

Abstract
Sirtuins, commonly known as NAD+-dependent class III histone deacetylase enzymes, have been extensively studied to evaluate their potential role in different disease states. Based on the published literature, sirtuins have been implicated in providing a myriad of intrinsic and extrinsic biological effects, which in turn may play an important role in the treatment of various disorders such as type II diabetes, obesity, cancer, aging and different neurodegenerative diseases. In particular, a number of studies have unequivocally supported the idea of sirtuins having therapeutic potential in neurodegenerative diseases such as stroke, ischemic brain injury, Alzheimer's disease, Parkinson's disease, Huntington's disease and amyotrophic lateral sclerosis. To exploit the therapeutic potential of sirtuins, their manipulation in terms of development of small-molecule modulators, inhibitors and analogs has increased dramatically since their inception, in both scientific and industrial worlds. Studies on the structure and catalytic core of sirtuins along with chemical mechanisms and substrate specificity have provided important input into the design and synthesis of sirtuin modulators. To study the role of sirtuins in the biological system, it has become extremely important to understand the molecular and chemical structure of sirtuins. In this review, we have discussed the biological role of sirtuins in various neurodegenerative diseases, and also provided an insight into their chemical structure.
Key Words: Sirtuins, Neurodegenerative diseases, Deacetylation, ADP ribosylation
Introduction
Sirtuins (‘sir-two-ins’) are a class of NAD+-dependent class III histone deacetylase enzymes that are phylogenetically conserved from archeobacteria to humans [1]. The yeast silent information regulator factor 2 (Sir2) was the first sirtuin to be discovered [2]. It has been a decade since the yeast Sir2 gene was shown to extend the lifespan of budding yeast by repressing genome instability [3], and since then research on the health-promoting and longevity effects of Sir2-like genes has been a matter of interest.
There are seven members in the mammalian sirtuin family that are homologs of the yeast Sir2 (SIRT1–SIRT7), with SIRT1 being evolutionarily the closest to the yeast Sir2. Sirtuins have been reported to possess two different activities: an ADP ribosyl transferase [4] and/or a deacetylase [5] activity. Sirtuins exert their deacetylase activity on a variety of targets, including histones, transcription factors and apoptotic modulators [6, 7]. Also, the discovery that their catalytic activity depends on NAD+, unlike Zn2+ in cases of other classes of deacetylases, demonstrates their potential role in responding to the energy status and modulating metabolic processes in the cell.
Of all the mammalian sirtuins, SIRT1 has been most extensively reviewed in the literature. SIRT1 removes acetyl groups from lysine side chains of proteins using NAD+ as cofactor, generating deacetylated protein, 2′-O-acetyl-ADP-ribose and nicotinamide as a consequence [8]. It deacetylates histones H1-Lys26, H3-Lys9, Lys14 and H4-Lys16, and reduces the methylation of histone H3-Lys79 [9, 10]. It also deacetylates and attenuates the activities of p53 [11], and has been observed to have the same effects on the forkhead transcription factor (FOXO), FOXO1 and FOXO4 [12, 13]. Under oxidative stress, SIRT1 increases the cell's resistance to stress and reduces the apoptotic ability of FOXO3 [14]. SIRT1 has also been linked to metabolic processes in adipose tissue, liver and muscle by interacting with peroxisome proliferator-activated receptor-gamma co-activator-1α (PGC-1α), a major modulator of the gluconeogenic pathway [15].
The discovery of such diverse effects of SIRT1 has sprouted a plethora of studies aimed towards development of small-molecule modulators of the enzyme. Some of these molecules/inhibitors (splitomycin, sirtinol and EX-527) as well as analogs of sirtinol have been observed to inhibit yeast SIR2, SIRT1 and SIRT2. SIRT1-activating compounds (STACs) include the natural polyphenolic compound resveratrol, SRT1720, SRT2183 and analogs of nicotinamide. Over last 10 years, research in this area has led to the synthesis of over 3,500 STACs. Resveratrol and other natural polyphenols with a similar structure, like fisetin and butein, have been demonstrated to extend lifespan in a wide variety of organisms, from yeast to flies to obese mice [16].
Sirtuins, Caloric Restriction and Aging
Sirtuins have been found to delay aging and promote longevity by regulating the activity of key cellular proteins like p53, FOXO and Ku70 that are involved in either apoptotic processes or cellular repair mechanisms. Sirtuins may thus promote health and longevity partly by either decelerating cell death and/or by boosting repair mechanisms in the cells [17].
It has become increasingly evident that the salutary effects of the dietary regimen, caloric restriction (CR), are in part due to the promotion of sirtuins [17]. CR involves reducing an organism's food consumption compared to normal (ad libitum) consumption. SIRT1 is expressed in the developing and adult brain [18] and the expression levels of SIRT1 increase upon CR in several rodent and human tissues, such as white adipose, liver, skeletal muscle, brain and kidney. It has been shown that the levels of NAD rise in liver cells under CR-like conditions, which in turn induces expression of SIRT1 [19]. SIRT1 ends up consuming NAD+ as a result of its deacetylase activity, generating nicotinamide, an inhibitor of its own activity. NAD+ is known to protect neurons [20] and thus by increasing the levels of NAD+, CR may retain the activity of SIRT1. SIRT1 also activates PGC1α, [19] which results in mitochondriogenesis [20]. A decline in mitochondrial activity is thought to be the causative factor in many age-related diseases [21, 22]. CR brings about similar improvements in the mitochondrial activity as SIRT1. Therefore, a possibility can be envisaged, wherein small-molecule modulators of SIRT1 may act on the same pathways modified by CR, and may have potential to mitigate age-related diseases [8].
