

Abstract
The synthesis and biological evaluation of the first members of a new series of designed bryostatin A-ring analogues (bryologs) are described. An advanced intermediate is produced that allows for step economical access to diverse analogs. The first of these analogues, bearing side chains of completely different polarities from alkyl to hydroxyl and carboxyl functionalities, were evaluated. All exhibit potent protein kinase C binding (54.7 to 2.4 nM) with affinities increasing with decreasing side chain polarity. This series of bryostatin analogues demonstrates that A ring surrogates can indeed be used for tuning pharmacophore and ADME characteristics as needed to improve bryolog function.
Keywords: Bryostatin, PKC, bioactivity, macrocycle
The bryostatins are a structurally and biologically unique family of macrocyclic lactones1 first collected from bryozoa2 in the Gulf of Mexico in 1968 by Pettit and coworkers.3 In 1982, the structure of bryostatin 1 (Figure 1), the lead member of this family, was established by X-ray crystallography.4 Subsequent studies revealed that bryostatin exhibits promising activity against several cancer cell lines. Significantly, bryostatin promotes apoptosis,5 reverses multidrug resistance,6 and synergizes with other anticancer agents.7 In contrast to many anticancer drugs, bryostatin stimulates the immune system, thus providing the basis for a promising immunotherapeutic approach to cancer.8 Bryostatin has also been found to facilitate learning and extend memory in animals9 and has recently been entered into a clinical trial for treating Alzheimer’s disease.10 Remarkably, bryostatin also induces activation of latent HIV reservoirs, thus serving as a lead in efforts to eradicate HIV/AIDS.11
Figure 1.
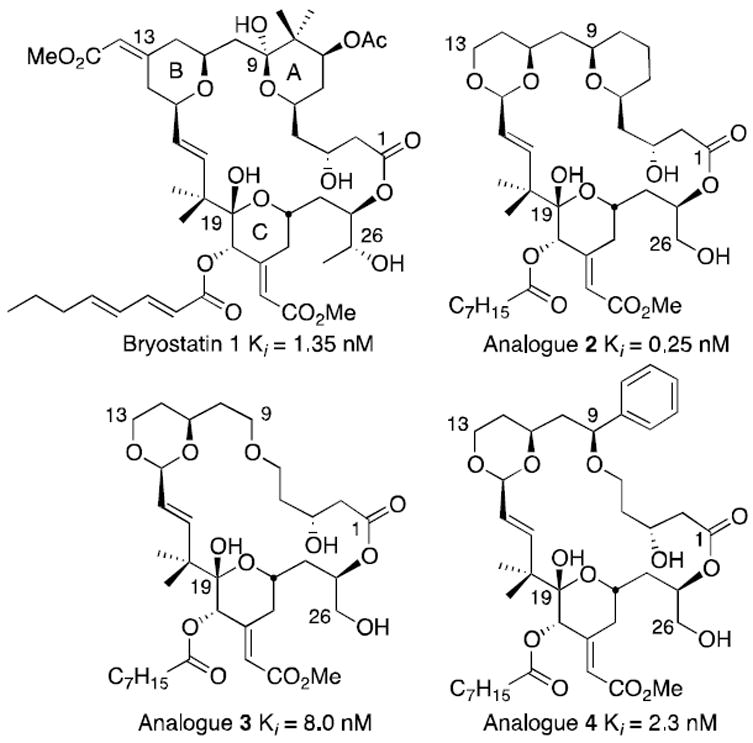
Bryostatin 1 and lead analogues (Ki = binding affinity to rat brain PKC).
Bryostatin’s activities arise from binding to the C1 domain of protein kinase C (PKC)12 and possibly other C1 domain targets.13 Unlike many kinase-targeting molecules that bind to the ATP binding site and thus serve only to inhibit function, ligands targeting the C1 domain can turn on or turn off function. Bryostatin binds with high affinity (<10 nM) to conventional and novel PKC isozymes.14 Since different isoforms are associated with various therapeutic indications,15 bryostatin is a promising lead for drug discovery.
Despite this unique portfolio of biological activities, the study and use of bryostatin has been impeded by its limited supply and challenges associated with systematic modification of its complex structure. Low isolation yields (18 grams are obtained from 14 tons of bryozoa16) and environmental concerns about larger scale marine harvesting make it impractical to obtain significant amounts from natural sources. Early research by the groups of Masamune, Evans and Yamamura led to impressive total syntheses of bryostatins 7, 2, and 3, respectively.17 These syntheses have, however, not been further advanced and are currently too long (>70 steps) to impact supply. Recent advances include a formal total synthesis of bryostatin 7 by the Hale Group18 and a total synthesis of bryostatin 16 by Trost and Dong19. The Keck Group20 has also reported a notable advance with an approximately 57-step synthesis of bryostatin 1 and recently the Krische Group21 described a 36-step synthesis of bryostatin 7. Adam Schrier (Wender Group) recently reported a scalable 42-step synthesis of bryostatin 9.22 Additional groups have contributed substantially to this field, including those of Thomas, Vandewalle, Roy, Burke, Hoffmann, and Yadav.23
While efforts to synthesize natural bryostatins continue to impressively advance, in the 1980s we24 took an alternative approach to the goal of achieving more time- and step-economical access to molecules with bryostatin activity that is generally applicable to many therapeutically promising complex molecules that are beyond the immediate reach of supply-impacting synthesis. Realizing that the demand for bryostatin, not unlike that of many natural products, is driven primarily by its function (i.e. activity) and not its structure and that function is neither evolved nor optimized for human therapy, we sought to use synthesis-informed design to create simpler structures that would be more readily accessible and yet exhibit similar if not superior activity to the natural lead. This function oriented synthesis (FOS) approach25 has yielded to date over 100 designed bryostatin analogues (bryologs), over 35 of which have comparable or better affinities than bryostatin in binding PKC and are synthetically accessible on scale in under 30 total steps.26 This FOS approach opens broad opportunities to explore the varied activities of tunable bryostatin-like compounds and is expected to be a preferred strategy for those interested in its function. Moreover, given supply and some off target clinical issues with bryostatin, these analogs and related approaches could offer more effective agents with fewer side effects. The potential of our approach has indeed been recognized in recent clinical studies.27
In computational studies of competitive PKC ligands, we proposed that the affinity of bryostatin could arise in part from the array of hydrogen bond donors and acceptors at C1, C19, and C26.24 Deletion of groups not in contact with PKC or that do not influence conformational populations led to bryolog 2 (Figure 1) with a PKC affinity better than bryostatin and accessible in <30 steps.28 Further simplification caused some loss in affinity (3) that was largely restored in 4 by introduction of a conformation influencing phenyl group at C9.29 Encouraged by the significant activity of 4, the current study sought to create a diversifiable, advanced intermediate 5 (Scheme 1) that would allow systematic investigation of how variations in this part of the molecule would affect PKC affinity with the eventual goal of using this information to design selective PKC regulators.30
Scheme 1.
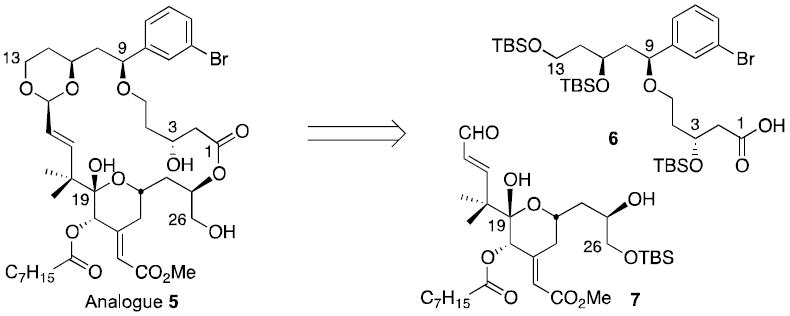
Retrosynthetic analysis.
Our A-ring analogues have been shown to translocate PKCδ-GFP from the cytosol to cellular membranes in RBL cells, an assay that correlates with biological function.31 Analogue 4 in particular shows a translocation profile similar to bryostatin.32 These significant findings warranted further study of how this A-ring region could be used to modulate activity and selectivity. It has been shown that non-polar side chains are tolerated in this region,33 but little is known about polar side chains. Polarity is however important, since bryostatins 1 (OAc) and 2 (OH) differ in PKC selectivity. Analogue 5 (Scheme 1) was chosen as a diversification node for accessing derivatives with varying polarity in the A-ring region. Based on previous modeling studies of 4 the meta-substitution of the phenyl-ring was expected to favor contact with PKC in the vicinity of bryostatin’s C7-acetate.
Analogue 5 is synthesized in a highly convergent fashion by coupling a top piece with a known bottom piece 728 in two steps using a macrotransacetalization procedure (Scheme 1). The synthesis of the C1-C13 spacer domain 6 (Scheme 2) began with asymmetric allylation34 of 3-bromobenzaldehyde to give an alcohol (92% enantiopurity, 76% yield) that on subsequent cyanoethylation with acrylonitrile provided nitrile 8 in 98% yield. Blaise-type conditions with Zn and catalytic Cp2TiCl2 converted the nitrile group in 8 into a β-ketoester.35,36 Ozonolysis followed by addition of ethyl diazoacetate and SnCl4 gave 9 via a Roskamp rearrangement.37 Asymmetric hydrogenation with (R)-BINOL-RuBr2 adjusted the oxidation state of C3 and C11. HPLC analysis established that 10 was obtained in 98% diastereomeric purity. Selective reduction of C13 was achieved with superhydride. Triol 10 was treated with excess TBSOTf / 2,6-lutidine, which silylated all three hydroxyl groups and cleaved the t-butyl ester functionality,38 affording acid 6 in 92% yield. The synthesis of the new top piece 6 was thus achieved in only 7 steps with an overall yield of 24%.
