Surg. 1987;205(1):82–87. doi: 10.1097/00000658-198701000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herndon DN, Barrow RE, Traber DL, et al. Extravascular lung water changes following smoke inhalation and massive burn injury. Surgery. 1987;102(2):341–349. [PubMed] [Google Scholar]
- 29.Abdi S, Herndon D, McGuire J, et al. Time course of alterations in lung lymph and bronchial blood flows after inhalation injury. J Burn Care Rehabil. 1990;11(6):510–515. doi: 10.1097/00004630-199011000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Soejima K, Schmalstieg FC, Sakurai H, et al. Pathophysiological analysis of combined burn and smoke inhalation injuries in sheep. Am J Physiol Lung Cell Mol Physiol. 2001;280(6):L1233–L1241. doi: 10.1152/ajplung.2001.280.6.L1233. [DOI] [PubMed] [Google Scholar]
- 31.Lange M, Hamahata A, Traber DL, et al. A murine model of sepsis following smoke inhalation injury. Biochem Biophys Res Commun. 2010;391(3):1555–1560. doi: 10.1016/j.bbrc.2009.12.124. [DOI] [PubMed] [Google Scholar]
- 32.Mizutani A, Enkhbaatar P, Esechie A, et al. Pulmonary changes in a mouse model of combined burn and smoke inhalation-induced injury. J Appl Physiol. 2008;105(2):678–684. doi: 10.1152/japplphysiol.00232.2007. [DOI] [PubMed] [Google Scholar]
- 33.Westphal M, Cox RA, Traber LD, et al. Combined burn and smoke inhalation injury impairs ovine hypoxic pulmonary vasoconstriction. Crit Care Med. 2006;34(5):1428–1436. doi: 10.1097/01.CCM.0000215828.00289.B9. [DOI] [PubMed] [Google Scholar]
- 34.Dai NT, Chen TM, Cheng TY, et al. The comparison of early fluid therapy in extensive flame burns between inhalation and noninhalation injuries. Burns. 1998;24(7):671–675. doi: 10.1016/s0305-4179(98)00092-8. [DOI] [PubMed] [Google Scholar]
- 35.Navar PD, Saffle JR, Warden GD. Effect of inhalation injury on fluid resuscitation requirements after thermal injury. Am J Surg. 1985;150(6):716–720. doi: 10.1016/0002-9610(85)90415-5. [DOI] [PubMed] [Google Scholar]
- 36.Wright MJ, Murphy JT. Smoke inhalation enhances early alveolar leukocyte responsiveness to endotoxin. J Trauma. 2005;59(1):64–70. doi: 10.1097/01.ta.0000171588.25618.87. [DOI] [PubMed] [Google Scholar]
- 37.Kurzius-Spencer M, Foster K, Littau S, et al. Tracheobronchial markers of lung injury in smoke inhalation victims. J Burn Care Res. 2008;29(2):311–318. doi: 10.1097/BCR.0b013e3181667991. [DOI] [PubMed] [Google Scholar]
- 38.Riyami BMS, Tree R, Kinsella J, et al. Changes in alveolar macrophage, monocyte, and neutrophil cell profiles after smoke inhalation injury. J Clin Pathol. 1990;43(1):43–45. doi: 10.1136/jcp.43.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albright JA, Davis CS, Bird MD, et al. Elevated Pulmonary Inflammatory Response in Patients Sustaining Greater Severity of Smoke Inhalation Injury. Cytokine. 2010;Vol 52(Issues 1–2):54. [Google Scholar]
- 40.Colohan SM. Predicting prognosis in thermal burns with associated inhalational injury: a systematic review of prognostic factors in adult burn victims. J Burn Care Res. 2010;31(4):529–539. doi: 10.1097/BCR.0b013e3181e4d680. [DOI] [PubMed] [Google Scholar]
- 41.Krob MJ, D'Amico FJ, Ross DL. Do trauma scores accurately predict outcomes for patients with burns? J Burn Care Rehabil. 1991;12(6):560–563. doi: 10.1097/00004630-199111000-00011. [DOI] [PubMed] [Google Scholar]
- 42.Osler T, Glance LG, Hosmer DW. Simplified estimates of the probability of death after burn injuries: extending and updating the baux score. J Trauma. 2010;68(3):690–697. doi: 10.1097/TA.0b013e3181c453b3. [DOI] [PubMed] [Google Scholar]
- 43.Hassan Z, Wong JK, Bush J, et al. Assessing the severity of inhalation injuries in adults. Burns. 2010;36(2):212–216. doi: 10.1016/j.burns.2009.06.205. [DOI] [PubMed] [Google Scholar]
- 44.Klein MB, Hayden D, Elson C, et al. The association between fluid administration and outcome following major burn: a multicenter study. Ann Surg. 2007;245(4):622–628. doi: 10.1097/01.sla.0000252572.50684.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodriguez JL, Miller CG, Garner WL, et al. Correlation of the local and systemic cytokine response with clinical outcome following thermal injury. J Trauma. 1993;34(5):684–694. doi: 10.1097/00005373-199305000-00011. discussion 694–5. [DOI] [PubMed] [Google Scholar]
- 46.Sakurai H, Soejima K, Schmalstieg FC, et al. Inhibition of lung permeability changes after burn and smoke inhalation by an anti-interleukin-8 antibody in sheep. Surg Today. 2009;39(5):399–406. doi: 10.1007/s00595-008-3879-3. [DOI] [PubMed] [Google Scholar]
- 47.Huebinger RM, Rivera-Chavez F, Chang LY, et al. IL-10 polymorphism associated with decreased risk for mortality after burn injury. J Surg Res. 2010;164(1):e141–e145. doi: 10.1016/j.jss.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mokart D, Capo C, Blache JL, et al. Early postoperative compensatory anti-inflammatory response syndrome is associated with septic complications after major surgical trauma in patients with cancer. Br J Surg. 2002;89(11):1450–1456. doi: 10.1046/j.1365-2168.2002.02218.x. [DOI] [PubMed] [Google Scholar]
- 49.Dries DJ, Perry JF., Jr Interferon-gamma: titration of inflammation. Crit Care Med. 2002;30(7):1663–1664. doi: 10.1097/00003246-200207000-00050. [DOI] [PubMed] [Google Scholar]
- 50.Nakos G, Malamou-Mitsi VD, Lachana A, et al. Immunoparalysis in patients with severe trauma and the effect of inhaled interferon-gamma. Crit Care Med. 2002;30(7):1488–1494. doi: 10.1097/00003246-200207000-00015. [DOI] [PubMed] [Google Scholar]