It has been established that aging is a known risk factor for many neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), Wallerian neurodegeneration, Huntington's disease and amyotrophic lateral sclerosis (ALS). The pathomechanisms involved in these disorders involve common biochemical pathways and processes, including protein misfolding, oligomerization and aggregation, proteolysis, post-translational modifications, mitochondrial dysfunction, abnormal metabolic processes, and proinflammatory and proapoptotic responses.
Sirtuins in Alzheimer's Disease
Abnormal Aβ deposition in discrete areas of the brain is one of the hallmarks of AD neuropathology. These Aβ peptides are generated by sequential proteolysis of amyloid precursor protein by β- and γ-secretases, which constitutes the ‘amyloidogenic pathway’. The generation of amyloidogenic Aβ peptides can be inhibited by another pathway initiated by the proteolytic cleavage of amyloid precursor protein by α-secretase, also called the ‘non- amyloidogenic pathway’. Recent epidemiological studies suggest that humans who maintain a low-calorie diet have a reduced risk of developing AD [23, 24, 25]. In various animal models, CR has been shown to prevent amyloid neuropathology as seen in AD [26, 27]. A study by Wang et al. [26] demonstrated that CR vitiated the Aβ-type neuropathology and cognitive impairment in the Tg2576 AD mouse model. Based on evidence linking some of the actions of CR and SIRT1 activation and the above-mentioned experimental evidence regarding AD, it is reasonable to suggest that CR might have beneficial effects in mitigating the AD-type amyloid neuropathology, at least in part, by activating SIRT1. In order to explore this possibility, the interplay between CR, sirtuins and the non-amyloidogenic pathway contributing to AD has been extensively investigated. It has been observed that neuronal cultures treated with SIRT1 activators, i.e. NAD+ and resveratrol, significantly reduced Aβ peptide accumulation by binding to amyloid fibrils and also increased cellular α-secretase activity, which in turn is known to activate the protective non-amyloidogenic pathway [17].
Research has suggested resveratrol to be a potent neuroprotective agent due to its chemical properties rather than targeted activation of SIRT1. In microglial cells, SIRT1 has been shown to protect against Aβ-induced neurotoxicity by inhibiting NF-κB signaling [28]. In studies involving cell-based models for AD and ALS, SIRT1 and resveratrol were shown to promote neuronal survival, where as in animal studies, resveratrol has been shown to reduce neurodegeneration and reverse learning impairment in inducible p25 transgenic mice. The possible mechanism of action has been, at least in part, attributed to deacetylation of p53 and PGC-1α, known substrates of SIRT1 [29].
Besides Aβ peptide, another protein called microtubule-associated protein tau (MAPT) has also been implicated to have a major role in the pathogenesis of AD. The hyperphosphorylated form of tau mainly found in neurofibrillary tangles is another hallmark of AD. Although the in vivo studies on SIRT2 in AD are still in the preliminary phase, some interesting data has surfaced on an AD transgenic model (3xTg-AD mice) exposed to nicotinamide, a well-known inhibitor of class III histone deacetylases. It has been observed that nicotinamide improves cognitive performance and reduces phosphorylation of protein tau at threonine 231, thus promoting its degradation [30]. Surprisingly, cross-breeding of 3xTg-AD and SIRT1 null mice produced the same effects of reduced tau phosphorylation, which leads to neuroprotection. This effect goes against the general belief that SIRT1 activation leads to a salutary effect in neurodegeneration. The seemingly opposite effects of SIRT1 on Aβ metabolism and tau phosphorylation gives an idea that SIRT1 downstream events could be a lot more complex than previously understood, and warrants detailed investigation of the molecular events following sirtuin activation.
Sirtuins in Parkinson's Disease
PD is yet another progressive neurodegenerative disorder affecting the central nervous system. The neuropathology mainly involves loss of dopaminergic neurons in the substantia nigra, and its symptoms include muscle rigidity, bradykinesia, resting tremor and postural instability, amongst others. Cytoplasmic inclusions called Lewy bodies containing the protein α-synuclein, some proteasomal and lysosomal subunits have been found on histological analysis. Although the reason for the neuronal cell loss still remains elusive, misfolding, oligomerization and α-synuclein aggregation have been implicated in the neuropathology of PD [31]. In PD models using Drosophila, it has been demonstrated that SIRT2 inhibition prevents α-synuclein toxicity in cultured cells and salvages the degeneration of dopaminergic neurons from the α-synuclein toxicity [32]. More recently, it has been shown that resveratrol, a SIRT1 activator, protects SK-N-BE cells from α-synuclein-induced toxicity associated with AD-related Aβ toxicity. This protective action was reversed upon the addition of sirtinol, a SIRT1 inhibitor, thus suggesting that SIRT1 activation has ameliorative effects on α-synuclein-triggered toxicity [33]. There has been speculation regarding the neuroprotective potential of sirtuins in PD. STACs, resveratrol and quercetin were observed to prevent the loss of dopaminergic neurons induced by 1-methyl-4-phenyl pyridinium (MPP) neurotoxicity in dopaminergic (organotypic midbrain) neuronal cell cultures. However, the protective effects of STACs were unaltered upon addition of known sirtuin inhibitors like sirtinol or nicotinamide, suggesting a non-sirtuin activating pathway for the potential protective effects of resveratrol. The authors suggested that the antioxidant properties of resveratrol explain its protective actions. It remains to be established by which pathway resveratrol induces its more potent neuroprotective effects, i.e. antioxidant and/or sirtuin activation [34].