Scheme 2.
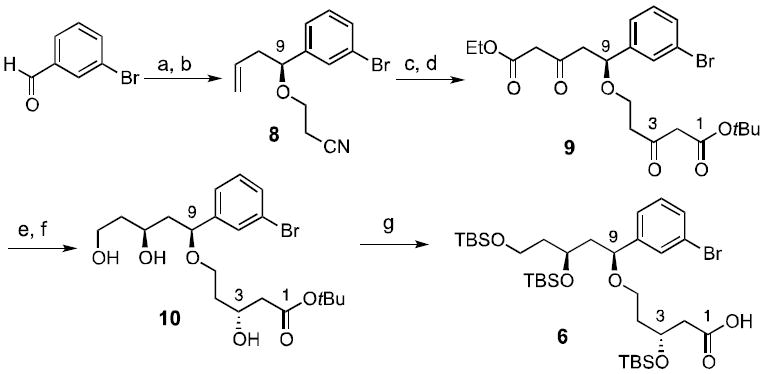
Top piece synthesis. (a) (S)-BINOL/Ti(OiPr)4 (10 mol%), B(OMe)3, Bu3Sn(allyl), 4Å MS, CH2Cl2, 76%; (b) acrylonitrile, TritonB in H2O, CH2Cl2, 98%; (c) tBu-bromoacetate, Zn, cat. Cp2TiCl2, THF, 85%; (d) i. O3, PPh3 ii. ethyl diazoacetate, SnCl4, 63%; (e) ((R)-BINAP)RuBr2, EtOH, H2, 80%; (f) Superhydride, THF, 81%; (g) TBSOTf, 2,6-lutidine, 92%.
The top and bottom pieces (6 and 7) were linked following the Yamaguchi protocol via the mixed anhydride (Scheme 3).39 Subsequent addition of HF/pyridine cleaved the silyl protecting groups and effected formation of the macrocyclic lactone 5 by transacetalization. Analogue 5 was synthesized in 19 steps (longest linear sequence) and 2% overall yield.
Scheme 3.
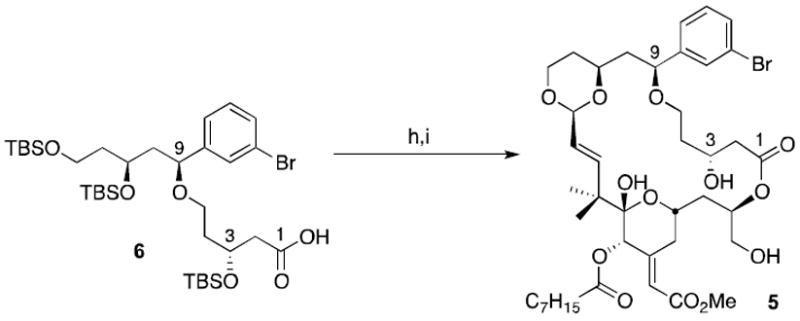
Coupling and ring closure. (h) Et3N, 2,4,6-tribenzoyl chloride, then 7, DMAP, toluene, 91%; (i) 70% HF/pyridine, 83%.
The diversification of bryolog 5 was addressed next. While aryl bromides are attractive groups for diversification, the presence in 5 of several sensitive groups including free alcohols, three esters, two olefins (one a Michael acceptor), two acetals and one benzyl ether puts severe constraints on reaction choice. Diversification reactions such as palladium promoted carbonylation, alkynylation and vinylation as well as radical allylations worked well for model systems but did not transfer to reactions with analogue 5. Alkenylation was finally achieved using a Suzuki coupling protocol employing the S-Phos ligand with Pd(OAc)2, CsF and trans-1-hexen-1-ylboronic acid (Scheme 4). The resulting vinylated analogue 11 is obtained in 70% yield.
Scheme 4.
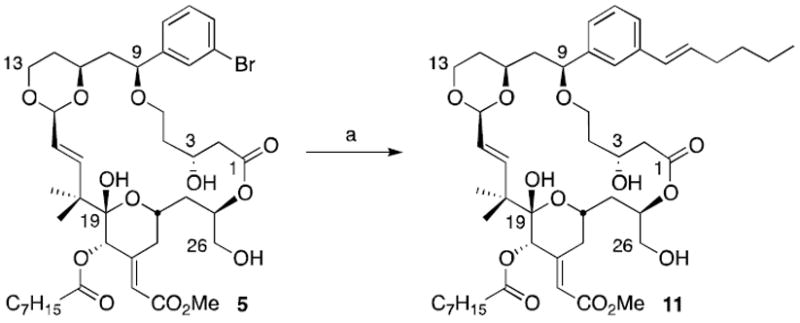
Suzuki coupling of analogue 5.a (a) Pd(OAc)2, S-Phos, CsF, trans-hex-1-en-1-ylboronic acid, dioxane, 60 °C, 70%.
Cleavage of the styrenyl double bond was accomplished chemoselectively with one equivalent of ozone or alternatively a dihydroxylation – diol cleavage strategy. Subsequent reduction of the resultant aldehyde gave the 9-(3-hydroxymethyl)aryl substituted bryolog (13) in 72% yield (Scheme 5). Alternatively, the aldehyde can be oxidized to carboxylic acid 14 upon treatment with mCPBA. Interestingly, the Baeyer-Villiger oxidation only furnishes the 9-(3-carboxy)-aryl substituted analogue (52% yield). The corresponding aryl migration product was not detected by NMR or HPLC.
Scheme 5.
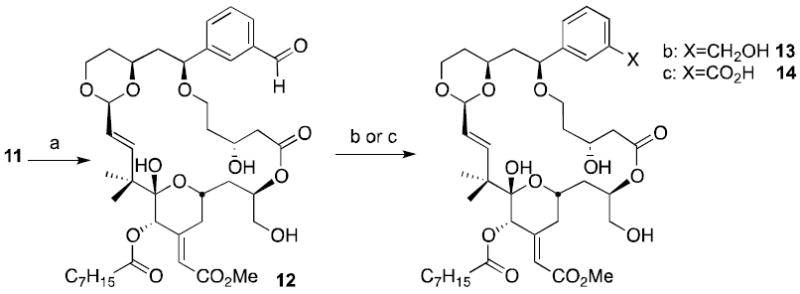
Late stage diversification. (a) OsO4, NaIO4, or O3 (b) NaBH4, MeOH, 72% (over two steps); (c) mCPBA, 52% (over two steps).
A competitive binding assay was performed with the new A-ring analogues 5, 11, 13 and 14 on a mixture of rat brain PKC isozymes (Table 1) used as a preliminary benchmark assay to determine whether further study is warranted. All compounds exhibit potent nanomolar binding affinity to this PKC isozyme mixture. Significantly, decreasing polarity in the side chain results in increased affinity: the parent analogue 4 and its bromo variant 5 have an affinity of 2.3-2.4 nM. The alkenyl substituted analogue 11 shows a similar (4.0 nM) binding affinity. Substitution of these non-polar groups with oxygen-containing functional groups reduces the binding affinity to 15.5 nM in case of the hydroxyl methyl analogue 13. Introduction of an even more polar carboxyl group still furnishes a potent compound but with a further reduction in binding affinity (54.7 nM). Although this number constitutes the lowest binding affinity in the series it is striking that such drastic changes in the polarity still led to highly potent compounds.
Table 1.
Binding affinities for new A-ring bryologs (rat brain PKC).
| Analogue | Ki | |
|---|---|---|
| 5 | Br | 2.4 nM |
| 11 |
|
4.0 nM |
| 13 |
|
15.5 nM |
| 14 |
|
54.7 nM |
These and previous studies show that simplified A-ring analogs made in uniquely step economical sequences exhibit bryostatin like binding and can be tuned as needed to improve function or suppress undesired side effects. This work provides the shortest synthetic sequence to natural or non-natural compounds with bryostatin-like PKC affinities. This FOS approach provides quantities of diversifiable intermediates that are being explored for preclinical candidacy.
Experimental section
Synthesis of (S)-1-(3-bromophenyl)but-3-en-1-ol
To a solution of 0.1 equiv. (S)-BINOL (1.0 mmol, 288 mg) in 4.6 ml CH2Cl2 4 g powdered 4Å MS were added followed by 0.05 equiv. Ti(OiPr)4 (0.5 mmol, 150 μl). The resulting orange mixture was refluxed for 1 h and then cooled in a water bath to rt. A solution of 1.0 equiv. 3-bromo benzcarboxaldehyde (10.0 mmol, 1.17 ml) in 1 ml CH2Cl2 followed by 1.2 equiv. B(OMe)3 (12 mmol, 1.34 ml) was added. 1.18 equiv. Bu3SnCH2CHCH2 (11.8 mmol, 3.7 ml) were added dropwise to the deep red suspension over 5 min. The resulting mixture was stirred at rt for 14 h, then the orange mixture was filtered through Celite into saturated NaHCO3 (50 ml). The Celite was washed with Et2O and the biphasic mixture was stirred for 1 h. Afterwards the aqueous layer was extracted with Et2O (3×30 ml) and the combined organic layers were dried with Na2SO4 and concentrated to an orange oil. The residue was purified by column chromatography (silica gel, Et2O/pentane 1:4) to yield the (S)-1-(3-bromophenyl)but-3-en-1-ol (1.723 g, 7.58 mmol, 76%) as a slightly yellow liquid. Rf = 0.55 (pentane/EtOAc = 10:1) – one blue spot with p-anisaldehyde stain.IR (thin film): 3368, 3077, 3009, 2979, 2907, 1642, 1595, 1570, 1476, 1429, 1344, 1298, 1193, 1092, 1070,997, 919, 882, 783, 687, 666 cm−1.1H-NMR (500MHz, CDCl3): δ 7.52 (mc, 1H, aryl-H); 7.42-7.37 (m, 1H, aryl-H); 7.29-7.18 (m, 2H, aryl-H); 5.78 (mc, 1H, H-2); 5.20-5.14 (m, 2H, H-1); 4.69 (mc, 1H, H-4); 2.54-2.40 (m, 2H, H-3); 2.21-2.16 (m, 1H, OH). 13C-NMR (125MHz, CDCl3): δ 146.1, 133.8, 130.5, 129.9, 128.9, 124.4, 122.5, 119.0, 72.4, 43.8.HRMS: Calculated for C10H11OBr [M]+: 225.9993; Found: 225.9990.[α]D23 = −44.5 in CHCl3, c = 0.93; op = 87%. Synthesis of nitrile 8.
