

Abstract
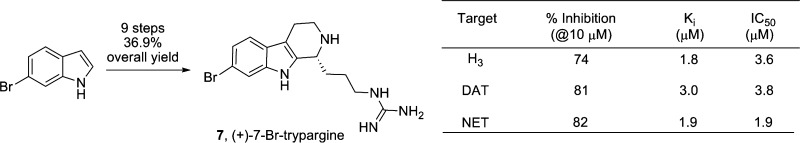
The first total synthesis of (+)-7-bromotrypargine, a β-carboline alkaloid from Ancornia sp. is reported. The synthesis proceeds in nine steps, eight steps longest linear sequence, in 36.9% overall yield. Biological characterization found that (+)-7-bromotrypargine is a H3 antagonist, and a selective inhibitor of the dopamine transporter (DAT) and norepinephrine transporter (NET), without inhibiting the serotonin transporter (SERT). Moreover, unlike electron rich congeners, (+)-7-bromotrypargine is not cytotoxic and thus represents an attractive starting point for chemical optimization; therefore, we piloted a number of chemistries for the synthesis of unnatural analogues.
Drug discovery based on natural products, and marine natural products in particular, has been a highly successful approach toward novel lead series and marketed therapeutics for both peripheral and central nervous system (CNS) indications.1,2 A number of marine natural products, such as aplysamine 1,3 verogamine 2,4 conessine 3,5 and dispyrin 4,6,7 have displayed both affinity for and inhibition of the histamine subtype 3 (H3) receptor (Figure 1).8−10 The H3 receptor is a presynaptic autoreceptor within the Class A GPCR family, but it also functions as a heteroreceptor modulating levels of neurotransmitters such as dopamine, acetylcholine, norepinephrine, serotonin, GABA, and glutamate.8−10 Thus, H3R has garnered a great deal of interest from the pharmaceutical industry for the possible treatment of obesity, epilepsy, sleep/wake, schizophrenia, Alzheimer’s disease, neuropathic pain, and ADHD.8−10 Importantly, conserved elements have been identified within small molecule H3 ligand scaffolds that resulted in a highly predictive pharmacophore model, and many marine natural products conform to this model.8−10
Figure 1.

Marine natural products 1–4 with significant affinity for and inhibition of H3, a target of interest for numerous CNS indications. Inset: refined H3 pharmacophore model, to which synthetic small molecule and natural product H3 antagonists conform.
β-Carboline alkaloids are a prevalent class of biologically active natural products from marine organisms. They exhibit diverse structural features and distinct neuropharmacological profiles.11,12 Our lab has worked extensively in this arena,13,14 and we were recently attracted to a class of β-carboline alkaloids represented by the trypargines 5–7 (Figure 2), as these alkaloids mapped well onto the H3 pharmacophore model and offered a synthetic challenge.15,16 Both trypargine 5(15) and 6-hydroxytrypargine 6(16) are highly toxic alkaloids. (+)-7-Bromotrypargine 7, was only recently isolated by Quinn et al. from the Australian marine sponge Ancornia sp. and found to possess antimalarial activity.17 6-Bromotyramine 8 was isolated along with 7 in similar quantities, and it is believed to be a key biosynthetic precursor.17 Based on the neuropharmacological profiles of β-carboline alkaloids, and the electron-deficient nature of 7, relative to the electron-rich congeners 5 and 6, which might diminish the cytotoxicity, the total synthesis of 7 and biological evaluation seemed warranted.
Figure 2.

Structures of the (+)-trypargines 5–7 and 6-bromotyramine 8.
Our retrosynthesis of 7 (Figure 3) afforded two approaches to close the tricyclic ring: either utilizing the recently reported Brønsted-acid-catalyzed, enantioselective Protio–Pictet–Spengler reaction18,19 or a Bischler–Napieralski reaction,14 followed by an asymmetric Noyori transfer hydrogenation protocol.20 Both routes required the synthesis of 6-bromotyramine 8. As shown in Scheme 1, nitro-olefination of 6-bromo-1H-indole 9, as prescribed by Büchi and Mak,21 proceeded smoothly affording 10 in 98% yield. Exhaustive reduction then provided natural 6-bromotyramine 8 in 76% yield.
Figure 3.
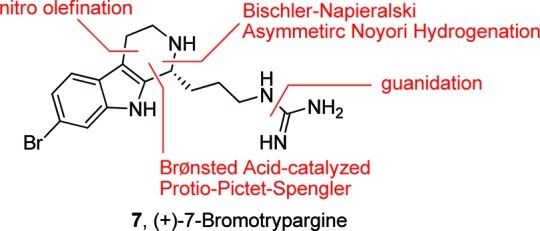
Retrosynthesis of (+)-7-bromotrypargine (7).
Scheme 1. Synthesis of 6-Bromotyramine (8).