Sirtuins in Amyotrophic Lateral Sclerosis
ALS is a debilitating progressive disorder of the nervous system that affects motor neurons responsible for voluntary muscle movement gradually leading to muscle weakness and atrophy. Although the causes of ALS are not clearly defined, it has been postulated that it may in part be due to mutations in the gene coding for the enzyme Cu/Zn superoxide dismutase (SOD1) that protects against superoxide-induced cellular toxicity. Thus, oxidative stress seems to play a pivotal role in this disorder too, suggesting a potential intervention of antioxidants or sirtuins to provide protection.
In a study conducted by Kim et al. [29], transgenic mice expressing a mutant form of superoxide dismutase 1 (SOD1G37R) showed severe motor neuron and axon degeneration in the spinal cord. SIRT1 was observed to be upregulated in ALS mouse models as well as in neuronal cell cultures treated with neurotoxins ionomycin and hydrogen peroxide. Intervention with resveratrol protected against neurotoxicity elicited by the SOD1G93A (mutant form of SOD1 linked to ALS), which was proportionate to the decrease in the level of acetylation of PGC-1α, suggesting that resveratrol is acting through the SIRT1 pathway in ALS models.
Sirtuins in Cerebral Ischemia
Ischemic brain injury is caused by a transient or permanent reduction of cerebral blood flow, leading to neuronal cell death. Unlike other disorders, it is not a progressive disorder, but causes neuronal degeneration. Hence, activation of SIRT1 as a neuroprotective strategy could be a promising approach. Resveratrol has been extensively investigated in this regard. It has been shown that resveratrol mimics ischemic preconditioning in the brain using an in vitro model of cerebral ischemia. Ischemic preconditioning has been previously demonstrated to have cardio- and cerebroprotective effects against ischemic insults [35]. A study by Raval et al. [36] not only reported that resveratrol emulated ischemic preconditioning, but also that the protective mechanism was SIRT1 mediated, as evidenced by the abolishment of the protection in presence of sirtinol, a SIRT1 inhibitor. More recently, using an in vivo rat model, it was reported that resveratrol pretreatment confers neuroprotection specifically via the SIRT1 target mitochondrial uncoupling protein 2 (UCP2), thus altering mitochondrial function [36]. Another study targeted the exploration of the JNK, reactive nitrogen and oxygen species pathways that are activated following ischemia/reperfusion injury. It was found that 2,3,5,4′-tetrahydroxystilbene-2-O-β-D-glucoside (TSG), a natural polyphenolic compound obtained from Polygonum multiflorum, exhibits anti-inflammatory and antioxidant effects in an in vitro ischemic model of oxygen-glucose deprivation. TSG was found to inhibit iNOS mRNA expression, which may be mediated by SIRT1 activation and inhibition of NF-κB activation [37]. Thus, SIRT1 seems to be one of the many protective pathways that TSG activates following ischemia/reperfusion injury.
The biological effects manifested by sirtuins would not be completely clear without detailed understanding of the factors that govern the reaction mechanisms, their catalytic elements and their substrate specificity. We discuss these aspects in the following sections.
Sirtuin Chemical Mechanisms
Detailed mechanistic studies on the actions of sirtuins are essential for designing drugs and molecular mapping agents targeting its enzymatic activity. As discussed earlier in the review, sirtuins possess deacetylation and/or ribosyltransferase activity. Deacetylation is carried out by means of a bi-ter kinetic mechanism in which the acetyl-lysine moiety binds prior to NAD+. Nicotinamide is released first, followed by the release of deacetylated lysine and O-acetyl-ADP-ribose (OAADPr) in a random order [38]. Two distinct mechanisms for the ribosyltransferase activity have been reported [39]. In one mechanism, acetyl-lysine and NAD+ react to form the O-alkylamidate intermediate which might react with the amino acid side chains to result in ADP ribosylation instead of deacetylation. In the other mechanism, the acetyl-lysine substrate and NAD+ proceed through deacetylation to form OAADPr, which then reacts nonenzymatically with the substrate to form the ADP-ribosylated product.