To a rt solution of 1.0 equiv. (S)-1-(3-bromophenyl)but-3-en-1-ol (7.39 mmol, 1.678 g) were added 5.0 equiv. acrylonitrile (36.95 mmol, 2.43 ml) followed by 0.1 equiv. Triton B (40% wt in H2O, 0.74 mmol, 0.29 ml), each via syringe. The reaction was monitored by GC, since the starting material (Rt = 12.15 min) and the product (Rt = 15.87 min) cospot. After 3.5 h the yellow solution was poured into saturated aq. NH4Cl solution (50 ml). The phases were separated and the aqueous layer was extracted with CH2Cl2. The combined organic phases were dried with Na2SO4 and concentrated in vacuo to furnish a yellow oil. The crude was purified by column chromatography (silica gel, pentane/Et2O 1:7 → 1:4) to give nitrile 8 (2.023 g, 7.22 mmol, 98%) as a colorless liquid. Rf = 0.24 (pentane/EtOAc = 6:1) – one purple-grey spot with p-anisaldehyde stain. IR (thin film): 3077, 3011, 2978, 2911, 2878, 2253, 1642, 1595, 1571, 1475, 1429, 1342, 1279, 1222, 1193, 1106, 1071, 997, 921, 787, 699, 666 cm−1. 1H-NMR (500MHz, CDCl3): δ 7.17-7.40 (m, 2H, aryl-H); 7.27-7.22 (m, 2H, aryl-H); 5.76 (mc, 1H, H-2); 5.08-5.03 (m, 1H, H-1); 4.28 (dd, J = 6.0, 7.5 Hz, 1H, H-4); 3.53 (dt, J = 6.5, 9.5 Hz, 1H, H-5); 3.49 (dt, J = 6.5, 9.5 Hz, 1H, H-5’); 2.60-2.53 (m, 3H, H-6/H-3’); 2.39 (dddt, J = 1.4, 6.0, 7.0, 13.0, 1H, H-3). 13C-NMR (125MHz, CDCl3): δ 143.4, 133.6, 131.0, 130.2, 129.5, 125.1, 122.6, 117.70, 117.68, 82.0, 63.4, 42.2, 18.9. HRMS: Calculated for C10H9NOBr [M − CH2CHCH2]+: 237.9868; Found: 237.9875. Elemental Analysis: Calculated for C13H14BrNO: C 55.73; H 5.04; N 5.00. Found: C 55.54; H 5.04; N 5.06. [α]D23 = −34.5 in CHCl3, c = 0.88.
Synthesis of di-β-ketoester 9
In a dry flask 1.0 equiv. nitrile 8 (5.25 mmol, 1.500 g) was dissolved in 53.5 ml dry THF under N2 before 15.0 equiv. Zn (80.25 mmol, 5.240 g), 0.03 equiv. Cp2TiCl2 (0.16 mmol, 40 mg) and t-butyl-bromoacetate (1 drop) were added. The green suspension was heated to 60°C and 10 equiv. t-butyl-bromoacetate (53.5 mmol, 7.22 ml) were added in portions over 1 h. The reaction was stirred for an additional 1.5 h and then allowed to cool to rt. The mixture was filtered into 30 ml Et2O through a pad of celite, which was washed with Et2O (a white precipitate initially formed and then dissolved). To the filtrate 70 ml 1N HCl were added and the biphasic mixture was stirred for 18 h. The layers were separated and the aqueous phase was extracted with Et2O (3×30 ml). The combined organic layers were dried with Na2SO4 and concentrated to give a yellow oil. The crude residue was purified by silica gel column chromatography (pentane/Et2O 10:1), which furnished the β-ketoester (1.800 g, 4.53 mmol, 85%) as a colourless oil. Rf = 0.66 (pentane/EtOAc = 6:1) – one brown spot with p-anisaldehyde stain. IR (thin film): 3078, 2980, 2934, 2904, 2873, 1734, 1715, 1644, 1595, 1571, 1476, 1452, 1394, 1369, 1316, 1255, 1149, 1101, 997, 951.4, 918.28, 840, 786, 699, 666 cm−1. 1H-NMR (500MHz, CDCl3): δ 7.45-7.37 (m, 2H, aryl-H); 7.23-7.17 (m, 2H, aryl-H); 5.71 (mc, 1H, H-2); 5.05-4.98 (m, 2H, H-1); 4.22 (dd, J = 6.0, 7.5 Hz, 1H, H-4); 3.57 (dt, J = 3.1, 6.0 Hz, 2H, H-5); 3.39 (s, 2H, H-8); 2.80 (dt, J = 6.5, 17.0 Hz, 1H, H-6); 2.73 (dt (J = 6.0, 17.0 Hz, 1H, H-6’); 2.49 (ddd, J = 7.0, 7.5, 14.5 Hz, 1H, H-3); 2.34 (ddd, J = 6.0, 6.5, 14.5 Hz, 1H, H-3’); 1.47 (s, 9H, tBu-H). Note: Spectrum shows 9% of the enol tautomer present in solution. 13C-NMR (125MHz, CDCl3): δ 201.6, 166.2, 144.1, 134.0, 130.6, 129.9, 129.5, 125.1, 122.4, 117.3, 81.8, 81.7, 63.7, 51.0, 42.8, 42.2, 27.8 (3C). HRMS: Calculated for C16H20O4Br [M – CH2CHCH2]+: 355.0545; Found: 355.0553. Elemental Analysis: Calculated for C19H25BrO4: C 57.44; H 6.34. Found: C 57.77; H 6.22. [α]D23 = −25.8 in CHCl3, c = 1.38.
1.0 equiv. β-ketoester (1.51 mmol, 600 mg) was dissolved in 15 ml CH2Cl2. Ozone was bubbled through the solution at −78°C (bath temperature) until the blue colour persisted (1min). Then oxygen was bubbled through until the blue colour disappeared (3min). To the cooled solution 1.5 equiv. solid triphenylphosphine (2.29 mmol, 600 mg) were added in one portion and the mixture was allowed to warm to rt. The mixture was stirred for 2 h at rt before another 0.48 equiv. solid triphenylphosphine (0.73 mmol, 191 mg) were added and the reaction was stirred another 1 h to complete reduction of the ozonide. The reaction mixture was recooled to −78 C and 3.0 equiv. ethyldiazoacetate (4.53 mmol, 0.65 ml) were added in one portion via syringe followed by 1.5 equiv. SnCl4 (1M in CH2Cl2, 2.27 mmol, 2.3ml). The mixture was allowed to slowly warm up. At −35°C the formation of bubbles in the solution was observed. At this temperature another 1.5 equiv. SnCl4 (1M in CH2Cl2, 2.27 mmol, 2.3ml) was added in small portions over 1 h until a bubble-free solution was observed. Afterwards the reaction mixture was gradually warmed to rt and poured into a mixture of 100 ml saturated aq. NaHCO3 solution and 50 ml H2O. The layers were separated and the aqueous layer was extracted with CH2Cl2 (5× 20ml). The combined organic layers were dried over Na2SO4, concentrated in vacuo and the remaining crude was chromatographed on silica (pentane:EtOAc 4:1→ 3:1) to give the di-β-ketoester 9 (462 mg, 0.95 mmol, 63%). Rf = 0.57 (35%OAc in pentane) – one red spot with p-anisaldehyde stain. IR (thin film): 3069, 2981, 2935, 1749, 1732, 1715, 1652, 1596, 1573, 1471, 1393, 1369, 1318 1255, 1151, 1032, 999, 951, 840, 788, 697, 665 cm–1. 1H-NMR (500MHz, CDCl3): δ 7.48-7.40 (m, 2H, aryl-H); 7.28-7.20 (m, 2H, aryl-H); 4.74 (dd, J = 7.5 Hz, 2H, Et); 4.18 (q, J = 7.5 Hz, 2H, Et); 3.61 (dt, J = 6.0, 9.5 Hz, 1H, H-6); 3.55 (dt, J = 6.0, 9.5 Hz, 1H, H-6’); 3.45 (s, 2H, H-2 or H-9); 3.06 (dd, J = 9.0, 16.0, 1H, H-4); 2.75 (dt, J = 1.9, 6.0; 2H, H-7); 2.68 (dd, J = 4.0, 16.0, 1H, H-4’); 1.46 (s, 9H, tBu); 1.32 (t, J = 7.5 Hz, 3H, Et). Note: spectrum shows >16% of enol tautomers present in solution. 13C-NMR (125MHz, CDCl3): δ 201.4, 200.1, 166.8, 166.2, 143.1, 131.2, 130.3, 129.4, 125.1, 122.8, 81.9, 77.4, 63.8, 61.4, 50.9, 50.8, 50.0, 42.6, 27.9, 14.1. HRMS: Calculated for C22H29O7NaBr [M + Na]+: 507.0994; Found: 507.0982. Elemental Analysis: Calculated for C22H29BrO7: C 54.44; H 6.02. Found: C 54.00; H 6.16. [α]D23 = −36.9 in CHCl3, c = 1.11.