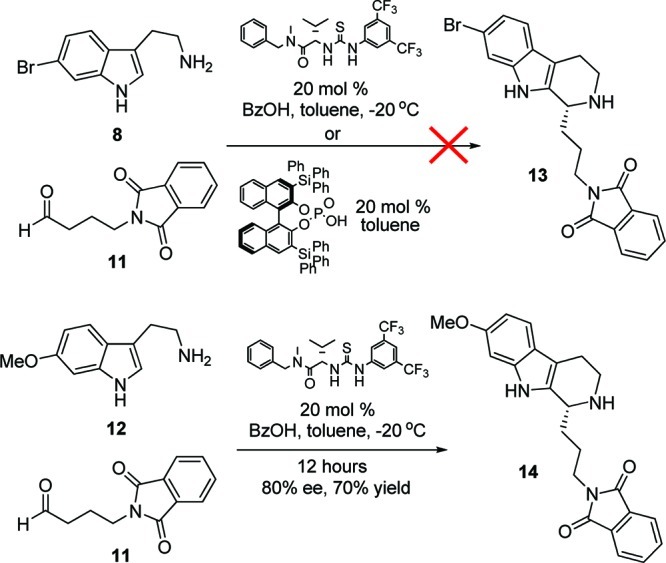
With large quantities of 8 in hand, we evaluated both the Jacobsen thiourea Brønsted-acid-catalyzed, enantioselective Protio–Pictet–Spengler reaction18 as well as the Dixon chiral BINOL-derived phosporic acid variation19 with known aldehyde 11 to produce the key, chiral β-carboline core 13 (Scheme 2). Interestingly, neither protocol afforded 13; however, electron rich congeners of 8, such as 12, proceeded smoothly, providing 14 in good % ee and conversion. Thus, the Brønsted-acid-catalyzed, enantioselective Protio–Pictet–Spengler approach with the more electron deficient 8 was not tractable. Indeed, the original reports by Jacobsen and Klausen, and Dixon et al. utilized only electron rich substrates.18,19 Conversion to 13 could be forced under both protocols (∼80% yield) at high temperatures, but enantioselectivity eroded. Thus, focus shifted toward a Bischler–Napieralski/Noyori asymmetric transfer hydrogenation approach.14,20
Scheme 2. Brønsted-Acid-Catalyzed, Enantioselective Protio–Pictet–Spengler Approach to β-Carboline Core 13.
Key acid 17 was prepared in 98% yield by treating 4-aminobutanoic acid 15 with anhyride 16 under microwave conditions (Scheme 3). Standard amide coupling conditions with 8 and 17 provided amide 18. The Bischler–Napieralski reaction14,20 was facilitated by treatment with POCl3 to imine 19 in 88% yield. Noyori asymmetric transfer hydrogenation with (S,S)-ruthenium catalyst 20 delivered the β-carboline core 13 in 80% yield and 90% ee.20,22 Removal of the phthalimide moiety with hydrazine to afford amine 21, guanidation, and removal of the Boc protecting groups proceeded in 84% yield over the three steps, delivering, for the first time, (+)-7-bromotrypargine 7. Synthetic 7 exhibited physical and spectoscopic data identical to that of natural 7,17,22 confirming the structure and absolute stereochemistry. Overall, the synthesis proceeds in nine steps, eight steps longest linear sequence, in 36.9% overall yield which provided significant material to enable detailed biological evaluation.
Scheme 3. Synthesis of (+)-7-Bromotrypargine (7).

As 5 and 6 are highly toxic alkaloids, we first evaluated 7 in a standard cytotoxicity assay and found 7 to be nontoxic up to 20 μM, suggesting the pharmacological profile might diverge from 5 and 6.23 This surprising result led us to study 7 in our standard HCT116 colon carcinoma cell viability assay.22−24 Here, as well, 7 had no affect on HCT116 cell viability after 48 h. In contrast, advanced intermediate 21, an unnatural analogue of 7, displayed an IC50 of 3 μM in this assay, completely killing the cells after 48 h. These data prompted examination of unnatural analogue 21 in additional colon carcinoma (SW620 and H520) cell lines.25 Interestingly, 21 had dramatic affects inhibiting proliferation in BrdU assays as well as significant activity on cell viability.22 In contrast, 7 was devoid of activity in all of these assays. Collectively, these data informed us of two important points: (1) the pharmacology of the more electron deficient (+)-7-bromotrypargine (7) is distinct from 5 and 6 and warrants further biological evaluation, as it lacks toxicity; and (2) unnatural analogue 21 possesses an intriguing pharmacological profile warranting the synthesis and characterization of additional unnatural analogues of 7.
