

Abstract
An effective, asymmetric total synthesis of the antibiotic (+)-sorangicin A (1) has been achieved. Central to this venture was the development of first and second generation syntheses of the signature dioxabicyclo[3.2.1]octane core, the first featuring chemo- and stereoselective epoxide ring openings facilitated by a Co2(CO)6-alkyne complex, the second involving a KHMDS-promoted epoxide ring formation/opening cascade. Additional highlights include effective construction of the dihydro- and tetrahydropyran ring systems, respectively via a stereoselective conjugate addition/α-oxygenation protocol and a thioketalization/hydrostannane reduction sequence. Late-stage achievements entailed two Julia–Kociénski olefinations to unite three advanced fragments with high E-stereoselectivity, followed by a modified Stille protocol to introduce the Z,Z,E trienoate moiety, thereby completing the carbon skeleton. Mukaiyama macrolactonization, followed by carefully orchestrated Lewis and protic acid-promoted deprotections that suppressed isomerization and/or destruction of the sensitive (Z,Z,E)-trienoate linkage completed the first, and to date only, total synthesis of (+)-sorangicin A (1)
Keywords: Sorangicin A, Antibiotic, Trienoate linkage, Julia-Kociénski olefination, Total synthesis
1. Introduction
In 1985 Höfle, Jansen, and co-workers reported the isolation and structural elucidation of a new class of macrolide antibiotics termed the sorangicins, from a fermentation broth derived from the myxobacteria Sorangium cellulosum (strain So ce 12).1 Sorangicin A (1, Figure 1), the most prevalent and potent congener, displayed extraordinary antibiotic activity against both Gram-positive and Gram-negative bacteria with minimum inhibitory concentrations (MIC) of 0.01–0.3 and 3–25 μg/mL, respectively.
Fig 1.

Structures of (+)-Sorangicin A (1) and Rifampicin (2)
Subsequently the Jansen group demonstrated that the mechanism of action entails inhibition of DNA-dependent RNA polymerase (RNAP) both in Escherichia coli (E. coli) and Staphylococcus aureus, while not affecting eukaryotic cells.2 Rats infected with virulent E. coli underwent marked improvement when dosed with sorangicin A. Comparison studies, in conjunction with the Darst group, of (+)-sorangicin A (1) and rifampicin (2, Figure 1), the latter a clinically used ansamycin antibiotic possessing a broad spectrum of activity, revealed that, despite being chemically unrelated, the two antibiotics possess remarkably similar overall molecular shapes, leading to the same mechanism of action, namely inhibition of RNA elongation by binding to the same β-subunit pocket of RNAP.3 Importantly, in cross-resistance studies (+)-sorangicin A displayed an advantage against rifampicin-resistant microbial mutants. This observation, based on molecular dynamic simulations, was proposed to arise from an increase in conformational flexibility of the C(14)–C(20) segment of (+)-sorangicin A, permitting better adaptation to mutational changes within the RNAP binding pocket. As such, conformational flexibility of sorangicin A and analogues thereof holds important implications for future drug design against rapidly mutating pathogenic bacteria.3
Architecturally (+)-sorangicin A (1) is comprised of a C(1)–C(8) carboxylic acid side chain, attached to a highly unsaturated 31-membered macrocyclic lactone, possessing 15 stereogenic centers. Structural elements inscribed within the macrocyclic ring include the signature dioxabicyclic[3.2.1]octane, a rare (Z,Z,E)-trienoate linkage, and di- and tetrahydropyran ring systems. Initially the backbone connectivity, relative stereochemistry and olefin geometries were assigned based on extensive one- and two-dimensional NMR experiments, along with molecular weight assignment by mass spectrometry. X-ray analysis confirmed the structure and stereochemistry, with the absolute configuration being verified via a Hamilton test.4 Of interest was the observation of dynamic disorder in the crystalline solid in the C(17)–C(18) region. Significant solvent and pH effects were also observed in the NMR spectra, presumably due to a hydrogen bond between the C(1) carboxyl oxygen and the C(21, 22, and 25)-triol moiety.4
In addition to (+)-sorangicin A (1), 12 additional sorangicin congeners have been reported, with 8 possessing varying degrees of isomerization of the C(37)–C(43) trienoate region, the latter suggesting instability of this linkage.5 That (+)-sorangicin A (1) possessed considerable instability leading to decomposition was demonstrated upon treatment with a variety of reagents (e.g., fluoride ion, DDQ, and the dissolving metal sodium amalgam).6 Taken together, the intricate and challenging architecture, in conjunction with the potent antibiotic activity, and novel mechanism of action has led to considerable interest in (+)-sorangicin A (1) by the synthetic community, including pioneering studies from the Morken,7 Schinzer,8 Lee,9 and Yadav10 research groups, with a recent (2011) formal total synthesis achieved by Crimmins et. al.11 Herein we provide a full account on the evolution of a synthetic strategy, that in 2009 led to the first, and to date only, total synthesis of this architecturally intriguing antibiotic.12
At the outset of this synthetic program, we relied on the structure of (+)-sorangicin A to possess the R-configuration at C(10) (Scheme 1, Structure 3) as depicted in reference 6, and not as originally reported correctly in reference 4 as S at C(10) (Figure 1, Structure 1). As will be described, this misunderstanding came to light late in our synthetic venture (vide infra).
Scheme 1.
2. Results and Discussion
2.1. Initial Synthetic Planning
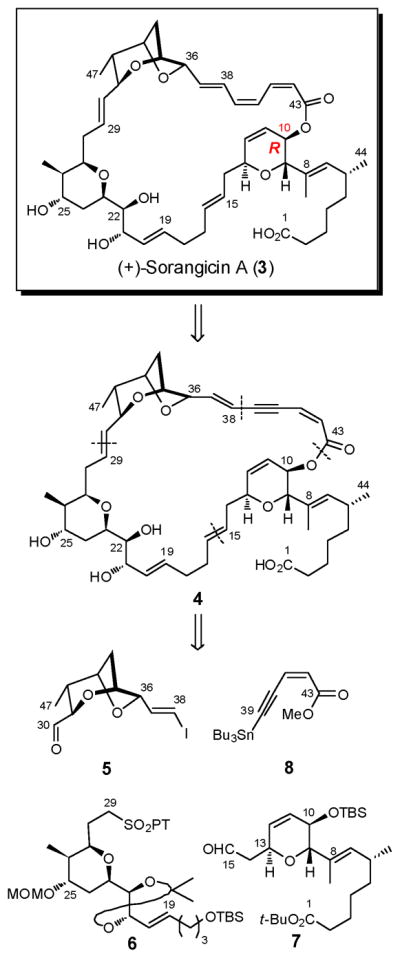
At the outset of the total synthesis that follows, a central tenet was to take full advantage of the reported chemical sensitivities of (+)-sorangicin A.6 Toward this end, we initially planned to mask the delicate (Z,Z,E)-trienoate moiety as dienyne (4) to attenuate the potential for isomerization and/or decomposition (Scheme 1). Disconnection of 4 at the macrocyclic lactone, the C(38)–C(39) σ-bond, and both the C(15)–C(16) and C(29)–C(30) trans disubstituted olefins revealed four potential advanced subtargets: the signature bicyclic aldehyde 5, tetrahydropyran 6, dihydropyran 7, and enyne 8. In the forward sense, we envisioned consecutive Julia–Kociénski unions of 6 with 5 and 7 to install the C(29)–C(30) and C(15)–C(16) trans olefins, thereby uniting three of the four advanced fragments. Subsequent Stille union of known enyne 813 with the strategically placed C(38) vinyl iodide in 5 was anticipated to complete construction of the full carbon skeleton possessing a dienyne moiety. Macrolactonization and semihydrogenation at a late stage would then be followed by global deprotection to reveal the labile (Z,Z,E)-triene. To mask the vicinal 21,22-diol in 6 during construction of the (+)-sorangicin A skeleton, we selected a dimethyl acetonide, since protection and deprotection of (+)-sorangicin A (1) employing the acetonide functionality had been successfully achieved by Höfle and co-workers in conjunction with the preparation of various sorangicin analogues.14 Protection of the C(25) hydroxyl as a MOM ether (illustrated in 6) and the C(1) carboxylic acid as a t-butyl ester (depicted in 7) were also envisioned, considering their acid lability. Importantly their removal would not require use of fluoride, oxidizing and/or reducing agents, conditions known to destroy the natural product.
Continuing with this analysis, the signature 2,6-dioxabicyclo[3.2.1]octane was anticipated to arise via an unprecedented chemo- and stereoselective epoxide ring-opening cascade of 9, facilitated by a Co2(CO)6-alkyne complex (Scheme 2). That is, upon formation of the Co2(CO)6-alkyne complex of bis-epoxide 10, a stereocontrolled epoxide ring-opening of the activated propargylic epoxide mediated by acid would generate the tetrahydrofuran ring; completion of the bicyclic skeleton would then entail a 6-exotet epoxide ring-opening via the derived C(35) secondary hydroxyl. For construction of sulfone 6, the presence of the 2,6-cis-disubstituted tetrahydropyran ring initially suggested application of the Petasis-Ferrier union/rearrangement protocol, a tactic developed in our laboratory,15 which would unite known β-hydroxy acid (+)-1115d with aldehyde (+)-12.16 Finally, construction of 2,3,6-trans, cis-trisubstituted dihydropyran 7 would call upon conjugate addition of vinyl bromide 14 to known dihydropyranone (−)-13,17 followed by α-oxygenation and elaboration of the C(11,12) double bond. Stereocontrol during construction of 7 would be provided by the stereogenicity at C(13) (Scheme 2).
Scheme 2.