Synthesis of triol 10
The Ruthenium catalyst was prepared as described by Blanc et al.40 1.0 equiv. (COD)Ru(2-methylallyl)2 (0.013 mmol, 4.21 mg, Acros) and 1.2 equiv. (R)-BINAP (0.016 mmol, 10.21 mg) were measured into a dry flask under N2 and a mixture of 0.9 ml degassed anhydrous acetone and 0.09 ml degassed methanol were added. Then 2.2 equiv. HBr (48% aq. HBr, 0.029 mmol, 3.16μl) were added and the brown solution was stirred for 1 h at rt. The solvent was blown off with N2 gas and the brown residue dried under high-vacuum for 2.5 h. The flask was refilled with N2 and used directly for the hydrogenation. To the prepared catalyst di-β-ketoester 9 (0.206 mmol, 100 mg) was added as a solution in degassed EtOH (1.5 ml, 200 proof, Gold Shield Chemical Company) and the reaction flask was placed in a high-pressure bomb, flushed with N2 and sealed tightly. The apparatus was purged with H2 (3×20bar) and then finally pressurized to 40 bar H2. The apparatus was placed into a 45°C oil bath on a stir plate and the mixture was stirred for 44 h. Afterwards the apparatus was removed from the bath, allowed to cool to rt and slowly depressurized. The solvent was blown off with N2 and the black residue was purified by silica gel chromatography (silica gel, pentane: EtOAc 3:2 → 1:1) to the diol (80.5 mg, 0.164 mmol, 80%) as a pale yellow oil. Rf = 0.23 (pentane:EtOAc = 3:2) – one black-blue spot with p-anisaldehyde stain. IR (thin film): 3445, 2980, 2931, 2873, 1732, 1715, 1595, 1571, 1471, 1428, 1394, 1369, 1300, 1258, 1155, 1098, 952, 845, 787, 700, 666 cm−1. 1H-NMR (500MHz, CDCl3): δ 7.48-7.39 (m, 2H, aryl-H); 7.28-7.20 (m, 2H, aryl-H); 4.49 (dd, J = 5.5, 8.5 Hz, 1H, H-5); 4.15 (dq, J = 1.2, 7.0, 2H, Et); 4.11 (mc, 1h, H-8); 4.08 (mc, 1H, H-3); 3.72 (d, J = 2.3 Hz, 1H, OH); 3.49 (ddd, J = 6.0, 6.5, 9.5, 1H, H-6); 3.44 (dt, J = 6.0, 9.5 Hz, 1H, H-6’); 3.40 (d, J = 3.6 Hz, 1H, OH), 2.51 (dd, J = 8.0, 16.0 Hz, 1H, H-2); 2.42 (dd, J = 4.8, 16.0 Hz, 1H, H-2’); 2.38 (dd, J = 4.5, 16.5, 1H, H-9); 2.33 (dd, J = 8.0, 16.5 Hz, 1H, H-9’); 2.01 (ddd, J = 3.5, 9.0, 14.0, 1H, H-4); 1.82-1.66 (m, 3H, H-4’/H-7); 1.46 (s, 9H, tBu); 1.26 (t, J = 7.0 Hz, 3H, Et). Note: The spectrum shows 5% of a second diastereomer. 13C-NMR (125MHz, CDCl3): δ 172.2, 172.1, 144.0, 131.0, 130.2, 129.6, 125.2, 122.8, 81.3, 81.1, 66.8, 66.0, 65.9, 60.7, 44.3, 42.2, 41.6, 36.1, 28.8 (3C), 14.1. HRMS: Calculated for C22H33O7NaBr [M + Na]+: 511.1307; Found: 511.1301. [α]D23 = −33.2 in CHCl3, c = 0.78.
7.0 equiv. Super Hydride (1.0 M solution of lithium triethylborohydride in THF, 0.147 mmol, 147μl) were added dropwise via 2 min to a solution of 1.0 equiv. of the diol (0.02 mmol, 9.8 mg) at 0°C. The reaction was stirred for 1 h at 0°C, gradually warmed to rt and then stirred additional 1.5 h at rt. Afterwards the reaction was recooled to 0°C followed by addition of H2O2 (30% in H2O, 50μl) and aq. NaOH (1M, 1 drop). The mixture was allowed to stir for 45 min before 2 ml NH4Cl and 1 ml H2O were added at 0°C. After warming the mixture to rt the layers were separated and the aqueous layer was extracted with EtOAc (3×5 ml). The combined organic layers were dried with Na2SO4 and concentrated in vacuo. The crude residue was purified by chromatography on silica (pentane:EtOAc 70:30 → EtOAc 100%) to give triol 10 (7.2 mg, 0.016 mmol, 81%) as a colourless oil. Rf = 0.54 EtOAc 100%) – one bright blue spot with p-anisaldehyde stain. IR (thin film): 3387, 2974, 2922, 2872, 1724, 1595, 1571, 1473, 1427, 1368, 1258, 1155, 1099, 997, 955, 885, 786, 700, 665 cm−1. 1H-NMR (500MHz, CDCl3): 7.46-7.40 (m, 2H, aryl-H); 7.27-7.22 (m, 2H, aryl-H); 4.48 (dd, J = 3.3, 10.0 Hz, 1H, H-5); 4.13 (m, 2H, H-3/H-8); 3.99 (s, 1H, OH); 3.83 (mc, 2H, H-1); 3.53-3.40 (m, 3H, H-6/OH), 2.80 (mc, 1H, OH(H-1)); 2.39 (dd, J = 4.0, 16.5 Hz); 2.34 (dd, J = 8.5, 16.5 Hz, 1H, H-9’); 1.99 (dt, J = 10.0, 15.0 Hz, 1H, H-4); 1.73-1.65 (m, 5H, H-4’/H-2/H-7); 1.46 (s, 9H, tBu). 13C-NMR (125MHz, CDCl3): δ 172.2, 144.1, 131.0, 130.3, 129.4, 124.9, 122.8, 82.6, 81.5, 71.6, 65.9, 65.5, 61.3, 45.3, 42.2, 38.6, 36.0, 28.1. HRMS: Calculated for C20H31O6NaBr [M + Na]+: 469.1202; Found: 469.1201. [α]D23 = −38.5 in CHCl3, c = 0.66.
Synthesis of carboxylic acid 6
30.0 equiv. 2,6-lutidine (1.95 mmol, 230 μl) were added to a solution of 1.0 equiv. triol 10 (0.065 mmol, 29 mg) in 1.5 ml CH2Cl2 under N2 atmosphere. The solution was cooled to 0°C and 10.0 equiv. TBSOTf (0.65 mmol, 149μl) were added dropwise via 30 seconds. The ice bath was removed and the solution was slowly concentrated by blowing with N2 gas until ~0.3M solution was obtained. The concentrated mixture was allowed to stir for 22 h at rt. Afterwards first 1.0 ml H2O and 1.0 ml CH2Cl2 were added to the reaction followed by NaOH (1N in MeOH:THF:H2O 3:1:1, 0.3 ml). The slightly yellow biphasic mixture was stirred for 2 h and solid KHSO4 was added until a pH of 7 of the aqueous phase was reached. The organic phase was extracted with EtOAc (4×10 ml) and the combined organic layers were dried with Na2SO4. After concentration in vacuo the crude residue was purified by chromatography on silica (pentane/EtOAc 80:20) to give the carboxylic acid 6 (43.7 mg, 0.06 mmol, 92%) as a colourless oil. Note: The product is acid sensitive; chromatography on silica with 1% AcOH in the solvent mixture leads to decomposition! Rf = 0.30 (pentane:EtOAc = 80:20) – one black spot with p-anisaldehyde stain. Note: The Rf value is strongly dependent on the concentration. IR (thin film): 2955 (br), 2929, 2856, 1712, 1594, 1571, 1471, 1429, 1361, 1256, 1185, 1097, 940, 836, 776, 699, 664 cm−1. 1H-NMR (500MHz, CDCl3): 7.39-7.35 (m, 2H, aryl-H); 7.21-7.15 (m, 2H, aryl-H); 4.26 (dd, J = 5.0, 8.5 Hz, 1H, H-5); 4.24 (quit, J = 5.5 Hz, 1H, H-8); 3.87 (tt, J = 5.5, 6.5 Hz, 1H, H-3); 3.65 (quint, J = 6.5 Hz, 1H, H-1); 3.64 (quint, J = 6.5 Hz, 1H, H-1’); 3.26 (mc, 2H, H-6); 2.49 (dd, J = 5.0, 15.5 Hz, 1H, H-9); 2.39 (dd, J = 6.0, 15.5 Hz, 1H, H-9’); 1.94 (ddd, J = 5.5, 8.5, 13.5 Hz, 1H, H-4); 1.85-1.63 (m, 5H, H-2/H-4’/H-7); 0.87 (s, 9H, OTBDMS); 0.84 (s, 9H, OTBDMS); 0.83 (s, 9H, OTBDMS); 0.07 (s, 3H, OTBDMS); 0.06 (s, 3H, OTBDMS); 0.02 (s, 3H, OTBDMS); 0.01 (s, 3H, OTBDMS); 0.00 (s, 3H, OTBDMS); −0.02 (s, 3H, OTBDMS). 13C-NMR (125MHz, CDCl3): δ 174.1, 145.1, 130.7, 130.2, 129.7, 125.2, 122.7, 79.0, 66.9, 66.5, 64.9, 59.7, 45.8, 41.5, 39.9, 37.1, 25.9 (×6), 25.7 (3C), 18.2, 18.0, 17.9, −0.01, −4.4, −4.7, −4.9, −5.4 (2C). HRMS: Calculated for C34H65O6BrSi3Na [M + Na]+: 755.3170; Found: 755.3167. [α]D23 = −24.1 (CHCl3, c = 0.82).