(+)-7-Bromotrypargine (7) was then evaluated in an external panel26 of 68 GPCRs, ion channels, and transporters in radioligand binding assays in an attempt to identify a discrete molecular target with therapeutic relevance, a strategy that has been highly successful.6,7 In this instance, 7 was found to have a very clean pharmacological profile, affording significant percent inhibition at only three targets: histamine H3 (74%@10 μM), the dopamine transporter, DAT (81%@10 μM), and the norepinephrine transporter, NET (82%@10 μM). Interestingly, these are all targets for important pathologies of the CNS, and 7 did prove to possess activity at the H3 receptor as envisioned. Indeed, 7 is now the fifth marine natural product with significant activity at H3, a target of interest for Alzheimer’s disease, schizophrenia, ADHD, and other indications.3−6 Full dose–response curves were generated for the three targets in both binding (Ki) and functional (IC50) assays,22 proving that 7 both bound to and inhibited the three targets with moderate potencies (Table 1).
Table 1. Pharmacological Profile of (+)-7-Bromotrypargine (7).
| target | % inhibitation (@10 μM) | Ki (μM) | IC50 (μM) |
|---|---|---|---|
| H3 | 74 | 1.8 | 3.6 |
| DAT | 81 | 3.0 | 3.8 |
| NET | 82 | 1.9 | 1.9 |
(+)-7-Bromotrypargine (7) possessed a unique and rare pharmacological profile. There are three monoamine transporters (MATS) that control the extracellular levels of the monoamine neurotransmitters.27 The three major MATS (SERT, DAT, and NET) control the reuptake of serotonin, dopamine, and norepinephrine, respectively. Drugs targeting MATS are blockbusters and target either serotonin reuptake (SSRIs), serotonin and norepinephrine reuptake (SNRIs), or uptake of all three (triple reuptake inhibitors).27 To date, only one drug, methylphenidate (Ritalin), targets only NET and DAT,28 making 7 a novel chemotype, and a most attractive starting point for further optimization for dual NET/DAT activity and/or H3 activity.
In order to optimze the dual NET/DAT activity, we would need to have reliable chemistry to prepare unnatural analogues of 7 (Figure 4) in short order, while also removing functionality, such as the guanidine, that would limit brain penetration. Thus, we piloted a number of “classical” reactions (reductive amination, acylation, sulfonylation, Suzuki coupling, etc.) that are mainstays in the pharmaceutical industry en route to unnatural analogues of 7 (Scheme 4). Prior to removal of the phthalimide moiety, the secondary amine 13 could undergo a reductive amination reaction to provide tertiary amine 22 in 90% yield. Removal of the phthalmide moiety liberated a primary amine 23 that was smoothly acylated 24, sulfonylated 25, or guanidated 26 to afford unnatural analogues of 7 in excellent isolated yields (>85%).
Figure 4.
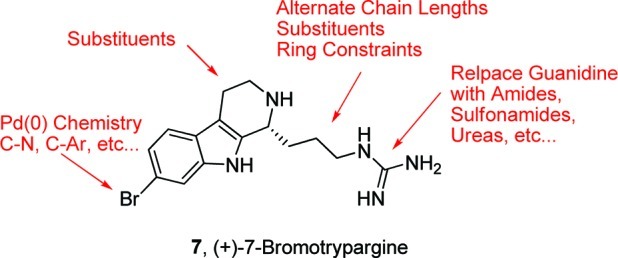
Diversity plan for unnatural analogues of (+)-7-bromotrypargine (7).
Scheme 4. Synthesis of Unnatural Analogues 24–26 of (+)-7-Bromotrypargine (7).

In summary, we have completed the first total synthesis of (+)-7-bromotrypargine (7) in nine steps (eight steps longest linear sequence) in 36.9% overall yield from commercial materials. In the course of this work, we found that the Brønsted-acid-catalyzed, enantioselective Protio–Pictet–Spengler reaction works well for electron-rich substrates but fails for electron-deficient cores, and thus, it was ineffective in accessing 7. Biological evaluation of 7 and an advanced intermediate 21 proved very exciting, with 7 displaying divergent pharmacology from related β-carbolines 5 and 6, while 21 was extremely cytotoxic in mutiple nontransformed and colon cancer cell lines. Importantly, receptor profiling efforts identified 7 as a moderately potent, dual NET/DAT inhibitor, and only the second known compound, and chemotype, to display such a pharmalogical profile devoid of serotonin transporter (SERT) activity. This finding was in addition to the anticipated activity as a H3 antagonist, based on the H3 pharmacophore model. The intriguing pharmacological profile led us to then explore chemistry to access unnatural analogues. These reactions will serve as the groundwork for a larger effort aimed at unnatural analogue synthesis to develop structure–activity relationships (SARs) around 7 and to optimize dual NET/DAT activity. Efforts toward these aims are in progress and will be reported in due course.
Methods
General
The general chemistry, experimental information, and spectral data of all new compounds are supplied in the Supporting Information. Purity of all final compounds was determined by HPLC analysis and is >98%.