The first chemical step involved in the catalytic activity of sirtuins upon the binding of the substrates is the nucleophilic attack of the 1′-carbon of the nicotinamide ribose to form the α-1′-O-alkylamidate intermediate (fig. 1) [38]. The formation of this intermediate has been supported on many fronts. The first line of support shows that the acetyl oxygen is directly transferred to the 1′-hydroxy of OAADPr [40]. Another line of support implicates a nicotinamide exchange [41]. Nicotinamide exchange and deacetylation reaction have one commonality of acetyl-lysine substrate observed to be essential for nicotinamide-ribosyl bond cleavage in both the cases. On the other hand, the formation of O-alkylamidate intermediate has been postulated to take place by SN1 or SN2 mechanisms via formation of an oxocarbenium intermediate, although the intermediate in the case of SN2 mechanism is found to be highly dissociative [38].
Fig. 1.

The proposed chemical mechanism of the deacetylation reaction of sirtuins. The first step involves the nucleophilic attack of the anomeric (1′C) carbon via SN1 or SN2 mechanism to form the O-alkylamidate intermediate. The second step involves activation of the 2′-hydroxyl of the ribose followed by subsequent formation of the 1,2′-cyclic intermediate, which undergoes several transformations that lead to the elimination of the deacetylated lysine and water addition to form 2′-OAADPr.
In the second step, the O-alkylamidate formation is followed by the activation of 2′-hydroxyl of nicotinamide ribose by an active-site histidine for the attack of O-alkylamidate carbon to form 1′,2′-cyclic intermediate (fig. 1). Finally, the deacetylated lysine is eliminated followed by the addition of water to form 2′-OAADPr. Upon release from the active site, OAADPr undergoes non-enzymatic interconversion between 2′-OAADPr and 3′-OAADPr [38]. Although sirtuins (class III HDACs) and class I/II/IV HDACs carry out the same catalytic reaction, their mechanisms are dissimilar. In sirtuins, the acetyl group is the nucleophile, whereas in the other classes it is the electrophile. This difference could be capitalized upon in developing selective deacetylase substrates.
In addition to acetylation, propionylation or butyrylation of lysine residues has also been reported. This includes histone as well as non-histone proteins, i.e. p53, p300 and CREB-binding protein. It has also been shown that Hst2, a Sirt2 homolog, Sirt1, Sirt2 and Sirt3 catalyze depropionylation and debutyrylation [42, 43]. The level and the relevance of propionylation or butyrylation need further investigation. It possibly provides another level of regulation at sites of known acetylation, either by promoting or inhibiting its action.
Architectural Basis for Sirtuin Function
The determination of the three-dimensional structure of sirtuins is imperative for the elucidation of its catalytic mechanism, substrate specificity and inhibitory mechanisms. The X-ray crystallographic structures of sirtuins along with biochemical data have provided an ocean of information regarding its activities. However, there is still a dearth of structural biological data on this class of enzymes.
Sequence alignment studies of the sirtuin proteins show that they contain a conserved catalytic core consisting of approximately 275 amino acid residues [44] and variable N- and C-terminal chains. The catalytic core exists in an elongated shape and consists of a large Rossmann fold domain (which is a hallmark of NAD+/NADH-binding proteins), a more structurally diverse but smaller zinc-binding domain and many loops connecting these domains. An extended cleft formed by these loops between the two domains forms an entry port for the NAD+ and the acetyl-lysine protein substrates, which enter and bind the enzyme from opposite sides. The distinct characteristics of the domains are discussed briefly below using the three-dimensional structure of S. cerevisiae Hst2 (PDB accession number 1Q14) as a model (fig. 2).
Fig. 2.

The three-dimensional protein structure of S. cerevisiae Hst2 depicting the major domains. The figure shows the large domain and the small domain connected by four loops, the largest of which forms the cofactor binding loop. The Zn+2 binding site is also clearly shown.
The Rossmann Fold Domain
This is the large domain of the catalytic core exhibiting the classical α/β Rossmann fold structure. The Rossmann fold is a protein structural motif found in proteins that bind nucleotides, especially the cofactor NAD. The structure is generally composed of three or more parallel β-strands linked by two α-helices in the topological order β-α-β-α-β. Six parallel β-strands form a central β-sheet sandwiched between many α-helices [45]. It has all the characteristics of a NAD-binding site including a Gly-X-Gly sequence for phosphate binding, a pocket for NAD+ and charged residues for the ribose moiety [46].
The Zn-Binding Domain
This small domain that results from two insertions in the Rossmann fold represents the most diverse region among sirtuin family members. Two structural modules have been suggested for the domain: a three-stranded antiparallel β-sheet and a variable α-helical region depending on the type of sirtuins [46]. A Zn2+ ion is held in a tetrahedral conformation by the sulfhydryl groups of two pairs of highly conserved cysteine residues in the β-sheet module [45], except for CobB, which is bound visibly by only two cysteine residues [44]. The Zn2+ is postulated to play a critical structural role of holding the β-strands together. One of the striking characteristics of this domain is a conserved salt bridge between an arginine/lysine and glutamic acid which serves to orient the small domain with respect to the large domain. Overall, the structural diversity observed in the small domain may potentially have a role in substrate specificity, protein-protein interactions, thus finally affecting the enzyme function.