Synthesis of analogue 5
To a solution of 1.0 equiv. carboxylic acid 6 (0.022 mmol, 16.0 mg) in 1.5 ml toluene were added 4.0 equiv. Et3N (0.088 mmol, 12.5μl) in one portion at r.t. under N2 followed by addition of a solution of 1.1 equiv. (0.024 mmol, 3.8μl) 2,4,6-trichlorobenzoyl chloride. The reaction mixture was allowed to stir at r.t. for 6 h. 5.0 equiv. DMAP (0.111 mmol, 13.7 mg) and 1.1 equiv. alcohol 7 (0.024 mmol, 14.3 mg) were dissolved in a second flask in 0.3 ml toluene and added dropwise over 5 min to the reaction mixture via syringe. The flask and the syringe were washed three times with 0.3 ml toluene. The cloudy mixture was stirred for 1.5 h at rt. Afterwards the solution was concentrated to ~30% by blowing with N2 gas and the concentrated mixture was directly purified by chromatography on silica (pentane/EtOAc 80:20). The enal (26.1 mg, 0.020 mmol, 91%) was furnished as a colourless oil. Rf = 0.84 (pentane:EtOAc = 80:20) – one black spot with p-anisaldehyde stain. IR (thin film): 3479 (w), 2954, 2920, 2857, 1724, 1692, 1435, 1387, 1361, 1255, 1155, 1104, 1036, 836, 776, 669, 665 cm−1. 1H-NMR (600MHz, CDCl3): 9.55 (d, J = 6.5 Hz 1H, H-11); 7.29 (d, J = 13.5 Hz, 1H, H-13); 7.28-7.32 (m, 2H, aryl-H); 7.19-7.14 (m, 2H, arxl-H); 6.00 (d, J = 1.6 Hz, 1H, H-23); 5.94 (dd, J = 6.5, 13.5 Hz, 1H, H-12); 5.19 (ddt, J = 1.0, 4.7, 9.0 Hz, 1H, H-21), 5.12 (s, 1H, H-16); 4.25 (dd, 4.7, 6.5 Hz, 1H, H-6); 4.22 (quint, J = 5.5 Hz, H-3); 3.85 (quint, J = 4.7 Hz, 1H, H-8); 3.79 (tt, J = 1.5, 9.5 Hz, 1H, H-19); 3.67 (s, 3H, OMe); 3.66-3.60 (m, 5H, H-10/H-18/H-22); 3.25 (dt, J = 5.5, 7.5 Hz, 1H, H-5); 3.21 (dt, J = 7.5, 8.7 Hz, 1H, H-5’); 3.07 (s, 1H, OH(C-15)); 2.35 (dd, J = 4.8, 5.5 Hz, 1H, H-2); 2.11-1.88 (m, 5H, H-7’/H-18/H-20/H-24/H-24’); 1.77-1.63 (m, 7H, H-4/H-7/H-9/H-20’); 1.47 (dt, J = 6.5, 12.5 Hz, 1H, H-25); 1.30-1.16 (m, 9H, H-12 – H-29); 1.20 (s, 3H, CH3); 1.14 (s, 3H, CH3); 0.88 (s, 9H, OTBDMS); 0.87 (s, 9H, OTBDMS); 0.85 (t, J = 6.0 Hz, 3H, H-30); 0.84 (s, 9H, OTBDMS); 0.78 (s, 9H, OTBDMS); 0.048 (s, 3H, OTBDMS); 0.044 (s, 3H, OTBDMS); 0.026 (s, 3H, OTBDMS); 0.014 (s, 3H, OTBDMS); 0.005 (s, 3H, OTBDMS); −0.012 (s, 3H, OTBDMS); −0.017 (s, 3H, OTBDMS); −0.021 (s, 3H, OTBDMS). 13C-NMR (125MHz, CDCl3): δ 194.6, 172.6, 171.7, 166.6, 166.4, 150.4, 145.3, 130.6, 130.2, 129.8, 127.4, 125.1, 122.6, 120.8, 99.5, 78.9, 72.5, 71.5, 66.52, 66.46, 66.2, 65.0, 64.9, 59.6, 51.2, 45.7 (×2), 42.2, 40.0, 37.5, 37.3, 34.5, 31.6, 30.9, 28.88, 28.86, 25.92 (3C), 25.90 (3C), 25.8 (3C), 25.7 (3C), 24.4, 23.0, 22.5, 20.0, 18.3, 18.2, 18.1, 18.0, 14.1, −4.2, −4.4, −4.7, −4.8, −5.30, −5.32; −5.4 (2C). HRMS: Calculated for C65H117O14NaSi4Br [M + Na]+: 1335.6602; Found: 1335.6584. [α]D23 = −27.3 in CHCl3, c = 1.22.
1.0 equiv. enal (0.02 mmol, 26 mg) was dissolved in 5.5 ml anhydrous THF in a polypropylene vial under N2 at 0°C. Then 2.8 equiv. HF·pyridine (0.055 mmol, 1.07 ml) were added via syringe and the reaction was stirred at 0°C for 1 h. Afterwards the bath was removed and the reaction allowed to stir additional 4 h. The reaction mixture was transferred to a separating funnel containing 8 ml H2O using 5 ml EtOAc. Saturated aq. NaHCO3 (~50 ml) was added in portions until the vigorous bubbling stopped. The layers were separated and the aqueous layer was extracted using Et2O (3×15 ml). The combined organic layers were dried with Na2SO4, filtered and concentrated in vacuo. The crude was purified by column chromatography on silica using pentane:EtOAc 40:60 → 20:80 as eluents. Purification yielded the bryostatin analogue 5 (14 mg, 0.017 mmol, 83%) as a white solid. Rf = 0.48 (pentane:EtOAc = 20:80) – one black spot with p-anisaldehyde stain. IR (thin film): 3465, 3333, 2926, 2856, 1731, 1715, 1667, 1595, 1573, 1469, 1434, 1403, 1378, 1362, 1285, 1259, 1230, 1159, 1136, 1105, 980, 940, 918, 879, 851, 807, 791, 791, 736, 705 cm−1. 1H-NMR (600MHz, CDCl3): 7.44-7.40 (m, 2H, aryl-H); 7.24-7.18 (m, 2H, aryl-H); 6.01 (d, J = 16.2 Hz, 1H, H-13); 6.01 (d, J = 1.7 Hz, 1H, H-23); 5.44 (dd, J = 7.4, 16.2 Hz, 1H, H-12); 5.43-5.38 (m, 1H, H-21); 5.16 (s, 1H H-16 or C15-OH); 5.11 (d, J = 7.6 Hz, 1H, H-11); 5.08 (s, 1H, H-16 or C15-OH); 4.35 (d, J = 12.0 Hz, 1H, C3-OH); 4.32 (dd, J = 2.6, 11.4 Hz, 1H, H-6); 4.32-4.26 (m, 1H, H-3); 4.12-4.01 (m, 3H, H-8/H-10/H-19); 3.90 (dt, J = 1.9, 12.0 Hz, 1H, H-10); 3.87 (mc, 1H, H-22); 3.72 (dd, J = 2.2, 13.8 Hz, 1H, H-18); 3.68 (s, 3H, OMe); 3.70-3.63 (m, 1H, H-22’); 3.45 (ddd, J = 1.1, 5.0, 9.6 Hz, 1H, H-5); 3.33 (ddd, J = 2.2, 9.6, 10.2 Hz, 1H, H-5’); 2.56 (dd, J = 10.8, 12.6 Hz, 1H, H-2); 2.52 (dd, J = 3.3, 12.6 Hz, 1H, H-2’); 2.30 (mc, 2H, H-24); 2.24 (ddd, J = 2.6, 5.2, 10.2, 15.6 Hz, 1H, H-4); 2.14-1.96 (m, 3H, H-7/H-20/H-18’); 1.81 (ddd, J = 2.6, 11.4, 13.8 Hz, 1H, H-20’); 1.76 (dq, J = 5.0, 12.6 Hz, 1H, H-9); 1.65-1.53 (m, 3H, H-7’/H-25); 1.51 (d (br), J = 15.6 Hz, 1H, H-4’); 1.39 (d (br), J = 12.6 Hz, 1H, H-9’); 1.32-1.21 (m, 8H, H-26 – 29); 1.20 (s, 3H, -C(CH3)2); 1.06 (s, 3H, -C(CH3)2); 0.87 (t, J = 6.6 Hz, 3H, H-30). 13C-NMR (125MHz, CDCl3): δ 172.3, 172.1, 167.0, 151.6, 143.6, 142.6 131.2, 130.4, 129.5, 125.9, 124.9, 122.8, 119.9, 102.2, 98.9, 83.3, 75.6, 74.1, 71.6, 68.7, 66.2, 66.0, 65.7, 64.5, 51.1, 45.5, 45.1, 42.1, 35.8, 34.6, 33.6, 32.3, 31.6, 31.0, 29.0, 28.9, 24.7, 24.3, 22.6, 19.4, 14.1. HRMS: Calculated for C41H59O13NaBr [M + Na]+: 861.3037; Found: 861.3024. [α]D23 = −72.7 in CDCl3, c = 0.43.