Total Synthesis of (+)-Bromotrypargine (7)
6-Bromo-3-(2-nitrovinyl)-1H-indole (10)
1-(Dimethylamino)-2-nitroethylene (1.6 g, 13.72 mmol) was dissolved in 20 mL of trifluoroacetic acid and stirred for 5 min. 6-Bromoindole (9) (3.23 g, 16.47 mmol) was dissolved in 15 mL of dichloromethane (DCM) and added to the stirring solution. The mixture was stirred for 15 min at room temperature and monitored for loss of starting indole by thin layer chromatography. After 15 min, the reaction was transferred to a 1 L separatory funnel, rinsing with DCM and then quenched with 200 mL water. The aqueous phase was extracted three times with DCM, and the combined organic phase was dried over MgSO4, filtered, and concentrated onto 10 g of silica. The product was purified on silica (EtOAc/hexanes 1:2) and yielded 3.61 g (98%) 6-bromo-3-(2-nitrovinyl)-1H-indole as a yellow solid. 1H NMR (600.1 MHz, DMSO) δ (ppm): 8.39 (d, J = 13.5 Hz, 1H) 8.25 (s, 1H), 8.02 (d, J = 13.5 Hz, 1H), 7.95 (d, J = 8.51 Hz, 1H), 7.71 (d, J = 1.75 Hz, 1H), 7.35 (dd, J = 1.85, 8.51 Hz, 1H). 13C NMR (150 MHz, DMSO) δ (ppm): 138.9, 137.1, 134.4, 132.3, 124.9, 124.1, 122.6, 116.3, 115.8, 108.6. HRMS (TOF, ES+) C10H8N2O2Br [M+H]+ calcd, 266.9769; found, 266.9769.
6-Bromotryptamine (8)
A 250 mL Schlenk flask equipped with a stir bar and septa was flame-dried under vacuum. LiAlH4 (2.03 g, 53.69 mmol) was added, and the flask then was sealed and vacuum purged three times, refilling with argon. Tetrahyrdofuran (THF; 89 mL) was added, and the solution was stirred 5 min at room temperature before being cooled to −78 °C for 10 min. 6-Bromo-3-(2-nitrovinyl)-1H-indole 10 (2.39 g, 8.94 mmol) was dissolved in 89 mL of THF and added dropwise via syringe to the stirred solution. The reaction was allowed to warm to room temperature overnight. The reaction was monitored by thin layer chromatography over 24 h. After 24 h, the flask was placed in an ice bath and cooled for 5 min before the careful addition of 2 mL of water, followed by 2 mL of 15% aq. NaOH, followed by 6 mL of water. The flask was stirred for 30 min, and a small amount of MgSO4 was added. The mixture was then filtered through a glass frit, washing the formed solid with dichloromethane. The resulting brown solution was then concentrated to afford a crude brown oil. The product was purified on silica (80:18:2 CHCl3/MeOH/NH4OH) and yielded 1.63 g (76%) 8 as a brown solid. 1H NMR (600.1 MHz, MeOD) δ (ppm): 7.56 (d, J = 1.65 Hz, 1 H), 7.51 (d, J = 8.51 Hz, 1H), 7.21 (s, 1H), 7.18 (d, J = 7.51 Hz, 1H), 3.24 (t, J = 7.63 Hz, 2H), 3.12 (t, J = 7.63 Hz, 2H). 13C NMR (150 MHz, MeOD) δ (ppm): 137.5, 126.2, 122.9, 121.2, 119.2, 114.3, 113.6, 112.4, 41.5, 27.8. HRMS (TOF, ES+) C10H12N2Br [M+H]+ calcd, 239.0184; found, 239.0182.
N-(2-(6-Bromo-1H-indol-3-yl)ethyl)-4-(1,3-dioxoisoindolin-2-yl)butanamide (18)
6-Bromotryptamine (8) (1.0 g, 4.18 mmol), 4-(1,3-dioxoisoindolin-2-yl)butanoic acid (17) (1.17 g, 5.01 mmol), EDC (1.60 g, 8.36 mmol), and HOBT (1.12 g, 8.36 mmol) were dissolved in 30 mL of 9:1 DCM;DIEA solution. The mixture was stirred for 12 h. Analysis by LCMS showed product conversion. The contents of the flask were transferred to a 500 mL separatory funnel, 150 mL of water was added, and the solution was extracted three times with DCM. The combined organic phase was dried over MgSO4 and concentrated under reduced pressure. The product was purified on silica (2% MeOH in DCM) and isolated 18 as a yellow solid 1.61 g (85%). 1H NMR (600.1 MHz, CDCl3) δ (ppm): 8.09 (s, 1H), 7.78 (dq, J = 8.45, 57.8 Hz, 4H), 7.52 (d, J = 1.65 Hz, 1H), 7.48 (d, J = 8.45 Hz, 1H), 7.22 (dd, J = 1.65, 8.45 Hz, 1H), 7.07 (d, J = 2.25 Hz, 1H), 5.93 (s, 1H), 3.69 (t, J = 6.36 Hz, 2H), 3.59 (q, J = 6.79, 6.10 Hz, 2H), 2.97 (t, J = 6.91 Hz, 2H), 2.17 (t, J = 7.13 Hz, 2H), 2.01 (p, J = 6.91 Hz, 2H), 1.58 (s, 1H). 13C NMR (150 MHz, MeOD) δ (ppm): 171.7, 168.5, 137.0, 134.0, 131.9, 126.3, 123.2, 122.7, 122.5, 120.0, 115.7, 114.0, 113.3, 39.6, 37.1, 33.8, 25.1, 24.8. HRMS (TOF, ES+) C22H21N3O3Br [M+H]+ calcd, 454.0766; found, 454.0764.