2.2. Construction of the Signature Dioxabicyclo[3.2.1]octane Fragment 5 (Figure 2): Application of a Cyclization Cascade Facilitated by a Co2(CO)6-Acetylene Complex
Fig 2.
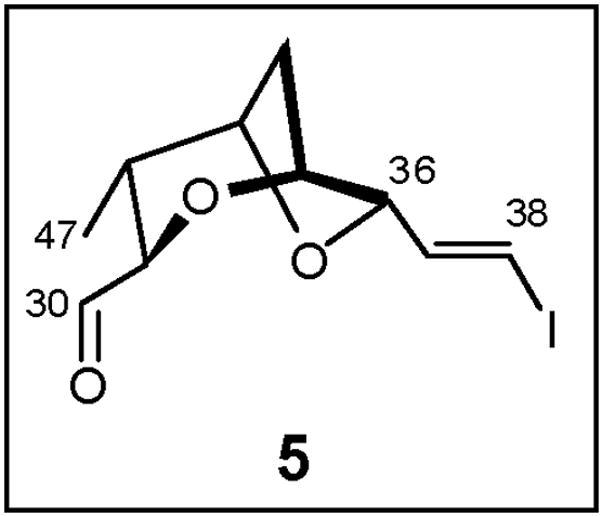
Structure of Bicyclic Fragment 5
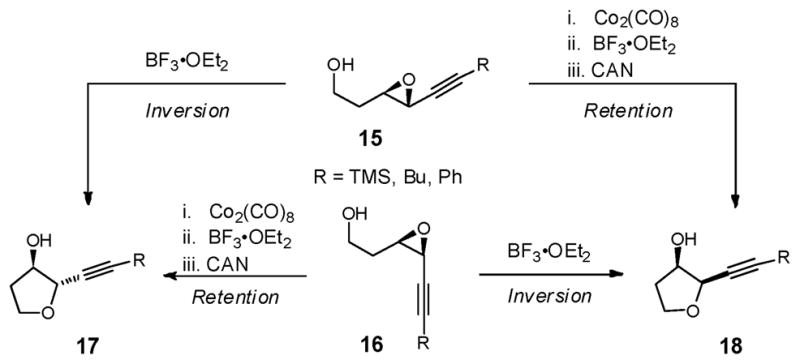
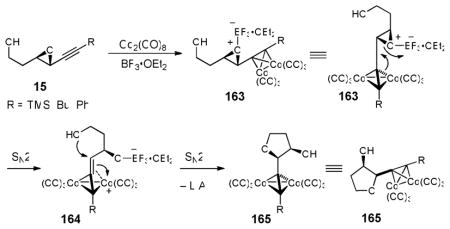
In 1994, Mukai and co-workers introduced stereocomplementary approaches to cis- and trans-2-ethynyl-3-hydroxytetrahydrofurans via epoxide ring-opening of 3,4-epoxy-6-substituted hex-5-yn-1-ols (Scheme 3).18 Specifically, exposure of either the trans- or cis-epoxy alcohol (15 or 16) to BF3•OEt2 resulted in intramolecular epoxide ring-opening with inversion of stereogenicity at the propargylic center to furnish respectively the trans- or cis-tetrahydrofurans (i.e., 15 → 17, or 16 → 18). Alternatively, addition of BF3•OEt2 to the corresponding Co2(CO)6-alkyne complexes induced “Nicholas” cyclizations19 to furnish respectively the cis- or trans-cobalt-complexed tetrahydrofurans. Importantly, epoxide ring-opening proceeded with overall retention of stereochemistry at the propargylic center. Subsequent demetallation with ceric ammonium nitrate (CAN) led to the respective tetrahydrofuran in a single-flask (i.e., 15 → 18, or 16 → 17). The observed retention of stereochemistry 7 for the epoxide ring-opening was rationalized to involve a double-SNII inversion.20
Scheme 3.
Based on this precedent, we became intrigued with the feasibility of constructing the (+)-sorangicin A signature bicycle subunit beginning with bis-epoxide 10 exploiting a four-step, one-flask reaction sequence (Scheme 4). If successful, a very rapid, highly stereocontrolled route to the signature subunit of (+)-sorangicin A would be in hand.
Scheme 4.