Synthesis of analogue 11
1.0 equiv. aryl bromide 5 (0.012mmol, 10.0mg) were measured in a dry vial, which was evacuated and refilled with N2. Then 7.0 equiv. CsF (0.083mmol, 12.6mg), which was dried in a 200°C vacuum oven, were added and the mixture was placed under high vacuum for 30min. The vial was refilled with N2 and kept under N2 atmosphere throughout the reaction. The mixture was dissolved in 0.1 ml dioxane. Then 1.25 equiv. Pd(OAc)2 (0.015mmol, 3.3 mg), 1.8 equiv. S-Phos (0.021 mmol, 8.8mg) and 6.0 equiv. trans-1-hexen-1-ylboronic acid were added via a stock solution in dioxane (0.1 ml solution). The orange-red reaction mixture was sealed with a 2-ply Teflon tape, followed by a plastic screw cap and finally the vial was sealed with parafilm. Afterwards the vial was vortexed and placed in a 60°C oil bath and stirred for 2h at 60°C. The black reaction mixture was filtered through a plug of silica using ethyl acetate and concentrated under a stream of N2. Purification of the yellow crude by preparative HPLC (MeCN/H2O 65% → 90% at 20ml/min) furnished the coupling product 11 as a white solid in 70% yield. Rf = 0.59 (pentane:EtOAc = 30:70) – one blue spot with p-anisaldehyde stain. IR (thin film): 3460, 3333, 2925, 2854, 1737, 1731, 1667, 1463, 1434, 1404, 1378, 1362, 1287, 1259, 1230, 1159, 1134, 1105, 1063, 1006, 979, 918, 887, 806, 737 cm−1. 1H-NMR (600MHz, CDCl3): 7.26-7.21 (m, 3H, aryl-H); 7.07-7.02 (m, 1H, aryl-H); 6.33 (d, J = 16.0Hz, 1H, H-31); 6.23 (d, J = 6.5Hz, 1H, H-32); 5.99 (d, J = 16.0 Hz, 1H, H-13); 5.99 (d, J = 1.4 Hz, 1H, H-23); 5.44 (dd, J = 7.5, 16.0 Hz, 1H, H-12); 5.41-5.36 (m, 1H, H-21); 5.14 (s, 1H, H-16 or C15-OH); 5.11 (s, 1H, H-16 or C15-OH); 5.09 (d, J = 7.5 Hz, 1H, H-11); 4.42 (d, J = 12.0 Hz, 1H, C3-OH); 4.31 (dd, J = 2.6, 12.0 Hz, 1H, H-6); 4.30-4.22 (m, 1H, H-3); 4.11-3.98 (m, 3H, H-8/H-10/H-19); 3.89 (mc, 1H, H-10); 3.85 (md, 1H, H-22); 3.70 (dd, J = 1.7, 13.5 Hz, 1H, H-18); 3.668 (s, 3H, OMe); 3.68-3.62 (m, 1H, H-22’); 3.44 (ddd, J = 1.4, 4.7, 9.5 Hz, 1H, H-5); 3.29 (ddd, J = 1.5, 9.5, 12.0 Hz, 1H, H-5’); 2.56 (t, J = 12.5 Hz, 1H, H-2); 2.50 (dd, J = 2.4, 12.5 Hz, 1H, H-2’); 2.28 (dt, J = 3.7, 7.2 Hz, 2H, H-24); 2.26-2.21 (m, 2H, H-33); 2.19 (mc, 1H, H-4); 2.12 (mc, 1H, H-7); 2.05-1.95 (m, 2H, H-20/H-18’); 1.79 (dt, J = 2.8, 12.0 Hz, 1H, H-20’); 1.73 (dq, J = 4.7, 12.5 Hz, 1H, H-9); 1.62-1.55 (m, 3H, H-7’/H-25); 1.49-1.41 (m, J = 15.6 Hz, 1H, H-4’); 1.36 (mc, 1H, H-9’); 1.30-1.19 (m, 12H, H-26 – 29/H-34/H-35); 1.18 (s, 3H, -C(CH3)2); 1.03 (s, 3H, -C(CH3)2); 0.91 (t, J = 7.5Hz, H-36); 0.85 (t, J = 6.5 Hz, 3H, H-30). 13C-NMR (125MHz, CDCl3): δ 172.5, 172.1, 167.0, 151.6, 142.5, 141.4, 138.4, 131.9, 129.2, 128.8, 126.0, 125.7, 124.8, 123.5, 119.9, 102.3, 98.9, 84.1, 76.0, 74.1, 71.6, 68.8, 66.3, 65.7 (2C), 64.5, 51.1, 45.5, 45.1, 42.1, 35.8, 34.6, 33.6, 32.7, 32.3, 31.6, 31.4, 31.0, 29.0, 28.9, 24.7, 24.3, 22.6, 22.3, 19.4, 14.1, 14.0. HRMS: Calculated for C47H70O13Na [M + Na]+: 865.4714; Found: 865.4701. [α]D23 = −74.3, CHCl3, c = 0.42.
Synthesis of analogue 13
1.0 equiv. of analogue 11 (0.0037 mmol, 3.1 mg) was dissolved in 300μl THF and 60 μl deionized H2O. Then 0.1 equiv. of OsO4 (0.0004 mmol, 2.5 μl) were added as a 4% stock solution in water via syringe followed by addition of 6.0 equiv. of solid NaIO4 (0.0222 mmol, 4.7 mg). The reaction mixture was stirred for 15 min at r.t. before the product was extracted from the solution using ethylacetate (3×5 ml). The combined organic layers were dried with Na2SO4, filtered and concentrated in vacuo. The crude was dried in vacuum for 1 h and was then dissolved in 1.0 ml anhydrous methanol and 0.2 ml anhydrous CH2Cl2. Afterwards the solution was cooled to 0°C via ice bath before 8.0 equiv. NaBH4 (0.0296 mmol, 1.2 mg) were added as a solid. The reaction mixture was stirred at 0°C for 30 min, then the ice bath was removed and the mixture was allowed to warm to r.t. and stirred for 1 h at r.t. The reaction mixture was transferred to a separating funnel which contained 5.0 ml concentrated NH4Cl solution in water. The layers were separated and the aqueous layer was extracted using CH2Cl2 (4×10 ml). The combined organic layers were dried with Na2SO4, filtered and concentrated in vacuo. The crude was filtered through a pad of silica gel using EtOAc/pentane 80:20 as eluent. All fractions containing product were further purified by reverse phase HPLC (see below for conditions) to give 2.1mg (0.0027 mmol, 72%) 13 as a white amorphous solid. Rf = 0.23 (pentane:EtOAc = 20:80) – one blue spot with p-anisaldehyde stain. IR (thin film): 3459, 3359, 3199, 2922, 2852, 1738, 1731, 1715, 1667, 1660, 1463, 1455, 1434, 1409, 1378, 1361, 1280, 1260, 1231, 1157, 1133, 1103, 1063, 1025, 980, 881, 861, 800, 737, 709 cm−1. 1H-NMR (600MHz, CDCl3): 7.36-7.33 (m, 1H, aryl-H); 7.31-7.26 (m, 2H, aryl-H); 7.21-7.19 (m, 1H, aryl-H); 6.01 (d, J = 15.6 Hz, 1H, H-13); 6.01 (d, J = 1.9 Hz, 1H, H-23); 5.45 (dd, J = 7.2, 15.6 Hz, 1H, H-12); 5.43-5.38 (m, 1H, H-21); 5.16 (s, 1H, H-16 or C15-OH); 5.11 (d, J = 7.2 Hz, 1H, H-11); 5.11 (s, 1H, H-16 or C15-OH); 4.70 (s, 2H, H-31); 4.42 (d, J = 12.0 Hz, 1H, C3-OH); 4.37 (dd, J = 2.8, 12.0 Hz, 1H, H-6); 4.32-4.26 (m, 1H, H-3); 4.12-4.02 (m, 3H, H-8/H-10/H-19); 3.91 (dt, J = 2.3, 12.0 Hz, 1H, H-10’); 3.87 (dd, J = 3.1, 12.0 Hz, 1H, H-22); 3.72 (dd, J = 2.0, 13.8 Hz, 1H, H-18); 3.68 (s, 3H, OMe); 3.68-3.63 (m, 1H, H-22’); 3.44 (ddd, J = 1.2, 4.7, 9.6 Hz, 1H, H-5); 3.32 (ddd, J = 2.5, 11.4, 15.6 Hz, 1H, H-5’); 2.56 (dd, J = 12.0, 12.6 Hz, 1H, H-2); 2.52 (dd, J = 2.7, 12.6 Hz, 1H, H-2’); 2.30 (dt, J = 4.3, 7.2 Hz, 2H, H-24); 2.27-2.20 (m, 1H, H-4); 2.14 (mc, 1H, H-7); 2.05 (mc, 2H, H-20/H-18’); 1.81 (ddd, J = 2.6, 11.4, 14.4 Hz, 1H, H-20’); 1.73 (dq, J = 4.9, 12.6 Hz, 1H, H-9); 1.67-1.53 (m, 3H, H-7’/H-25); 1.48 (d (br), J = 15.6 Hz, 1H, H-4’); 1.39 (d (br), J = 13.8 Hz, 1H, H-9’); 1.32-1.22 (m, 8H, H-26 – 29); 1.20 (s, 3H, -C(CH3)2); 1.06 (s, 3H, -C(CH3)2); 0.87 (t, J = 6.6 Hz, 3H, H-30). 13C-NMR (125MHz, CDCl3): δ 172.4, 172.1, 167.0, 151.6, 142.5, 141.6, 141.3, 129.0, 126.6, 125.9, 125.6, 124.8, 119.9, 102.3, 98.9, 83.9, 75.8, 74.1, 71.6, 68.8, 66.2, 65.8, 65.7, 65.1, 64.6, 51.1, 45.6, 45.1, 42.1, 35.9, 34.6, 33.7, 32.3, 31.6, 31.0, 29.0, 28.9, 24.7, 24.3, 22.6, 19.4, 14.1. HRMS: Calculated for C42H62O14Na [M + Na]+: 813.4037; Found: 813.4028. [α]D23 = −38.9 in CHCl3, c = 0.22.