2-(3-(7-Bromo-4,9-dihydro-3H-pyrido[3,4-b]indol-1-yl)propyl)isoindoline-1,3-dione (19)
N-(2-(6-Bromo-1H-indol-3-yl)ethyl)-4-(1,3-dioxoisoindolin-2-yl)butanamide (18) (1.23 g, 2.71 mmol) was placed in a 250 round-bottom flask. Acetonitrile (25 mL) was added, followed by POCl3 (2.08 g, 1.67 mmol). The flask was then brought to reflux for 1 h. Starting material loss and product conversion were monitored by TLC and LCMS. After 1 h, the flask was removed from the heating bath, and the excess POCl3 was evaporated under reduced pressure. The flask contents were then transferred to a 500 mL separatory funnel, washing with DCM, 100 mL of water was added, and the solution was extracted three times with DCM. The combined organic phased was concentrated and purified on silica (30:1 CHCl3/MeOH) to afford 1.043 g (88%) 19. 1H NMR (600.1 MHz, MeOD) δ (ppm): 7.73 (s, 4H), 7.66 (d, J = 1.49 Hz, 1H), 7.54 (d, J = 8.74 Hz, 1H), 7.27 (dd, J = 1.51 Hz, 8.74, 1H), 3.94 (t, J = 8.97 Hz, 2H), 3.83 (t, J = 6.49 Hz, 2H), 3.15 (t, J = 9.08 Hz, 2H), 3.12 (t, J = 8.93 Hz, 1H), 2.28 (p, J = 7.03 Hz, 2H). 13C NMR (150 MHz, MeOD) δ (ppm): 169.5, 168.2, 141.8, 134.0, 131.6, 125.8, 135.1, 125.0, 122.9, 122.85, 122.81,122.6, 115.5, 42.2, 36.5, 29.9, 25.5, 18.2. HRMS (TOF, ES+) C22H19N3O2Br [M+H]+ calcd, 436.0661; found, 436.0663.
(R)-2-(3-(7-Bromo-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)propyl)isoindoline-1,3-dione (13)
A 100 mL round-bottom flask was charged with 18 (893 mg, 2.046 mmol) and 20 (118.4 mg, 0.204 mmol), and DMF (7.8 mL) was added. The flask was cooled to 0 °C, and a solution of 5:2 formic acid/triethylamine (1.75 mL) was added. The flask was sealed, stirred, and allowed to warm overnight. Reaction progress was monitored by LCMS. After 12 h, the flask contents were transferred to a 250 mL separatory funnel, washing with DCM. Water (50 mL) was added, and the solution was extracted three times with DCM. The combined organic phase was dried over MgSO4 and concentrated. The product was purified on silica (30:1 CHCl3/MeOH) and yielded 717.4 mg (80%) 13 as a golden-brown solid. [α]D23 35.5° (c = 1.0, MeOH). 1H NMR (600.1 MHz, MeOD) δ (ppm): 7.85 (m, 4H), 7.49 (d, J = 1.56 Hz, 1H), 7.38 (d, J = 8.50 Hz, 1H), 7.17 (dd, J = 1.56, 8.50 Hz, 1H), 4.75 (m, 1H), 3.84 (t, J = 6.51 Hz, 2H), 3.73 (m, 1H) 3.44 (m, 1H), 3.04 (m, 2H), 2.30 (m, 1H), 1.96 (m, 3H). 13C NMR (150 MHz, MeOD) δ (ppm): 168.4, 137.5, 134.0, 131.8, 129.3, 124.8, 122.7, 122.3, 119.1, 115.4, 113.7, 106.2, 52.8, 41.3, 36.6, 29.0, 23.9, 17.8. HRMS (TOF, ES+) C22H21N3O2Br [M+H]+ calcd, 438.0817; found, 438.0814.
(R)-3-(7-Bromo-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)propan-1-amine (21)
A 100 mL round-bottom flask was charged with 13 (514 mg, 1.17 mmol). EtOH was added (10 mL), followed by hydrazine hydrate (368 uL, 11.72 mmol). The flask was brought to reflux for 45 min, monitoring by LCMS. After 45 min, the flask was cooled and the contents were concentrated to remove ethanol. The product was purified on silica (80:18:2 CHCl3/MeOH/NH4OH) and yielded 21 317.3 mg (98%) as a brown solid. [α]D23 28.5° (c = 2.0, MeOH). 1H NMR (600.1 MHz, MeOD) δ (ppm): 7.56 (d, J = 1.58 Hz, 1H), 7.39 (d, J = 8.43 Hz, 1H), 7.17 (dd, J = 1.58, 8.43 Hz, 1H), 4.77 (q, J = 4.41 Hz, 1H), 3.75 (m, 1H), 3.49 (m, 1H), 3.07 (m, 4H), 2.34 (m, 1H), 2.11 (m, 1H), 1.97 (m, 2H). 13C NMR (150 MHz, MeOD) δ (ppm): 161.1, 160.9, 137.6, 129.1, 124.9, 122.4, 119.2, 115.4, 113.9, 106.5, 52.5, 41.2, 38.7, 28.6, 22.8, 17.8. HRMS (TOF, ES+) C14H19N3Br [M+H]+ calcd, 308.0862; found, 308.0759.