The Cofactor Loop Region
The cleft formed by the four loops connecting large and small domains forms the enzyme active site, wherein both the acetyl-lysine substrate and the NAD+ bind [44]. This region is considered to have the highest sequence homology and the mutation of several residues in this region leads to disruption of deacetylase activity, thus highlighting the importance of this region. The largest of these four loops, referred to as cofactor binding loop, binds to the cofactor NAD+ and is conformationally dynamic as it is mainly dependent on the conformation of the bound NAD+. The loop exists in a relaxed flexible state (closed) when unbound, and transforms into an ordered state (open) when NAD+ binds to it [47]. The conformational switch between open and closed states seems to be contingent on the presence of bound alkylamidate intermediate formed upon the nucleophilic attack of acetyl-lysine on an NAD+ derived intermediate [48] and not upon the nicotinamide cleavage, as the loop is observed to maintain the open conformation even under conditions designed to mimic nicotinamide cleavage [44].
Substrate Binding Sites for Deacetylase Reactions
NAD+ Binding Site
The NAD+ binding pocket is formed by the Rossmann fold domain, which is the bottom of the binding site, and the NAD+ binding loop forms the remainder of the pocket [44]. An unusual characteristic of this family of enzymes is that the NAD+ binds in an inverted direction relative to most NAD dehydrogenases [44]. In this mode, the adenine binds to the C-terminal half of the β-sheet and the nicotinamide group binds to the N-terminal half. The NAD+ binding site can be segregated into three sites, i.e. sites A, B and C. (fig. 3). Site A is shallow and hydrophobic and serves as the adenine-ribose binding site. The hydrophobic residues include a few conserved glycines. The amide nitrogen of the base forms hydrogen bonds with a glutamate or serine residue and the hydroxyl groups of the ribose ring with asparagine residues. The B site is the nicotinamide ribose binding site with the cofactor binding loop forming its ceiling, whose conformation could also affect the nicotinamide ribose binding. Different conformations of the nicotinamide ribose could possibly spell different reaction mechanisms for the dissociation of the nicotinamide moiety from the rest of the molecule [47]. The site C binds the nicotinamide moiety of a productively bound NAD+. In this conformation, the nicotinamide moiety is compatible with both acetyl-lysine binding and catalysis. A highly conserved serine residue is essential for catalytic activity of the enzyme [45]. The serine serves to maintain the enzyme-nicotinamide interactions mediated by the NAD+ via a network of direct and water-mediated hydrogen bonds with invariant residues involved in the nicotinamide group binding [47].
Fig. 3.
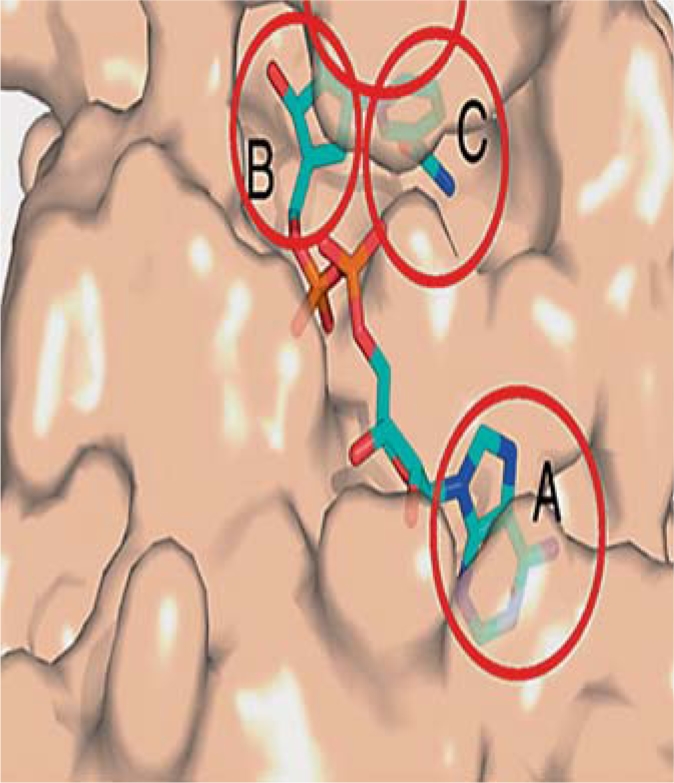
NAD+ binding pocket with Hst2 shown in tan surface representation. A, B and C binding sites are indicated by circles. Site A: adenine-ribose binding site; site B: nicotinamide ribose binding site (the cofactor binding loop forms the ceiling of site B); site C: nicotinamide moiety binding site. Figure reproduced and modified from Sanders et al. [44].
Acetyl-Lysine Binding Site
The peptide substrate binds to the cleft between the small and large domains with the acetyl-lysine side chain sliding into a hydrophobic tunnel within the cleft. The peptide backbone of the acetyl-lysine chain forms β-sheet-like interactions flanked by two strands in the enzyme to form a three-stranded antiparallel motif called a β-staple [49]. Studies have shown that there is a significant shift in the position of the small domain with respect to the large domain, bringing the domains closer to form the β-staple, positioning the conserved residues to form the acetyl-lysine binding tunnel [50]. It has been suggested that NAD+ and peptide may be binding co-operatively, considering that the region around the peptide binding site adopts the more closed conformation in the presence and not in the absence of bound NAD+[49]. The β-staple motif and the residues involved in peptide binding seem to be conserved aspects of acetyl-lysine peptide binding [50].