Synthesis of analogue 12
1.0 equiv. of analogue 11 (0.008 mmol, 6.8 mg) was dissolved in 500μl THF and 100 μl deionized H2O. Then 0.1 equiv. of OsO4 (0.0008 mmol, 5.4 μl) were added as a 4% stock solution in water via syringe followed by addition of 6.0 equiv. of solid NaIO4 (0.048 mmol, 10.3 mg). The reaction mixture was stirred for 15 min at r.t. before the product was extracted from the solution using ethyl acetate (3×5 ml). The combined organic layers were dried with Na2SO4, filtered and concentrated in vacuo. The crude was purified by column chromatography on silica gel using ethyl acetate/pentane 90:10 as eluent. The purification yielded the bryostatin analogue 12 (4.8 mg, 0.006 mmol, 75%) as a white solid. Rf = 0.45 (pentane:EtOAc = 20:80) – one blue spot with p-anisaldehyde stain. IR (thin film): 3460, 3359, 2960, 2922, 2851, 1715, 1737, 1731, 1667, 1633, 1463, 1455, 1434, 1403, 1378, 1362, 1280, 1260, 1230, 1157, 1133, 1102, 1063, 1022, 980, 863, 801, 722, 704, 665 cm−1. 1H-NMR (500MHz, CDCl3): 10.02 (s, 1H, H-31); 7.84.7.76 (m, 2H, aryl-H); 7.60-7.52 (m, 2H, aryl-H); 6.02 (d, J = 16.0 Hz, 1H, H-13); 6.01 (d, J = 1.9 Hz, 1H, H-23); 5.45 (dd, J = 7.5, 16.0 Hz, 1H, H-12); 5.44-5.38 (m, 1H, H-21); 5.16 (s, 1H, H-16 or C15-OH); 5.12 (d, J = 7.0 Hz, 1H, H-11); 5.09 (s, 1H, H-16 or C15-OH); 4.46 (dd, J = 2.8, 12.0 Hz, 1H, H-6); 4.37 (d, J = 12.0 Hz, 1H, C3-OH); 4.34-4.26 (m, 1H, H-3); 4.14-4.03 (m, 3H, H-8/H-10/H-19); 3.92 (dt, J = 1.8, 11.5 Hz, 1H, H-10’); 3.87 (dt, J = 3.7, 11.5 Hz, 1H, H-22); 3.72 (dd, J = 2.5, 14.0 Hz, 1H, H-18); 3.70-3.64 (m, 1H, H-22’); 3.68 (s, 3H, OMe); 3.44-3.39 (m, 1H, H-5); 3.39-3.33 (m, 1H, H-5’); 2.57 (dd, J = 10.5, 12.5 Hz, 1H, H-2); 2.53 (dd, J = 3.4, 12.5 Hz, 1H, H-2’); 2.30 (dt, J = 3.4, 7.5 Hz, 2H, H-24); 2.24 (m, 1H, H-4); 2.13 (mc, 1H, H-7); 2.09-1.98 (m, 2H, H-20/H-18’); 1.81 (ddd, J = 3.0, 10.0, 14.0 Hz, 1H, H-20’); 1.76 (dq, J = 4.2, 12.0 Hz, 1H, H-9); 1.66-1.56 (m, 3H, H-7’/H-25); 1.51 (d (br), J = 15.0 Hz, 1H, H-4’); 1.41 (d (br), J = 13.0 Hz, 1H, H-9’); 1.35-1.17 (m, 8H, H-26 – 29); 1.20 (s, 3H, -C(CH3)2); 1.06 (s, 3H, -C(CH3)2); 0.87 (t, J = 7.0 Hz, 3H, H-30). 13C-NMR (125MHz, CDCl3): δ 191.9, 172.3, 172.1, 166.9, 151.6, 142.6, 142.5, 136.8, 132.1, 129.7 (2C), 127.4, 125.9, 119.9, 102.2, 98.9, 83.4, 75.6, 74.1, 71.6, 68.7, 66.2, 66.1, 65.7, 64.6, 51.1, 45.5, 45.1, 42.2, 35.8, 34.6, 33.7, 32.3, 31.6, 31.0, 29.0, 28.9, 24.7, 24.3, 22.6, 19.4, 14.1. HRMS: Calculated for C42H60O14Na [M + Na]+: 811.3881; Found: 811.3874. [α]D23 = −68.8 in CDCl3, c = 0.40.
Synthesis of analogue 14
1.0 equiv. of analogue 12 (0.0036 mmol, 2.8 mg) was dissolved in 100μl acetonitrile (HPLC grade). Then 10 equiv. mCPBA (0.035 mmol, 6mg) were added as a solid in three single portions over 5 hours. The conversion of the reaction was observed by Maldi-MS. The reaction was stirred at r.t. After 7 h 0.1 ml of a saturated solution of sodium thiosulfate in water were added to the reaction mixture and the mixture was further diluted with 1.0 ml water and 1.0 ml acetonitrile. The layers were separated and the aqueous layer was extracted using CH2Cl2 (4×5 ml). The combined organic layers were dried with Na2SO4, filtered and concentrated in vacuo. The crude was filtered through a pad of silica gel using ethyl acetate as eluent. All fractions containing product were further purified by reverse phase HPLC to give 1.5 mg (0.002 mmol, 52%) 14 as a white amorphous solid. Rf = 0.11 (pentane:EtOAc = 20:80) – one blue spot with p-anisaldehyde stain; Rf shows a strong concentration dependency. IR (thin film): 3460, 3424, 2925, 2855, 1722, 1715, 1434, 1403, 1378, 1362, 1288, 1259, 1231, 1160, 1136, 1104, 1063, 1025, 1006, 980, 917, 885, 859, 834, 806, 760, 734, 703, 665 cm−1. 1H-NMR (500MHz, CDCl3): 8.06-8.02 (m, 1H, aryl-H); 7.99-7.97 (m, 1H, aryl-H); 7.58-7.55 (m, 1H, aryl-H); 7.51-7.47 (m, 1H, aryl-H); 6.25 (d, J = 16.0 Hz, 1H, H-13); 6.01 (d, J = 2.0 Hz, 1H, H-23); 5.45 (dd, J = 7.5, 16.0 Hz, 1H, H-12); 5.45-5.39 (m, 1H, H-21); 5.17 (s, 1H, H-16 or C15-OH); 5.12 (d, J = 7.5 Hz, 1H, H-11); 5.11 (s, 1H, H-16 or C15-OH); 4.45 (dd, J = 2.4, 12.0 Hz, 1H, H-6); 4.39 (d, J = 12.0 Hz, 1H, C3-OH); 4.34-4.26 (m, 1H, H-3); 4.14-4.02 (m, 3H, H-8/H-10/H-19); 3.92 (dt, J = 1.8, 12.0 Hz, 1H, H-10’); 3.88 (dd, J = 3.2, 12.0 Hz, 1H, H-22); 3.68 (s, 3H, OMe); 3.73 (dd, J = 1.9, 13.5 Hz, 1H, H-18); 3.70-3.65 (m, 1H, H-22’); 3.45-3.40 (m, 1H, H-5); 3.36 (mc, 1H, H-5’); 2.59 (dd, J = 11.0, 12.5 Hz, 1H, H-2); 2.53 (dd, J = 2.9, 12.5 Hz, 1H, H-2’); 2.30 (dt, J = 3.3, 7.5 Hz, 2H, H-24); 2.27-2.22 (m, 1H, H-4); 2.15 (mc, 1H, H-7); 2.10-1.98 (mc, 2H, H-20/H-18’); 1.82 (ddd, J = 2.8, 12.0, 13.5 Hz, 1H, H-20’); 1.76 (dq, J = 4.3, 12.0 Hz, 1H, H-9); 1.67-1.55 (m, 3H, H-7’/H-25); 1.50 (d (br), J = 15.0 Hz, 1H, H-4’); 1.40 (d (br), J = 13.0 Hz, 1H, H-9’); 1.31-1.23 (m, 8H, H-26 – 29); 1.20 (s, 3H, -C(CH3)2); 1.06 (s, 3H, -C(CH3)2); 0.87 (t, J = 7.0 Hz, 3H, H-30). 13C-NMR (125MHz, CDCl3): δ 172.4, 172.1, 169.4, 167.0, 151.6, 142.6, 141.9, 131.4, 130.0, 129.5, 129.2, 128.3, 125.9, 119.9, 102.3, 99.0, 83.6, 75.7, 74.1, 71.6, 68.8, 66.2, 66.0, 65.7, 64.6, 51.1, 45.5, 45.1, 42.2, 35.8, 34.6, 33.6, 32.3, 31.6, 31.0, 29.0, 28.9, 24.7, 24.3, 22.6, 19.4, 14.1. HRMS: Calculated for C42H60O15Na [M + Na]+: 827.3830; Found: 827.3828. [α]D24 = −100.8 in CHCl3, c = 0.15.
Acknowledgments
This manuscript is dedicated to Gilbert Stork, whose towering contributions to science and education has enriched us all. J. R. gratefully acknowledges support by a Feodor-Lynen Fellowship (Humboldt foundation). We thank the Mochly-Rosen group (Stanford University) for access to assay materials. HRMS analysis for some compounds was performed at UCSF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.For pertinent reviews, see: Wender PA, Baryza JL, Hilinski MK, Horan JC, Kan C, Verma VA. In: Drug Discovery Research: New Frontiers in the Post-Genomic Era. Huang Z, editor. Wiley; Hoboken: 2007. pp. 127–162.. Mutter R, Willis M. Biorg Med Chem. 2000;8:1841. doi: 10.1016/s0968-0896(00)00150-4.; Hale KJ, Hummersone MG, Manaviazar S, Frigerio M. Nat Prod Rep. 2002;19:413. doi: 10.1039/b009211h.