(+)-7-Bromotrypargine (7)
A 50 mL round-bottom flask was charged with 21 (231 mg, 0.749 mmol) and DCM (3 mL). N,N′-Bis(Boc)-1H-pyrazole-1-carboxamidine (255.8 mg, 0.824 mmol) and triethylamine (156 uL, 1.12 mmol) were then added. The flask was stirred for 3 h and monitored by TLC and LCMS. After 3 h, 5 mL of trifluoroacetic acid was added and the reaction was stirred 4 h. Reaction progress was monitored by LCMS. After 4 h, the reaction was concentrated and the product was purified by reverse phase high performance liquid chromatography using acetonitrile and 0.1% TFA/water (gradient: 10:90 to 90:10) and yielded 225 mg (86% over two steps) 3 as a glassy yellow solid. [α]D23 40.0° (c = 1.0, MeOH). 1H NMR (600.1 MHz, DMSO) δ (ppm): 11.3 (s, 1H), 9.49 (s, 1H), 9.00 (s, 1H), 7.73 (br s, 1H), 7.56 (d, J = 1.68 Hz, 1H), 7.46 (d, J = 8.50 Hz, 1H), 7.20 (br s, 2H), 7.18 (br s, 2H), 7.18 (dd, J = 1.68, 8.50 Hz, 1H), 4.69 (s, 1H), 3.58 (m, 1H), 3.34 (m, 1H), 3.18 (m, 2H), 2.93 (t, J = 5.5 Hz, 2H), 2.11 (m, 1H), 1.90 (m, 1H), 1.69 (m, 1H). 13C NMR (150 MHz, DMSO) δ (ppm): 157.2, 137.5, 131.4, 125.2, 122.4, 120.3, 118.5, 114.9, 114.3, 106.6, 52.2, 40.9, 40.7, 28.9, 24.7, 18.4. HRMS (TOF, ES+) C15H21N5Br [M+H]+ calcd, 350.0980; found, 350.0982.
Cell Culture and HCT116 Viability Assay
HCT116 cells (ATCC CCL-247, passage 8–18, mycoplasma-negative on 07/14/09 by Lonza MycoAlert mycoplasma detection assay) were grown at 37 °C in a humidified incubator with 5% CO2 in DMEM + 10% FBS. Cells were plated in growth medium at 8000 cells per well (100 μL) in 96-well plates and allowed to attach for 24 h. No cells were plated in the first column. Fresh DMEM + 10% FBS + penicillin/streptomycin (100 μL) containing the final concentrations of dimethyl sulfoxide (DMSO) or epi-Lucentamycin A or podophyllotoxin dilutions in DMSO (DMSO concentration maintained at 0.1%) was replaced in each well. After 48 h of cell growth with respective treatments, WST-1 Cell Proliferation Reagent (Roche, 11644807001) was added (10 μL) to each well for 45–60 min at 37 °C. The plates were read (Molecular Devices) at 450 nm for the formazan product and at 690 nm as a reference. Background absorbance (medium only plus WST-1, first column) was averaged and subtracted from all values. Six replicates were used in the calculations per treatment concentration in duplicate experiments. Data was plotted as a percentage of metabolism of WST-1 in the DMSO-treated cells. A colorectal adenocarcinoma cell line, SW620, and a squamous cell lung carcinoma cell line, H520, were obtained from the American Type Culture Collection (Manassas, VA) and maintained in a humidified atmosphere of 5% CO2 in air at 37 °C. The cells were routinely cultured in RPMI 1640 supplemented with 10% fetal bovine serum (Atlanta Biologicals, GA) and 100 μg/mL penicillin-streptomycin.
Proliferation Analysis
SW620 and H520 cells (2.5 × 104/100 μL) were seeded in 96-well microtiter plates prior to treatment. Cells were treated with 10 μM concentration of synthesized compound in triplicate for 48 h in RPMI 1640 supplemented medium and 100 μg/mL penicillin-streptomycin. The CycLex Cellular BrdU ELISA Kit from MBL International (Woburn, MA) was used to measure proliferation. The RPMI medium was removed and replaced with 100 μL of 1× BrdU label mix in RPMI media for 2 h at 37 °C in 5% CO2 in the air. The BrdU label mix was removed, and 200 μL of the fix/denature solution was added and incubated for 30 min at room temperature. The plate was drained, incubated with 50 μL of primary antibody for 1 h at room temperature, rinsed with wash buffer, and incubated with 50 μL of secondary antibody. The wells were rinsed with wash buffer followed by a single rinse with phosphate-buffered saline and drained. Fifty microliters of substrate solution was added and incubated for 6 min followed immediately by 50 μL of stop solution. The change in proliferation was quantified by measuring the absorbance of the dye solution at 450 nm on a microtiter plate reader.