Substrate Specificity of Sirtuins
The crux of peptide binding to sirtuins is said to be mainly controlled by the insertion of the acetyl lysine into the conserved hydrophobic tunnel and formation of the β-staple as discussed previously in the article. The substrate specificity of sirtuins is more or less thought to lie outside the highly conserved catalytic core of the enzyme. However, it was observed that some sirtuins could distinguish between substrates that have multiple acetylated lysine residues, suggesting that the catalytic core could be sufficient for substrate specificity [51, 52, 53]. However, a consensus regarding the basis of substrate specificity has not been reached yet.
Many studies have been carried out towards determining specificity of substrates [54]. In a relatively recent study by Cosgrove et al. [50], on peptide recognition by sirtuins carried out from a thermodynamic and structural perspective, the authors used the bacterial sirtuin, Sir2Tm from Thermatoga maritima using structures wherein acetylated peptides, unacetylated peptide and propylene glycol, which resembles an apoenzyme, were used as ligands. This study of the crystal structures unearthed a set of complementary side chain interactions that had not been discovered then. A comparative analysis of the structures led to the conclusion that these interactions could be mediated by hydrogen bond and van der Walls interactions with amino acids at the positions −1 and +2 of the peptide substrate, with the position −1 exhibiting a striking effect. It was found that the residue at position 165 was highly variable in an otherwise conserved domain, and could play an important role in the recognition of position −1 of the substrate peptide. The position 165 thus offers specificity as the likelihood of producing favorable interactions with the enzyme depends on the identity of the amino acid at that position [50]. This study gives a general scaffold for identifying sirtuin substrates and distinguishing among several acetyl-lysine targets.
The results obtained from these studies serve as a backdrop for future studies to be undertaken in this direction to discern the substrate parameters of sirtuins. One such study showed that SIRT1 could tolerate bulky acyl groups and that the side chain acetyl group of acetyl-lysine can be extended to substrates even bulkier than a butyryl group [55]. Recently, another study explored the effect of structural variation in the side chain of L-acetyl lysine between the α-carbon and the acetamide on the deacetylation reaction. For this purpose, many L-acetyl lysine analogs with different distances between the α-carbon and the acetamido group on the side chain were tested on SIRT1 to investigate if there is a significant difference in enzyme reactivity [56]. These results suggest that the distance between them is indeed very important for the activity especially with L-N∊-acetyl-lysine that shows the most optimal activity.
Sirtuins in Clinical Practice
Sirtuin-activating compounds have not yet been proven to be clinically useful for the treatment of neurodegenerative diseases. However, preclinical data are available on various other disease models, like diabetes, inflammation and fatty liver disease [57]. The information obtained from such studies, along-with the in vitro results of neurodegenerative disease models, could prove to be valuable for designing sirtuin-activating molecules that may have higher chances of clinical success. The discovery of such molecules is becoming increasingly important, considering the limitations of genetic manipulations and the lack of unequivocal evidence of specific SIRT1 activation by prototype molecules like resveratrol in physiological settings [58].
The task of discovering small molecules that selectively activate SIRT1 has been targeted using several rational design strategies based on the available protein structure and the catalytic pathway of sirtuins [58]. One of the strategies involves designing resveratrol-like molecules, which has not yielded successful results as the in vivo mechanism of SIRT1 activation has not been fully proven. Another approach aims at increasing the cellular levels of NAD+ as a means to activate sirtuin function. This approach has the advantage of harnessing a natural metabolic pathway to enhance sirtuin functions. Also, naturally occurring metabolites present the least risk of toxicity. However, the efficacies of agents that have been used to enhance NAD+ are still questionable. Moreover, NAD+ enhancement affects a host of other physiological pathways, thus making the approach non-specific to SIRT1. A third strategy currently in the proof of principle stage is called nicotinamide derepression. This concept is based on the catalytic mechanism of sirtuins, hence specific to the class of enzymes. This approach is based on countering the inhibitory effect of nicotinamide on sirtuins by designing molecules that are antagonistic to nicotinamide. This approach is still in its infancy and has not provided compounds with desired potency, but is an attractive line of approach to develop further. The drug discovery efforts toward SIRT1-activating molecules have recently been comprehensively reviewed by Blum et al. [57] and are beyond the scope of this review.
Conclusion
Although, sirtuins have been proven to be neuroprotective in numerous studies, it is clear from the diverse pathological mechanisms manifested in neurodegenerative disorders that the role of sirtuins needs to be investigated in detail. The availability of crystal structures and detailed mechanistic analysis are helpful in discovering sirtuin modulators, but would be of limited value if they fail to reach clinical trials, thus underlining the importance of developing robust animal models for investigating molecular mechanisms involved in sirtuin activation. Also, the potential negative effects of SIRT1 activation and energy depletion need to be investigated in animal models further. The path to the clinical success of STACs in neurodegenerative diseases relies overwhelmingly on developing new strategies and designing molecules based on the nuances of sirtuin chemistry and molecular pathways activated by this fascinating class of enzymes. In summary, there is no doubt that sirtuins hold promising therapeutic potential in neurodegenerative disorders like AD, PD, stroke and ischemic brain injury.