- 2.For a lead reference, see: Trindade-Silva AE, Lim-Fong GE, Sharp KH, Haygood MG. Curr Opin Biotechnol. 2010;21:834. doi: 10.1016/j.copbio.2010.09.018.
- 3.Pettit GR, Day JF, Hartwell JL, Wood HB. Nature. 1970;227:962–963. doi: 10.1038/227962a0. [DOI] [PubMed] [Google Scholar]
- 4.Pettit GR, Herald CL, Doubek DL, Herald DL, Arnold E, Clardy J. J Am Chem Soc. 1982;104:6846. [Google Scholar]
- 5.Wall NR, Mohammad RM, Al-Katib AM. Leukemia Res. 1999;23:881–888. doi: 10.1016/s0145-2126(99)00108-3. [DOI] [PubMed] [Google Scholar]
- 6.Spitaler M, Utz I, Hilbe W, Hofmann J, Grunicke HH. Biochem Pharmacol. 1998;56:861–869. doi: 10.1016/s0006-2952(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 7.(a) Dowlati A, et al. Clin Cancer Res. 2003;9:5929–5935. [PubMed] [Google Scholar]; (b) Barr PM, Lazarus HM, Cooper BW, Schluchter MD, Panneerselvam A, Jacobberger JW, Hsu JW, Janakiraman N, Simic A, Dowlati A, Remick SC. Am J Hematol. 2009;84:484–487. doi: 10.1002/ajh.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ajani JA. Invest New Drugs. 2006;24:353–357. doi: 10.1007/s10637-006-6452-1. [DOI] [PubMed] [Google Scholar]
- 8.For a recent study and lead references, see: Shaha SP, Tomic J, Shi Y, Pham T, Mero P, White D, He L, Baryza JL, Wender PA, Booth JW, Spaner DE. Clin Exp Immunol. 2009;158:186. doi: 10.1111/j.1365-2249.2009.04003.x.
- 9.Hongpaisan J, Alkon DL. Proc Natl Acad Sci U S A. 2007;104:19571. doi: 10.1073/pnas.0709311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For current information, see: http://clinicaltrials.gov.
- 11.Mehla R, Bivalkar-Mehla S, Zhang R, Handy I, Albrecht H, Giri S, Nagarkatti P, Nagarkatti M, Chauhan A. PLoS ONE. 2010;5:e11160. doi: 10.1371/journal.pone.0011160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For reviews of PKC, see: Newton AC. Chem Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801.; Gallegos LL, Newton AC. IUBMB Life. 2008;60:782–789. doi: 10.1002/iub.122.
- 13.Stang SL, Lopez-Campistrous A, Song X, Dower NA, Blumberg PM, Wender PA, Stone JC. Exp Hematol. 2009;37:122–134. doi: 10.1016/j.exphem.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kazanietz MG. Mol Pharmacol. 2002;61:759. doi: 10.1124/mol.61.4.759. [DOI] [PubMed] [Google Scholar]
- 15.Mackay HJ, Twelves CJ. Nature Rev Cancer. 2007;7:554. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- 16.Schaufelberger DE, Koleck MP, Beutler JA, Vatakis AM, Alvarado AB, Andrews P, Marzo LV, Muschik GM, Roach J, Ross JT, Lebherz WB, Reeves MP, Eberwein RM, Rodgers LL, Testerman RP, Snader KM, Forenza S. J Nat Prod. 1991;54:1265–1270. doi: 10.1021/np50077a004. [DOI] [PubMed] [Google Scholar]
- 17.(a) Kageyama M, Tamura T, Nantz MH, Roberts JC, Somfai P, Whritenour DC, Masamune S. J Am Chem Soc. 1990;112:7407–7408. [Google Scholar]; (b) Evans DA, Carter PH, Carreira EM, Prunet JA, Charette AB, Lautens M. Angew Chem Int Ed. 1998;37:2354–2359. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2354::AID-ANIE2354>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]; (c) Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M. J Am Chem Soc. 1999;121:7540–7552. [Google Scholar]; (d) Ohmori K, Ogawa Y, Obitsu T, Ishikawa Y, Nishiyama S, Yamamura S. Angew Chem Int Ed. 2000;39:2290–2294. [PubMed] [Google Scholar]
- 18.Frigerio M. Nat Prod Rep. 2002;19:413–453. doi: 10.1039/b009211h. [DOI] [PubMed] [Google Scholar]; b) Hale KJ, Manaviazar S. Chem Asian J. 2010;5:704–754. doi: 10.1002/asia.200900634. [DOI] [PubMed] [Google Scholar]
- 19.Trost BM, Dong G. Nature. 2008;456:485–488. doi: 10.1038/nature07543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keck GE, Poudel YB, Cummins TJ, Rudra A, Covel JA. J Am Chem Soc. 2011;133:744–747. doi: 10.1021/ja110198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Y, Woo S, Krische MJ. J Am Chem Soc. 2011;133 doi: 10.1021/ja205673e. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wender PA, Schrier AJ. J Am Chem Soc. 2011;133:9228–9231. doi: 10.1021/ja203034k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For a review, see: Hale KJ, Manaviazar S. Chem Asian J. 2010;5:704. doi: 10.1002/asia.200900634.. For lead references, see the following and references therein: Allen JV, Green AP, Hardy S, Heron NM, Lee ATL, Thomas EJ. Tetrahedron Lett. 2008;49:6352.; De Brabander J, Vandewalle M. Pure Appl Chem. 1996;68:715.; Roy R, Rey AW, Charron M, Molino R. Chem Commun. 1989:1308.; Voight EA, Seradj H, Roethle PA, Burke SD. Org Lett. 2004;6:4045. doi: 10.1021/ol0483044.; Vakalopoulos A, Lampe TJF, Hoffmann HMR. Org Lett. 2001;3:929. doi: 10.1021/ol015551o.; Yadav JS, Bandyopadhyay A, Kunwar AC. Tetrahedron Lett. 2001;42:4907.
- 24.Wender PA, Cribbs CM, Koehler KF, Sharkey NA, Herald CL, Kamano Y, Pettit GR, Blumberg PM. Proc Natl Acad Sci U S A. 1988;85:7197. doi: 10.1073/pnas.85.19.7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc Chem Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]; (b) Wender PA, Baryza JL, Brenner SE, DeChristopher BA, Loy BA, Schrier AJ, Verma VA. Proc Natl Acad Sci U S A. 2011;108:6721–6726. doi: 10.1073/pnas.1015270108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wender PA, Loy BA, Schrier AJ. Isr J Chem. 2011;51:453–472. doi: 10.1002/ijch.201100020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wender PA, et al. Beyond natural products: Synthetic analogs of bryostatin 1. In: Huang Z, editor. Drug Discovery Research: New Frontiers in the Post-Genomic Era. Wiley-VCH; Hoboken, NJ: 2007. pp. 127–162. [Google Scholar]
- 27.Barr PM, Lazarus HM, Cooper BW, Schluchter MD, Panneerselvam A, Jacobberger JW, Hsu JW, Janakiraman N, Simic A, Dowlati A, Remick SC. Am J Hematol. 2009;84:484. doi: 10.1002/ajh.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wender PA, Baryza JL, Bennett CE, Bi FC, Brenner SE, Clarke MO, Horan JC, Kan C, LaCote E, Lippa BS, Nell PG, Turner T. J Am Chem Soc. 2002;124:13648–49. doi: 10.1021/ja027509+. [DOI] [PubMed] [Google Scholar]
- 29.Wender PA, Clarke MO, Horan JC. Org Lett. 2005;7:1995–8. doi: 10.1021/ol0504650. [DOI] [PubMed] [Google Scholar]
- 30.Wender PA, Baryza JL, Brenner SE, Clarke MO, Craske ML, Horan JC, Meyer T. Curr Drug Disc Technol. 2004;1:1–11. doi: 10.2174/1570163043484888. [DOI] [PubMed] [Google Scholar]
- 31.Baryza JL, Brenner SE, Craske ML, Meyer T, Wender PA. Chem Biol. 2004;11:1261–7. doi: 10.1016/j.chembiol.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Horan JC. Ph D Thesis. Stanford University; 2005. [Google Scholar]
- 33.Wender MO, Horan JC. Org Lett. 2006;8:4581–4584. doi: 10.1021/ol0618149. [DOI] [PubMed] [Google Scholar]
- 34.(a) Keck GE, Krishnamurthy D. Org Synth. 1998;75:12–18. [Google Scholar]; (b) Yu CM, Choi HS, Yoon SK, Jung WH. Synlett. 1997:889–890. [Google Scholar]
- 35.Cason J, Rinehart KL, Thornton SD. J Org Chem. 1953;18:1594–1600. [Google Scholar]
- 36.(a) Ding Y, Zhao G. J Chem Soc Chem Commun. 1992:941–942. [Google Scholar]; (b) Parrish JD, Shelton DR, Little RD. Org Lett. 2003;5:3615–3617. doi: 10.1021/ol035269c. [DOI] [PubMed] [Google Scholar]
- 37.(a) Holmquist CR, Roskamp EJ. J Org Chem. 1889;54:3258–3260. [Google Scholar]; (b) Holmquist CR, Roskamp EJ. Tetrahedron Lett. 1992;33:1131–1134. [Google Scholar]
- 38.(a) Emde H, Simchen G. Synthesis. 1977:867–869. [Google Scholar]; (b) Borgulya J, Bernauer K. Synthesis. 1980:545–547. [Google Scholar]; (c) Bou V, Vilarasa J. Tetrahedron Lett. 1990;31:567–568. [Google Scholar]; (d) Franck X, Figadere B, Cave A. Tetrahedron Lett. 1995;36:711–714. [Google Scholar]
- 39.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 40.Blanc D, Ratovelomanana-Vidal V, Marinetti A, Genet JP. Synlett. 1999:480–279. [Google Scholar]