Our synthetic analysis of bis-epoxide 10 (Scheme 5) thus began with removal of both epoxides, and in turn scission of the C(34)–C(35) σ-bond to afford epoxy alcohol 23 and known vinyl bromide 24.21 In the forward sense, we envisioned that epoxide ring-opening of 23 via an organometallic reagent derived from 24 would furnish enyne 22; removal of the PMB group, ring-closure to furnish the terminal epoxide, and application of the elegant Shi oxidation protocol would provide bis-epoxide 10, the substrate for the proposed four-step construction of 21. Continuing with this analysis, removal of the epoxide moiety in 23 leads to a homoallylic alcohol,22 which upon application of a Brown crotylation retron, yields known aldehyde 25.23
Scheme 5.

We began the synthesis of 21 with aldehyde 25. Initially, bis-PMB ether 26,24 prepared from cis-2-butene-1,4-diol, was subjected to ozonolysis, in the presence of Sudan III as indicator,25 followed by reductive workup (PPh3). Kugelrohr distillation reproducibly furnished aldehyde 25 in 53% yield (Scheme 6). On larger scale (ca. 35 g), yields however were variable. As an alternative, ozonolysis of 26, followed by reduction of the ozonide, recrystallization and basic extractive workup of the derived bisulfite adduct 2726 provided aldehyde 25 on 35 g scale in 69% yield (3 steps) from cis-2-butene-1,4-diol, without chromatography or distillation.
Scheme 6.

Brown crotylation,27 followed by Sharpless dihydroxylation,28 next furnished triol (+)-29 in 74% yield for the two steps as a separable mixture (9:1) of diastereomers (Scheme 7). The absolute configuration of (+)-28 was confirmed via Mosher ester analysis, exploiting the Kakisawa test,29 while the relative stereochemistry of the major diastereomer [(+)-29], obtained via Sharpless dihydroxylation, was verified via the Rychnovsky-Evans empirical 13C acetonide tactic.30 Application of the Fraser-Reid epoxide ring construction furnished epoxide (+)-23 in modest yield (42–44%), after separation of the minor diastereomer, along with 15–16% trisylated product (−)-30.31 To eliminate over-trisylation, 1.05 equivalents of KHMDS could be used, employing slow addition of the trisylimid (1.5 h) as reported by Forsyth and coworkers.32 Although these conditions eliminated over trisylation, the yield of (+)-23 remained modest (48%).
Scheme 7.

With a route, albeit non-optimal, to epoxide (+)-23 established, we turned to union with 24 (Scheme 8). To our dismay, efforts to prepare the vinyl Grignard derived from 24,21 followed by addition to epoxide (+)-23, led only to recovery of starting materials.33
Scheme 8.

We therefore turned to installation of the E-enyne moiety via the anion derived from commercially available 1,4-bis(trimethylsilyl)-1,3-butadiyne (33) (Scheme 9),34 followed by chemo- and stereoselective reduction of the internal triple bond, directed by the resulting homoallylic alcohol.35 However to proceed with this scenario, improvement of the low efficiency of the conversion of triol (+)-29 to epoxide (+)-23 was required (i.e., 48% yield, Scheme 7). The hydroxyl group in (+)-28 was therefore protected as a PMB ether with the view of circumventing potential over-trisylation in the epoxide ring-closure. Sharpless dihydroxylation28 followed by application of the Fraser-Reid protocol31 then led to epoxide (−)-32 in excellent yield (98%) as a single diastereomer (Scheme 9).
Scheme 9.

With ample quantities of epoxide (−)-32 in hand, we examined the ring-opening with lithium trimethylsilylbutadiyne.36 Pleasingly generation of lithium trimethylsilylbutadiyne (2 equiv), followed by inverse addition of epoxide (−)-32 in THF containing BF3•OEt2 furnished alcohol (−)-34 in 92% yield on a 20 g scale (Scheme 9). Pre-cooling the THF/BF3•OEt2 solution of epoxide (−)-32 to −78 °C, prior to addition to the −78 °C THF solution of lithium trimethylsilylbutadiyne, and executing an inverse reaction quench with NH4Cl (Sat.) proved critical to achieving a high yield of (−)-34. Reduction with LiAlH4, followed by oxidative removal of both PMB groups furnished in turn alcohol (−)-35 and triol (+)-36, both in excellent yield as single E-isomers.35 Presumably, the chemo- and E-stereoselectivity arises via generation of an alkenyl aluminate (i.e., 37).37,38
What remained to complete the synthesis of bis-epoxide 10 was to form the internal and terminal epoxides. Unfortunately, closure of triol (+)-36 to terminal epoxide (+)-38 led irreproducibly to (+)-38 under a variety of conditions (Scheme 10A). Alternatively, protection of (−)-35 as the tert-butyl-diphenylsilyl (BPS) ether (Scheme 10B), followed by oxidative removal of both PMB groups and epoxide formation, employing the Mitsunobu conditions, reproducibly furnished terminal epoxide (−)-39 in 66% yield (three steps on a 4 g scale).
Scheme 10.

Initially the critical Shi epoxidation39 of alcohol (+)-38 led to (+)-10 with modest efficiency (Scheme 11). Better results were obtained with (−)-39, when the oxidation was carried out at low concentration (0.016 M) with addition of oxone and K2CO3 over 5 h; bis-epoxide (−)-40 was reproducibly formed in 66% yield, along with 22% recovered starting material.
Scheme 11.

Our goal, with bis-epoxides (+)-10 and (−)-40 now available, was to construct the signature 2,6-dioxabicyclo[3.2.1]octane ring system of (+)-sorangicin A in a single-flask via the proposed sequential bis-epoxide ring-opening cascade (see Scheme 4). To define the landscape of this cyclization process, we initially investigated a stepwise protocol. Treatment of bis-epoxide (+)-10 with 1.1 equiv Co2(CO)8 in CH2Cl2 at ambient temperature, followed by 0.10 equiv BF3•OEt2 at −78 °C, furnished cobalt complex (+)-19 in 75% yield as a single cis-isomer (Scheme 12). Pleasingly, the reaction proceeded with complete chemoselectivity at the activated propargylic epoxide. Single-crystal X-ray analysis verified that the epoxide opening had occurred with retention of stereochemistry.18
Scheme 12.