Acknowledgements
This work was supported by National Institutes of Health grant (R00AT004197) to Z.A.S. The authors would like to thank Charisse N. Montgomery for her assistance in the manuscript editing.
References
- 1.Shoba B, Lwin ZM, Ling LS, Bay BH, Yip GW, Dinesh S. Function of sirtuins in biological tissues. Anat Rec. 2009;292:536–543. doi: 10.1002/ar.20875. [DOI] [PubMed] [Google Scholar]
- 2.Shore D, Squire M, Nasmyth KA. Characterization of two genes required for the position-effect control of yeast mating-type genes. EMBO J. 1984;3:2817–2823. doi: 10.1002/j.1460-2075.1984.tb02214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaeberlein M, McVey M, Guarente L. The sir2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liszt G, Ford E, Kurtev M, Guarente L. Mouse SIR2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem. 2005;280:21313–21320. doi: 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- 5.Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci USA. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med. 2007;39:335–345. doi: 10.1080/07853890701408194. [DOI] [PubMed] [Google Scholar]
- 8.Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins – novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008;7:841–853. doi: 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- 9.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 10.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 11.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR22(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of foxo1 activity by fhl2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 14.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of foxo transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 15.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 16.Haigis MC, Sinclair DA. Mammalian sirtuins: Biological insights and disease relevance. Annu Rev Pathol Mech Dis. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Fivecoat H, Ho L, Pan Y, Ling E, Pasinetti GM. The role of SIRT1: at the crossroad between promotion of longevity and protection against Alzheimer's disease neuropathology. Biochim Biophys Acta. 2010;1804:1690–1694. doi: 10.1016/j.bbapap.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 18.Hisahara S, Chiba S, Matsumoto H, Horio Y. Transcriptional regulation of neuronal genes and its effect on neural functions: Nad-dependent histone deacetylase SIRT1 (SIR2alpha) J Pharmacol Sci. 2005;98:200–204. doi: 10.1254/jphs.fmj05001x2. [DOI] [PubMed] [Google Scholar]
- 19.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 20.Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by sirt1 may endanger energetically compromised neurons. Neuromolecular Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh KK. Mitochondrial dysfunction is a common phenotype in aging and cancer. Ann NY Acad Sci. 2004;1019:260–264. doi: 10.1196/annals.1297.043. [DOI] [PubMed] [Google Scholar]
- 23.Luchsinger JA, Tang MX, Shea S, Mayeux R. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59:1258–1263. doi: 10.1001/archneur.59.8.1258. [DOI] [PubMed] [Google Scholar]
- 24.Hendrie HC, Ogunniyi A, Hall KS, Baiyewu O, Unverzagt FW, Gureje O, Gao S, Evans RM, Ogunseyinde AO, Adeyinka AO, Musick B, Hui SL. Incidence of dementia and Alzheimer disease in 2 communities: Yoruba residing in Ibadan, Nigeria, and African Americans residing in Indianapolis, Indiana. JAMA. 2001;285:739–747. doi: 10.1001/jama.285.6.739. [DOI] [PubMed] [Google Scholar]
- 25.Mattson MP. Will caloric restriction and folate protect against AD and PD? Neurology. 2003;60:690–695. doi: 10.1212/01.wnl.0000042785.02850.11. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Ho L, Qin W, Rocher AB, Seror I, Humala N, Maniar K, Dolios G, Wang R, Hof PR, Pasinetti GM. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer's disease. FASEB J. 2005;19:659–661. doi: 10.1096/fj.04-3182fje. [DOI] [PubMed] [Google Scholar]
- 27.Patel NV, Gordon MN, Connor KE, Good RA, Engelman RW, Mason J, Morgan DG, Morgan TE, Finch CE. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol Aging. 2005;26:995–1000. doi: 10.1016/j.neurobiolaging.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 28.Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280:40364–40374. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- 29.Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM, LaFerla FM. Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-Phosphotau. J Neurosci. 2008;28:11500–11510. doi: 10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Outeiro TF, Marques O, Kazantsev A. Therapeutic role of sirtuins in neurodegenerative disease. Biochim Biophys Acta. 2008;1782:363–369. doi: 10.1016/j.bbadis.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 33.Albani D, Polito L, Batelli S, De Mauro S, Fracasso C, Martelli G, Colombo L, Manzoni C, Salmona M, Caccia S, Negro A, Forloni G. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1–42) peptide. J Neurochem. 2009;110:1445–1456. doi: 10.1111/j.1471-4159.2009.06228.x. [DOI] [PubMed] [Google Scholar]