Invasion Analysis
SW620 cells (2.5 × 105/ mL) were seeded in 6-well plates prior to treatment. Cells were treated with 10 μM concentration of synthesized compound for 24 h in RPMI 1640 supplemented medium and 100 μg/mL penicillin-streptomycin. An amount of 40 μL (2.5 mg/mL) of BD Matrigel Basement Membrane Matrix (BD Biosciences, Bedford, MA) was added to top of Fluoroblok invasion chambers (BD Biosciences). Then the cells were trypsinized and 3 × 105/250 μL cells were added to the top of the chamber in serum-free RPMI medium, and 1 mL of RPMI medium with 10% FBS was added to the bottom of the well. Then the plates were incubated for 72 h at 37 °C in 5% CO2 in air. Invading cells were stained with Calcein AM as a fluorescent dye to count invading cells using a fluorometer (Molecular Devices Spectramax M5). All experiments were done in triplicate with a total of three independent replicates.
Radioligand Binding and Fucntional Assays
All biological assays were conducted at Ricerca Pharma according to published protocols.26
Acknowledgments
Vanderbilt is a member of the MLPCN and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development.
Supporting Information Available
Experimental procedures and spectroscopic data for selected compounds and detailed pharmacology methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
C.W.L. conceived and directed the project and wrote the manuscript. J.T.B. performed all chemical synthesis. S.L.S. and B.C.C. performed cytotoxicity assays, and L.J.M. directed the efforts of S.L.S. and B.C.C.
This work was supported, in part, by the Department of Pharmacology and Vanderbilt University. Funding for the NMR instrumentation was provided in part by a grant from NIH (S10 RR019022).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Koehn F. E.; Carter G. T. (2005) The evolving role of natural products in drug discovery. Nat. Rev. Drug Discovery 8, 69–85. [DOI] [PubMed] [Google Scholar]
- Molinski T. F.; Dalisay D. S.; Lieven S. L.; Sauldes J. P. (2009) Drug development from marine natural products. Nat. Rev. Drug Discovery 8, 69–85. [DOI] [PubMed] [Google Scholar]
- Swanson D. M.; Wilson S. J.; Boggs J. D.; Xiao W.; Apodaca R.; Barbier A. J.; Lovenberg T. W.; Carruthers N. I. (2006) Aplysamine-1 and related analogs as histamine H3 receptor antagonists. Bioorg. Med. Chem. Lett. 16, 897–900. [DOI] [PubMed] [Google Scholar]
- Mierzwa R.; King A.; Conover M. A.; Tozzi S.; Puar M. S.; Patel M.; Coval S. J.; Pomponi S. A. (1994) Verongamine, a novel bromotyrosine-derived histamine H3-antagonist from the marine sponge Verongula gigantea. J. Nat. Prod. 57, 175–177. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Sun M.; Bennani Y. L.; Gopalakrishnan S. M.; Witte D. G.; Miller T. R.; Krueger K. M.; Browman K. E.; Thiffault C.; Wetter J.; Marsh K. C.; Hancock A. A.; Esbenshade T. A.; Cowart M. D. (2008) The alkaloid conessine and analogues as potent histamine H3 receptor antagonists. J. Med. Chem. 51, 5423–5430. [DOI] [PubMed] [Google Scholar]
- Kennedy J. P.; Brogan J. T.; Lindsley C. W. (2008) Total synthesis and biological evaluation of the marine bromopyrrole alkaloid dispyrin: elucidation of discrete molecular targets with therapeutic potential. J. Nat. Prod. 71, 1783–1788. [DOI] [PubMed] [Google Scholar]
- Kennedy J. P.; Conn P. J.; Lindsley C. W. (2009) A novel class of H3 antagonists derived from the natural product guided synthesis of unnatural analogs of the marine bromopyrrole alkaloid dispyrin. Bioorg. Med. Chem. Lett. 19, 3204–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leurs R.; Bakker R. A.; Timmerman H.; de Esch I. J. P. (2005) The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nat. Rev. Drug Discovery 4, 107–120. [DOI] [PubMed] [Google Scholar]
- Hudkins R. L.; Raddatz R. (2008) Recent advances in drug discovery of histamine H3 antagonist. Annu. Rep. Med. Chem. 42, 49–63. [Google Scholar]
- LeBois E. P.; Jones C. K.; Lindsley C. W. (2011) The Evolution of Histamine H3 Antagonists/Inverse Agonists’. Curr. Top. Med. Chem. 11, 648–660. [DOI] [PubMed] [Google Scholar]