Turning to the second epoxide-ring opening in the same flask, treatment of (+)-19 with a variety of acids and bases failed to produce the desired bicycle 20 (Scheme 12). Instead, selective formation of bicycle (−)-41, arising from a 7-endo-tet cyclization, was observed with camphorsulfuric acid (CSA), while with BF3•OEt2, mixtures of (−)-41, along with respectively 6- and 7-membered bicycles (−)-42 and (−)-43 were formed, both having undergone epimerization (!) at the propargylic center. The structure and stereochemistry of bicycles (−)-41, (−)-42 and (−)-43 were assigned via 1H, COSY and D2O exchange experiments. Reexposure of the individual bicycles to BF3•OEt2 did not result in any improvement, suggesting that the cyclizations proceeded via kinetic control.
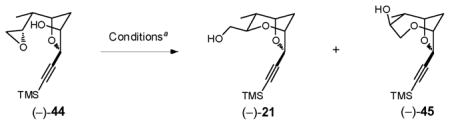
Based on the X-ray crystal structure of (+)-19 (Scheme 12), we reasoned that the steric bulk of the cobalt complex disfavored the desired 6-exo-tet ring-opening process, thereby preventing a one-flask cascade. We therefore removed the cobalt moiety from the alkyne prior to attempting the second cyclization (Scheme 13). A three-step operation, performed in a single-flask, furnished epoxide (−)-44 in 88% yield.
Scheme 13.

Reducing the steric bulk of alkyne did indeed facilitate the 6-exo-tet pathway, furnishing the desired bicycle (−)-21, upon treatment of epoxide (−)-44 with 0.10 equiv CSA in CH2Cl2 at ambient temperature. An equal amount of the undesired 7-endo-tet bicycle (−)-45 was also formed (Table 1, Entry 1). Importantly, no epimerized products were observed. Employing BF3•OEt2 at higher temperatures increased both the reaction rate and the amount of the desired 6-exo-tet bicycle (compare Entries 2 and 3; 5 and 6). Best results were obtained with 0.1 eq BF3•OEt2 at 40°C to afford a 1:1 ratio of (−)-21 and (−)-45 (Entry 4). Attempts to increase the yield of (−)-21 with other Lewis acids (e.g., TiCl4 or CeCl3), or under basic conditions (e.g., KH, DMSO) proved unsuccessful.40
Table 1.
Cyclization of (−)-44
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Acid | Equiv | Temp (°C) | Time (h) | Ratiob 21/45 | Conv.b (%) |
| 1 | CSA | 0.1 | 25 | 28 | 1:1 | 97 |
| 2 | BF3•OEt2 | 0.1 | −78 | 25 | 1:6 | 10 |
| 3 | BF3•OEt2 | 0.1 | 0 | 1 | 1:1 | 100 |
| 4 | BF3•OEt2 | 0.1 | 40 | 0.5 | 1:1 | 100 |
| 5 | BF3•OEt2 | 1.0 | −78 | 1 | 1:3 | 80 |
| 6 | BF3•OEt2 | 2.0 | 0 | 0.3 | 1:1 | 100 |
Entries 1–6 were run in CH2Cl2.
Ratios and conversions were determined by 1H NMR.
Having achieved access to bicycle (−)-21, we envisioned further enhancing the ratio of the 6-exo-tet derived bicycle by additional reduction of steric bulk at the alkyne. Accordingly, the TMS group of (−)-44 was removed to furnish (−)-46. Conditions identical to those employed in Entry 3 (Table 1) did modestly enhance delivery of the desired tricycle (−)-47 (1.4:1, Entry 1, Table 2). Single crystal X-ray analyses verified the structures and stereochemistries of both (−)-47 and (−)-48. We next performed the cyclization by addition of (−)-46 to a solution of CH2Cl2 at reflux for 5 minutes, containing 10.0 equiv BF3•OEt2. Again modest improvement in the product ratio (ca. 2.1:1) was achieved (Entry 2). Solvents such as benzene or acetonitrile at a variety of temperatures however led to no improvement. Notably, uncatalyzed bicycle formation was observed upon storing neat (−)-46 at –20 °C, favoring the undesired 7-endo-tet product (1:2, Entry 6).
Table 2.
Cyclization of (−)-46
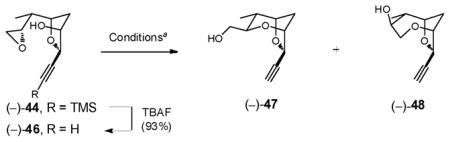 | ||||||
|---|---|---|---|---|---|---|
| Entry | Acid | Equiv | Temp (°C) | Time (min) | Ratiob 47/48 | Conv.b (%) |
| 1 | BF3•OEt2 | 0.1 | 0 | 60 | 1.4:1 | 100 |
| 2 | BF3•OEt2 | 10 | 40 | 5 | 2.1:1 | 100c |
| 3 | TfOH | 3.0 | 0 | 1 | 2.5:1 | 100 |
| 4 | FSO3H | 0.1 | 0 | 5 | 2:1 | 100 |
| 5 | SO3H:SbF5(1:1)d | NA | −20 | 5 | > 2:1 | 100 |
| 6 | None | NA | −20 | weeks | 1:2 | NA |
Entries 1–4 were run in CH2Cl2.
Ratios and conversions were determined by 1H NMR.
Isolated yield for (−)-47 was 65%.
Entry 5 was run in SO2.
We also explored a series of acids, stronger than BF3•OEt2 (Table 2). The best ratio of desired to undesired bicycle was achieved with TfOH (2.5:1, Entry 3), however the BF3•OEt2 conditions (Entry 2) provided a higher overall yield (65%) of (−)-47. The conditions in Entry 2 were therefore employed to advance material to (−)-47.
Having achieved construction of bicycle (−)-47, we were of course cognizant that preparation and epoxidation of enyne (+)-38 was less efficient than the synthesis and epoxidation of BPS-protected enyne (−)-39 (see Schemes 10 and 11). For material advancement, we therefore utilized the BPS-protected bis-epoxide (−)-40 to furnish (−)-47 in gram quantities (Scheme 14).41
Scheme 14.