- 34.Okawara M, Katsuki H, Kurimoto E, Shibata H, Kume T, Akaike A. Resveratrol protects dopaminergic neurons in midbrain slice culture from multiple insults. Biochem Pharmacol. 2007;73:550–560. doi: 10.1016/j.bcp.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Raval AP, Dave KR, Perez-Pinzon MA. Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2006;26:1141–1147. doi: 10.1038/sj.jcbfm.9600262. [DOI] [PubMed] [Google Scholar]
- 36.Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience. 2009;159:993–1002. doi: 10.1016/j.neuroscience.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang T, Gu J, Wu PF, Wang F, Xiong Z, Yang YJ, Wu WN, Dong LD, Chen JG. Protection by tetrahydroxystilbene glucoside against cerebral ischemia: involvement of JNK, SIRT1, and NF-kappaB pathways and inhibition of intracellular ROS/RNS generation. Free Radic Biol Med. 2009;47:229–240. doi: 10.1016/j.freeradbiomed.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 38.Smith BC, Hallows WC, Denu JM. Mechanisms and molecular probes of sirtuins. Chem Biol. 2008;15:1002–1013. doi: 10.1016/j.chembiol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kowieski TM, Lee S, Denu JM. Acetylation-dependent ADP-ribosylation by Trypanosoma brucei SIR2. J Biol Chem. 2008;283:5317–5326. doi: 10.1074/jbc.M707613200. [DOI] [PubMed] [Google Scholar]
- 40.Smith BC, Denu JM. SIR2 protein deacetylases: evidence for chemical intermediates and functions of a conserved histidine. Biochemistry. 2006;45:272–282. doi: 10.1021/bi052014t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sauve AA, Schramm VL. SIR2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry. 2003;42:9249–9256. doi: 10.1021/bi034959l. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng Z, Tang Y, Chen Y, Kim S, Liu H, Li SS, Gu W, Zhao Y. Molecular characterization of propionyllysines in non-histone proteins. Mol Cell Proteomics. 2009;8:45–52. doi: 10.1074/mcp.M800224-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanders BD, Jackson B, Marmorstein R. Structural basis for sirtuin function: what we know and what we don't. Biochim Biophys Acta. 2010;1804:1604–1616. doi: 10.1016/j.bbapap.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Min J, Landry J, Sternglanz R, Xu RM. Crystal structure of a SIR2 homolog-NAD complex. Cell. 2001;105:269–279. doi: 10.1016/s0092-8674(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 46.Finnin MS, Donigian JR, Pavletich NP. Structure of the histone deacetylase SIR2. Nat Struct Biol. 2001;8:621–625. doi: 10.1038/89668. [DOI] [PubMed] [Google Scholar]
- 47.Zhao K, Harshaw R, Chai X, Marmorstein R. Structural basis for nicotinamide cleavage and ADP-ribose transfer by NAD(+)-dependent SIR2 histone/protein deacetylases. Proc Natl Acad Sci USA. 2004;101:8563–8568. doi: 10.1073/pnas.0401057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hawse WF, Hoff KG, Fatkins DG, Daines A, Zubkova OV, Schramm VL, Zheng W, Wolberger C. Structural insights into intermediate steps in the SIR2 deacetylation reaction. Structure. 2008;16:1368–1377. doi: 10.1016/j.str.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Avalos JL, Celic I, Muhammad S, Cosgrove MS, Boeke JD, Wolberger C. Structure of a SIR2 enzyme bound to an acetylated p53 peptide. Mol Cell. 2002;10:523–535. doi: 10.1016/s1097-2765(02)00628-7. [DOI] [PubMed] [Google Scholar]
- 50.Cosgrove MS, Bever K, Avalos JL, Muhammad S, Zhang X, Wolberger C. The structural basis of sirtuin substrate affinity. Biochemistry. 2006;45:7511–7521. doi: 10.1021/bi0526332. [DOI] [PubMed] [Google Scholar]
- 51.Garske AL, Denu JM. Sirt1 top 40 hits: use of one-bead, one-compound acetyl-peptide libraries and quantum dots to probe deacetylase specificity. Biochemistry. 2006;45:94–101. doi: 10.1021/bi052015l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao K, Chai X, Marmorstein R. Structure and substrate binding properties of cobB, a SIR2 homolog protein deacetylase from Escherichia coli. J Mol Biol. 2004;337:731–741. doi: 10.1016/j.jmb.2004.01.060. [DOI] [PubMed] [Google Scholar]
- 53.Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry. 2004;43:9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- 54.Blander G, Guarente L. The SIR2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 55.Asaba T, Suzuki T, Ueda R, Tsumoto H, Nakagawa H, Miyata N. Inhibition of human sirtuins by in situ generation of an acetylated lysine-ADP-ribose conjugate. J Am Chem Soc. 2009;131:6989–6996. doi: 10.1021/ja807083y. [DOI] [PubMed] [Google Scholar]
- 56.Jamonnak N, Hirsch BM, Pang Y, Zheng W. Substrate specificity of SIRT1-catalyzed lysine Nepsilon-deacetylation reaction probed with the side chain modified Nepsilon-acetyl-lysine analogs. Bioorg Chem. 2010;38:17–25. doi: 10.1016/j.bioorg.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Blum CA, Ellis JL, Loh C, PY NG, Perni RB, Stein RL. SIRT1 modulation as a novel approach to the treatment of diseases of aging. J Med Chem. 2011;54:417–432. doi: 10.1021/jm100861p. [DOI] [PubMed] [Google Scholar]
- 58.Sauve AA. Pharmaceutical strategies for activating sirtuins. Curr Pharm. 2009;15:45–56. doi: 10.2174/138161209787185797. [DOI] [PubMed] [Google Scholar]