- Cain M.; Weber R. W.; Guzman F.; Cook J. M.; Barker S. A.; Rice K. C.; Crawley J. N.; Paul S. M.; Skolnick P. (1982) β-Carbolines: synthesis and neurochemical and pharmacological actions on brain benzodiazepine receptors. J. Med. Chem. 25, 1081–1091. [DOI] [PubMed] [Google Scholar]
- Phuong N. M.; Van Sung T.; Porzel A.; Schmidt J.; Merzweiler K.; Adam G. (1999) β-Carboline alkaloids from Hedyotis capitellata. Phytochemistry 52, 1725–1731. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Wisnoski D. D.; Leister W. H.; Wang Y.; Zhao Z. (2003) A ‘one pot’ microwave-mediated synthesis of the basic canthine skeleton: expedient access to unnatural β-carboline alkaloids. Tetrahedron Lett. 44, 4495–4498. [Google Scholar]
- Kennedy J. P.; Breininger M. L.; Lindsley C. W. (2009) Total synthesis of Eudistomins Y1-Y6. Tetrahedron Lett. 50, 7067–7069. [Google Scholar]
- Akizawa T.; Yamazaki K.; Yasuhara T.; Nakajima T.; Roseghini M.; Erspamer G. F.; Erspamer V. (1982) Trypargine, a new tetrahydro-β-carboline of animal origin: isolation and chemical characterization from the skin of the African rhacophorid frog, Kassina senegalensis. Biomed. Res. 3, 232–234. [Google Scholar]
- Cesar L. M.; Mendes M. A.; Tormena C. F.; Marques M. R.; de Souza B. M.; Saidemberg D. M.; Bittencourt J. C.; Palma M. S. (2005) Isolation and chemical characterization of PwTx-II: A novel alkaloid toxin from the venom of the spider Parawixia bistriata (Araneidae, Araneae). Toxicon 46, 786–796. [DOI] [PubMed] [Google Scholar]
- Davis R. A.; Duffy S.; Avery V. M.; Camp D.; Hooper J. N. A.; Quinn R. J. (2010) (+)-7-Bromotrypargine: an antimalarial β-carboline from the Australian marine sponge Ancorina sp. Tetrahedron Lett. 51, 583–585. [Google Scholar]
- Klausen R. S.; Jacobsen E. N. (2009) Weak Brønsted Acid-Thiourea Co-catalysis: Enantioselective, Catalytic Protio-Pictet-Spengler Reactions. Org. Lett. 11, 887–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway C. A.; Muratore M. E.; Storer R. I.; Dixon D. J. (2010) Direct Enantioselective Brønsted Acid Catalyzed N-Acyliminium Cyclization Cascades of Tryptamines and Ketoacids. Org. Lett. 12, 4720–4723. [DOI] [PubMed] [Google Scholar]
- Czarnocki S. J.; Wojtasiewicz K.; Jozwiak A. P.; Maurin J. K.; Czarnocki Z.; Drabowicz J. (2008) Enantioselective synthesis of (+)-trypargine and (+)-crispine E. Tetrahedron 64, 3176–3182. [Google Scholar]
- Büchi G.; Mak C.-P. (1977) Nitro olefination of indoles and some substituted benzenes with 1-dimethylamino-2-nitroethylene. J. Org. Chem. 42, 1784–1786. [Google Scholar]
- See the Supporting Information for complete details.
- Daniels R. N.; Melancon B. J.; Wang E. A.; Crews B. S.; Marnett L. M.; Sulikowski G. A.; Lindsley C. W. (2009) Progress towards the total synthesis of Lucentamycin A: Total synthesis and biological evaluation of 8-epi-Lucentamycin A. J. Org. Chem. 74, 8852–8855. [DOI] [PubMed] [Google Scholar]
- Aldrich L. N.; Stoops S. L.; Crews B. C.; Marnett L. J.; Lindsley C. W. (2010) Total synthesis and biological evaluation of tabjamine K and a library of unnatural analogs. Bioorg. Med. Chem. Lett. 20, 5207–5211. [DOI] [PubMed] [Google Scholar]
- Stoops S. L.; Pearson S. A.; Weaver C.; Waterson A. G.; Days E.; Farmer C.; Brady S.; Weaver C. D.; Beauchamp R. D.; Lindsley C. W. (2011) Identification and optimization of small molecules that restore E-cadherin expression and reduce invasion in colorectal cells’. ACS Chem. Biol. 6, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For information on the Lead Profile Screen, see: www.ricerca.com.
- Howell L. L.; Kimmel H. L. (2008) Monoamine transporters and psychostimulant addiction. Biochem. Pharmacol. 75, 196–217. [DOI] [PubMed] [Google Scholar]
- Fone K.; Nutt D. J. (2005) Stimulants: use and abuse in the treatment of attention deficit hyperactivity disorder. Curr. Opin. Pharmacol. 5, 87–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.