Completion of the synthesis of (−)-5, the signature core fragment for (+)-sorangicin A (1), was achieved via radical induced hydrostannylation/iodination42 of (−)-47, followed by Dess-Martin oxidation.43 Interestingly, application of a palladium-catalyzed hydrostannylation protocol,44 followed by iodination, furnished the undesired α-iodide as the major regioisomer, while use of iodine in place of NIS in the AIBN promoted hydrostannylation led to significant protodestannylation, presumably due to the formation of HI upon addition of iodine to the excess Bu3SnH. In the end, the signature core fragment (−)-5 for (+)-sorangicin A could be prepared in 15 steps (longest linear sequence) and in 3% overall yield utilizing the Co2(CO)6-alkyne complex approach.12a
2.3. A Second-Generation Synthesis of the Bicycle (−)-5: Development of a KHMDS-Promoted Cyclization Cascade
Although effective, the first-generation synthesis of (−)-5 was not considered sufficiently efficient vis-à-vis material advancement; a second generation approach based on precedence from the Crimmins laboratory11a was therefore developed. Early on we had recognized that the pyran portion of bicyclic aldehyde (−)-5 shares the same 2,6-trans-relationship as dihydropyran 7 (Scheme 1). We therefore envisioned a route featuring a similar substrate-controlled, stereoselective conjugate addition of a Michael donor to dihydropyranone (−)-13 (vide infra), followed by electrophilic trapping to install the C(47) methyl group (Scheme 15). In retrospect, the synthesis of (+)-sorangicin A would be significantly streamlined if two of the major subtargets would arise from the same homochiral starting material. The second-generation route would also be differentiated from the Co2(CO)6-alkyne approach by constructing the tetrahydropyran ring in advance of the tetrahydrofuran moiety, a tactic that was envisioned to avoid the 7-endo-tet cyclization.
Scheme 15.

Construction of dihydropyranone (−)-1345 began by exploiting a hetero Diels-Alder (HDA) reaction between the Danishefsky diene46 and aldehyde 51,47 promoted by chromium(III) complex 5248 (Scheme 16). Both the yield and enantioselectivity proved excellent (98%; 20:1 e.r.). A three-component conjugate addition/alkylation sequence involving dihydropyranone (−)-13, a suitable Michael donor, disguised as an aldehyde synthon, and methyl iodide, were employed to fashion the requisite 2,3,6-trans, cis-configuration. Initially, dihydropyranone (−)-13 was expected to be a somewhat reluctant Michael acceptor, as the ring oxygen would deactivate the β position.49 Indeed, efforts to add a mixed zincate, derived from BnOCH2SnBu3, resulted only in recovery of the starting enone, even with promoters such as Cu(OTf)2 and P(OEt)3.50 These observations however proved to be more of a donor problem, as α-alkoxycopper reagents lack reactivity toward Michael additions.51
Scheme 16.

Recourse was thus made to the commercially available β-bromostyrene (53), a surrogate addend, with a view to achieving oxidative cleavage of the styrene olefin at a later stage (Scheme 17). Pleasingly, the mixed zincate, generated via lithiation of β-bromostyrene (53) and transmetallation with Me2Zn, underwent effective conjugate addition to dihydropyranone (−)-13 to furnish the zinc enolate, which upon quenching with excess MeI and HMPA furnished adduct 54 (62%).52 Although commercial β-bromostyrene is a cis/trans isomeric mixture (ca. 1:9), 54 was obtained as a single diastereomer (confirmed by nOe studies), the result of excellent substrate control, as well as low reactivity of the minor cis-derived zincate.53
Scheme 17.

Reduction of tetrahydropyranone 54 with L-Selectride then furnished a single diastereomer; again the newly generated stereogenetic center was verified via nOe correlations. Protection of the secondary hydroxyl 55 was next envisioned, followed by desilylation and Grieco elimination to furnish diene 56, which was projected to undergo stereoselective dihydroxylation to furnish diol 57. Attack by the C(33) hydroxyl that proceeds with inversion of the C(36) stereogenicity would be required to construct the bicyclic skeleton possessing the S configuration at C(36). A critical question remained: could the requisite chemoselective oxidation of the terminal alkene be achieved in the presence of the electron rich styrene olefin?
With this issue in mind, further analysis revealed that the synthesis of subtarget (−)-5 could be significantly streamlined if the S-configuration at C(36) was installed early on. Dihydropyranone (−)-5954 (Scheme 18) was therefore selected as a more advanced Michael acceptor, possessing the S-configuration at C(36). Pleasingly, this Michael acceptor could be readily prepared via cyclocondensation between the Danishefsky diene46 and aldehyde (−)-58,55 employing the same Jacobsen catalyst 52.48 An analogous three-component coupling protocol furnished adduct (+)-61 (51%), again as a single diastereomer via Zn-enolate 60. Careful examination of the reaction mixture, however, led to the isolation and identification of α,α′-bismethylated product (+)-62 (ca. 20%). The mechanism of this side reaction is ascribed to the unusual reactivity of Zn-enolate 60; a similar observation was recorded by Alexakis et al.56 The hightened reactivity of the Zn-enolate (60) could be mitigated by addition of CuI•PBu3 just prior to the addition of MeI. The result was a slower, albeit effective reaction process furnishing (+)-61 in 73% yield. Other copper species, including Cu(OTf)2•P(OEt)3 and (2-Th)Cu(CN)Li, also proved effective.
Scheme 18.

Having assembled (+)-61 in two steps, L-Selectride reduction followed by acid promoted deprotection led to triol (−)-63, setting the stage for the critical tetrahydrofuran ring construction (Scheme 19).
Scheme 19.

Regioselective sulfonylation of the least hindered hydroxyl in (−)-63 was readily achieved with KHMDS (1 equiv), followed by the slow addition of the bulky Trisylimid (1 equiv), which in turn was treated with an additional 2 equiv KHMDS to promote the expected cyclization cascade, involving epoxide ring formation followed by epoxide opening to generate bicycle (−)-66.32 The yield of (−)-66 however was disappointing (33%); the major side product proved to be over-sulfonylation to furnish (−)-67 (36%). Lowering the reaction temperature or use of potassium tert-butoxide in tert-butanol32 did not affect an improvement. We therefore turned to an alternative stepwise protocol developed by the Tanaka group.57 Treatment of (−)-63 with triisopropyl-benzenesulfonyl chloride (TrisylCl) in pyridine/CH2Cl2 at room temperature (Scheme 20) selectively furnished the primary sulfonate (−)-68 in 77% yield, which proved stable to regular laboratory handling. Addition of 1.2 equiv KHMDS then delivered (−)-66 in excellent yield (91%). Thus, the signature dioxabicyclo[3.2.1]octane core of (+)-sorangicin A (1) could now be elaborated in 6 steps and 35% overall yield from (−)-58.
Scheme 20.

To arrive at advanced bicycle (−)-5 (Scheme 20), installation of the trans vinyl iodide and access to the C(30) aldehyde remained. Toward this end, (−)-66 was subjected to Parikh-Doering oxidation,58 and without purification, the resultant sensitive aldehyde subjected immediately to Takai olefination conditions.59 To our surprise, a diastereomeric mixture of Z/E isomers (1:3.2) was obtained as evidenced by the observed 1H NMR olefin coupling constants (8 Hz and 15.8 Hz). This selectivity was contrary to the general trend, in which α-alkoxy-aldehydes are reported to furnish nearly complete E-selectivity in the Takai olefination.60 Switching the reaction medium from THF to dioxane/THF61 did not alter the E/Z ratio, but did improve the scalability of the process; under these conditions (−)-69 and (−)-70 could be obtained in 16% and 52% yield on half gram scale after flash chromatography.
The challenge at this stage was to differentiate the two olefins in (−)-70. From the outset we speculated that the vinyl substituents, an electron-withdrawing iodide and an electron-donating phenyl group, might introduce sufficient differences in electron-density to permit the required chemoselectivity. In the event, Sharpless dihydroxylation28 led only to reaction at the styrene olefin in (−)-70 to furnish the corresponding diol, which upon NaIO4 treatment with pH 7 buffer generated the desired advanced aldehyde (−)-5, now available in 10 steps and 11% overall yield from (−)-58.12c
2.4. Construction of Advanced Linchpin Fragment 6
As noted earlier, the 2,6-cis-disubstituted tetrahydropyran embedded in fragment 6 (Figure 3) suggested application of the Petasis-Ferrier union/rearrangement tactic, ideally exploiting known β-hydroxy acid (+)-1115d and aldehyde (+)-1216 (Scheme 21). This strategy, however, was not without risk. First, the Petasis-Ferrier rearrangement had not previously been employed with either an α-oxygenated aldehyde or a resident acetonide moiety on the aldehyde. Second, there were no examples in a tetrahydro-pyranone endowed with a 2,3,6-cis-trisubstitution substituent pattern of selective reduction of a C(4) carbonyl to the corresponding axial alcohol (vide infra).
Fig 3.
Structure of THP Fragment 6
Scheme 21.

With this overview, we prepared known β-hydroxy acid (+)-1115d and aldehyde (+)-12,16 and examined the union/rearrangement tactic. Silylation of (+)-11, followed by condensation with (+)-12, promoted by TMSOTf,62 furnished dioxanone (+)-71; Petasis-Tebbe olefination63 then provided enol ether (+)-72 in 79% yield over the three steps without purification (Scheme 21). Exposure of (+)-72 to our optimized Lewis acid (Me2AlCl) protocol, to trigger the Petasis-Ferrier rearrangement, pleasingly furnished tetrahydropyranone (+)-73 in 70% yield as a single diastereomer. The cis stereochemistry was established based on nOe enhancements between H(23) and H(27). This transformation represented the first validated example of an α-oxygenated aldehyde in a Petasis-Ferrier union/rearrangement sequence.64
Reduction of the ketone in (+)-73 to the requisite C(25) axial alcohol however proved to be a major road block. A wide variety of reducing conditions, including K- and L-Selectride, NaBH4/CeCl3, the CBS reagents, the latter employing either the (R) and (S) enantiomers, delivered only the undesired C(25) equatorial alcohol as the major product. Best results were obtained with DIBAL-H, albeit a mixture (1:1) of diastereomers resulted. Equally daunting, Mitsunobu inversion of the undesired diastereomer led only to elimination. At the time that these results were recorded, a nearly identical observation, including Mitsunobu elimination, was reported by Funk and Cossey65 for an analogous 2,3,6-cis-cis-trisubstituted tetrahydropyranone. More recently, Yadav and co-workers10a corroborated both observations in their synthesis of an advanced THP fragment for (+)-sorangicin A.
Although we were able to arrive at the required 2,3,6-cis,cis-trisubstituted tetrahydropyranone system employing the Petasis-Ferrier union/rearrangement tactic, advancing materials through a sequence involving an unselective tetrahydropyranone reduction was viewed as less than optimal. A second-generation strategy was therefore developed.
2.5. Linchpin 6: A Second-Generation Synthesis
At this stage, we opted to explore a strategy that would first construct a linear precursor for the tetrahydropyran ring system 6, that would permit installation of the C(25) hydroxyl group with the requisite stereogenicity prior to ring formation (Scheme 22). An aldol tactic between aldehyde 78 and methyl ketone 79 became our focus (Scheme 22). Cyclization of 77 to ketal 75, followed in turn by reductive deoxygenation, application of a Suzuki–Miyaura union with alkyl boronate 76, and installation of the phenyltetrazolyl-sulfone (PT) would deliver the required advanced THP fragment 6 after protecting group adjustments. Installation of the C(25) hydroxyl in a stereoselective fashion to furnish 77 was viewed as the principle issue. Boron-mediated aldol reactions involving methyl ketones, governed respectively by the α- or β-alkoxy substituent of the methyl ketone, are known to give rise to the 1,4-anti or 1,5-anti adducts.66 For ketone 79 however the α- and β-benzyloxy groups would operate in an opposite sense. Which structural feature would dominate however was not clear.
Scheme 22.

With this question in mind, we constructed coupling partners 78 and 79. Protection of known alcohol (+)-8015d as the TES ether, followed by removal of the chiral auxiliary and oxidation with SO3•py58 furnished aldehyde (+)-78 in 91% yield over the three steps (Scheme 23A). Methyl ketone 79 was next prepared in five steps (Scheme 23B). Reduction of known lactone (+)-8167 with DIBAL-H followed by exposure of the resulting lactol to TMSCHN2/LDA68 provided alkynyl alcohol (+)-82 in good overall yield (85%, 2 steps). Parikh-Doering oxidation58 was then followed by methylation of the resulting aldehyde with AlMe3; a second Parikh-Doering oxidation led to the corresponding methyl ketone, albeit with partial racemization at the α-stereogenic center during the methylation. The less basic nucleophile generated from MeMgBr and anhydrous CeCl3 furnished a pair of diastereomeric secondary alcohols without epimerization. Oxidation then led to methyl ketone (+)-79 in 74% yield. Overall, the five-step sequence to (+)-79 proceeded in 62% yield.
Scheme 23.

With both coupling partners in hand, reaction of aldehyde (+)-78 (Scheme 24) with the boron enolate derived from methyl ketone (+)-79 via dicyclohexylboron chloride (Chx2BCl) led to a separable mixture of C(25) diastereomers (3.2:1), in favor of the desired β-alcohol, demonstrating that in this case, when the α- and β-alkoxy substituents act in opposite fashion, 1,4-anti selectivity dominates over 1,5-anti selectivity. Treatment of the alcohol mixture with PPh3•HBr in MeOH to effect desilylation followed by cyclization furnished both (+)-75, the desired methyl ketal, and the C(25) epimer (+)-83 in good yield. Although the aldol diastereoselectivity had proven to be only modest (3.2:1), this route held considerable appeal since the undesired methyl ketal (+)-83 could be oxidized to tetrahydropyranone (+)-84, and then reduced with L-Selectride to deliver (+)-75 as a single diastereomer (89% yield, 2 steps), permitting advancement of all synthetic material. The difference in stereochemical outcome vis-à-vis the selectivity observed with reductions of (+)-73 and (+)-84 is illustrated in Figure 4. In the case of tetrahydropyranone (+)-73, axial attack of the hydride is generally favored, aided by the steric hindrance of the C(26) axial methyl group. In ketone (+)-84, the axial trajectory however is highly unfavored due to the axial C(23) methoxy group, leading instead to the desired α-alcohol (+)-75.
Scheme 24.

Fig 4.
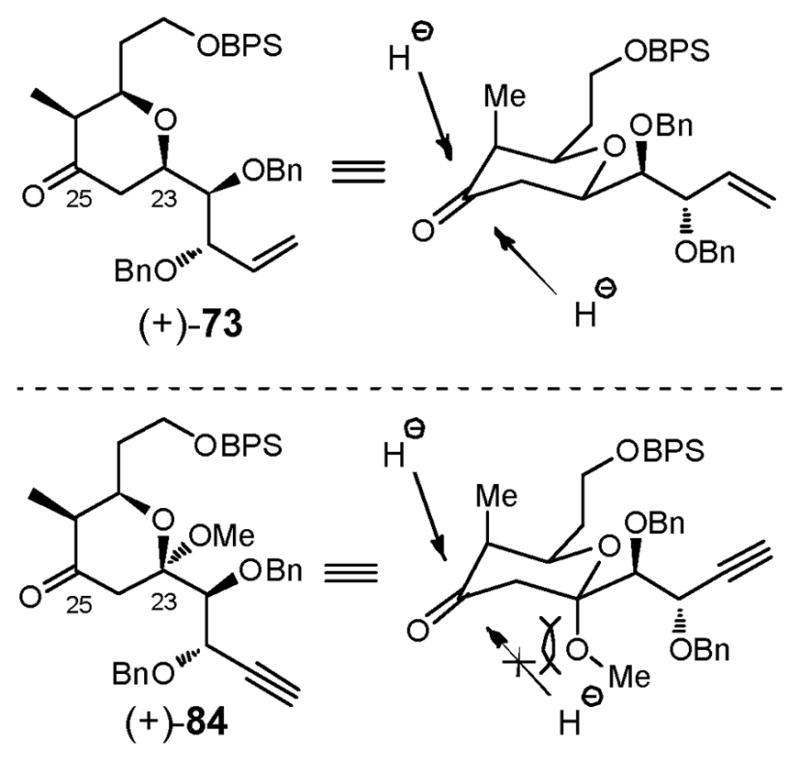
Rationale for observed stereoselectivity in the reduction of (+)-73 and (+)-84
Synthesis of linchpin 6 continued via reductive deoxygenation of the methyl ketal in (+)-75, with Et3SiH and TMSOTf, followed by MOM protection to furnish tetrahydropyran (+)-85 in 93% yield for the two steps (Scheme 25). The stereochemical outcome in (+)-85 was verified by nOe correlations. Installation of the trans vinyl iodide moiety was next achieved via a hydrozirconation/iodination protocol,69 which in turn permitted Suzuki–Miyaura cross coupling70 with alkyl boronate 7671 to provide olefin (+)-86 as a single E-isomer, possessing the full carbon skeleton of THP fragment 6. Debenzylation via treatment with lithium 4,4′-di-tert-butylbiphenolide (LiDBB), followed by acetonide formation and selective removal of the primary BPS group with KOH/DMPU72 in acetonitrile then furnished alcohol (+)-87 in 59% yield for the three steps. The phenyltetrazolyl sulfone (PT) was next installed to arrive at fragment (−)-6. Overall (−)-6 was constructed in 17 steps beginning with commercially available D-erythronolactone.12b
Scheme 25.

2.6. An Augmented Synthesis of Advanced Linchpin (−)-6
Upon scaling the first-generation synthesis of (−)-6, a number of issues were encountered. First, the C(25) axial hydroxy in (+)-75 was prone to elimination during the Lewis acid promoted reductive deoxygenation. To solve the issue, we envisioned that thioketalization would provide an intermediate that could be reduced under conditions that would not activate the C(25) hydroxyl toward elimination (e.g., radical reduction). A second issue related to removal of the primary BPS-ether in the presence of the primary TBS-ether to afford (+)-87 employing strong basic conditions. An orthogonal protecting group strategy would address this problem. Aldehyde (+)-89 (Scheme 26A) possessing a terminal benzyl ether, was therefore prepared from known amide (+)-8873 in 2 steps (80%).
Scheme 26.

To improve convergency, we also decided to introduce the vinyl iodide moiety at an earlier stage. After a number of experiments, hydrozirconation of 90, protected as the TES ether, (Scheme 26B) with the Schwartz reagent [Cp2Zr(H)Cl]69 and I2, furnished vinyl iodide 91 in 59% yield along with alkene 92 (12%). Speculating that the Schwartz reagent was reacting to generate HI with the excess iodine, which in turn would quench the intermediate vinylzirconocene to generate 92, NIS was employed to remove zirconium. These conditions furnished vinyl iodide (+)-93 in 65% yield along with concomitant desilylation. This sequence could be further streamlined by conducting the hydrozirconation directly on alcohol (+)-82, employing LiEt3BH to achieve deprotonation prior to treatment with the in situ generated Schwartz reagent.69 These conditions produced (+)-93 in 87% yield. The resulting vinyl iodide was then converted to methyl ketone (+)-94 in similar fashion to the conversion of (+)-82 to (+)-79 (see Scheme 23).
With both coupling partners in hand, realization of the aldol condensation became our focus. Initial NMR experiments in CD2Cl2 indicated that the previously employed Lewis acid Chx2BCl did not efficiently generate the desired enol-boronate. Consequently, the more reactive Lewis acid, Chx2BBr,74 was employed. Moderate diastereoselectivity (β:α = 3:1) was obtained using a slight excess of the Lewis acid (1.25 equiv), albeit at the expense of yield (33%, Scheme 27). The reaction could be driven to completion by increasing the amount of Lewis acid (3.5 equiv), albeit nearly equal quantities of the α- and β-adducts were obtained, which fortunately could be separated by flash chromatography. The observed modest diastereoselectivity is in line with the expectation that the substituents on (+)-94 act in the opposite sense to direct the anti-selective aldol (vide ante). Given that the undesired α-diastereomer was also a viable intermediate, via an oxidation/reduction sequence en route to sulfone (−)-6, further optimization of the aldol reaction was not pursued. Rather both acyclic precursors were utilized in the subsequent thioketalization studies.
Scheme 27.

Beginning with adduct (+)-95β, desilylation was successfully achieved with Et3N•3HF giving rise to 96, which exists as a mixture of hemiketal tautomers 96/97 (Scheme 28A). Treatment with a catalytic amount of BF3•OEt2 (0.33 equiv) in CH2Cl2/EtSH at –15 °C effected thioketalization to produce the desired thioketal (+)-98 (33%), along with competitive elimination of the C(25) hydroxyl to afford near equal quantities of (+)-99 (34%). Attempts with different temperature and Lewis acids régimes did not improve the matter. Diastereomer (+)-95α faired better under the same thioketalization conditions, resulting in less dehydration and in higher efficiency (Scheme 28B). This was not surprising considering that the equatorial C(25) hydroxyl group in (+)-102, based on stereoelectronic effects, would be expected to be less prone to elimination as compared to the C(25) axial diastereomer (+)-98.
Scheme 28.

Gratifyingly, a further screen of solvents with additional optimization revealed that slow addition of a catalytic amount of BF3•OEt2 (0.08–0.16 equiv) in MeCN via syringe pump to solutions of both 96 and 100 in MeCN/EtSH furnished the desired thioketals in 82–83% yield, with <10% of dehydration product (+)-99 observed (Scheme 29).75 A similar two-step oxidation/reduction protocol (see Scheme 24) then transformed thioketal (+)-102 into the desired (+)-98 with good overall efficiency (83% yield, 2 steps).
Scheme 29.

At this point, attempts to protect the C(25) hydroxyl of (+)-98 as the corresponding MOM-ether led only to recovered starting material, suggesting that in the presence of the axial thioethyl group the same steric factor permitting effective conversion of (+)-102 to (+)-98 hinders ether formation. Fortunately, extension of the side-chain of (+)-98 without protection of the C(25)-hydroxyl could be achieved via Suzuki union70 with boronate 76, to furnish (+)-103 in good yield (85%, Scheme 30).
Scheme 30.

Initial experiments to reduce the thioketal employing excess Bu3SnH as the radical reductant provided the desired (+)-105, albeit in low yield (ca. 21–28%, Scheme 30). As anticipated the reaction was not plagued by C(25) hydroxyl elimination, however competitive deprotection of the primary benzyl ether occurred, leading to (+)-104; yields ranged from 34–52%. Attempts to reprotect the free 1°-hydroxyl as a benzyl ether in the presence of the secondary alcohol were not rewarding.
We reasoned that if the highly reactive tributyltin radicals were responsible for removal of the primary benzyl ether, a less reactive and hence more stable radical source might suppress the debenzylation process. To this end, we turned to Ph3SnH to furnish (+)-105 in 65% yield, with only modest loss of the benzyl group (15%, Scheme 31).
Scheme 31.

With the axial thioethyl group removed, the C(25) hydroxyl of (+)-105 was protected as the MOM ether, and in turn, triol (+)-106 was generated upon treatment with excess lithium 4,4′-di-tert-butylbiphenylide (LiDBB) (Scheme 31). Selective conversion of the primary alcohol to the corresponding mesylate (+)-107 (80%) was then achieved using carefully defined conditions (MsCl, collidine) to avoid loss of the TBS group. The resultant vicinal diol in (+)-107 was then protected as the acetonide, using 2-methoxypropene and catalytic PPTS, to furnish (+)-108 in near quantitative yield.
Installation of the Julia-Kociénski sulfone, was next achieved via displacement of the mesylate in (+)-108 with cesium arylthiolate derived from phenyltetrazole-thiol (PTSH) and Cs2CO3 in DMF to produce (−)-109 (97%). Use of the more readily available sodium thiolate on the other hand led to significant β-elimination to furnish the terminal olefin. The Julia-Kociénski sulfone (−)-6 was then generated in excellent yield (96%) upon oxidation with the ammonium molybdate-hydrogen peroxide oxidative system [(NH4)6Mo7O24•4H2O-H2O2].76 Overall the thioketalization-based approach produced the requisite advanced THP linchpin (−)-6 in 17 steps (longest linear sequence) and in 9.2% overall yield.
2.7. Construction of Advanced Dihydropyran Fragment 7 (Figure 5)
Fig 5.
Structure of DHP Fragment 7
As earlier illustrated (Scheme 2), disconnection of the C(8)–C(9) sp2-sp3 carbon-carbon bond in 7 revealed dihydropyranone (−)-1317 and vinyl bromide 14 (Scheme 32). In the synthetic direction, we envisioned that the C(13) stereocenter in (−)-13 would permit control of the stereochemical outcome at both C(9) and C(10). To this end, 1,4-conjugate addition of the cuprate derived from vinyl bromide 14 and enone (−)-13 (Scheme 16), the latter prepared during the synthesis of bicycle aldehyde (−)-5, would furnish the 2,6-trans-disubstituted tetrahydropyranone enolate 110. Direct capture of this enolate, or that derived from the silyl enol ether with an oxygen source would in turn set the C(10) stereogenicity. Installation of the requisite C(11)–C(12) unsaturation via reduction of the kinetic enol triflate obtained from the C(11) ketone, followed by oxidation state adjustments, would then complete construction of advanced fragment 7.
Scheme 32.

Preparation of vinyl bromide 14 entailed Myers alkylation77 of the lithium enolate derived from amide (+)-111 with alkyl iodide 112, the latter prepared in two-steps and in 93% yield from 1,5-pentanediol (Scheme 33). Amide (+)-113 was produced both in excellent yield and with high diastereoselectivity (99%, d.r. >20:1). In turn, reduction with LiHAl(OEt)3 generated in situ, led directly to aldehyde (−)-114 in good yield after hydrolysis of the intermediate pseudoephedrine aminal.77,78 A Corey–Fuchs homologation/methylation sequence,79 followed by hydrozirconation with Schwartz reagent,69 and bromination with N-bromosuccinimide (NBS) then led to vinyl bromide (−)-14 as a single stereo- and regioisomer in excellent yield. Overall, vinyl bromide (−)-14 was prepared in 7 steps (longest linear sequence) and in 55% overall yield from commercially available starting materials.12b
Scheme 33.

Union of dihydropyranone (−)-13 with vinyl bromide (−)-14 entailed addition of the derived cuprate to the Michael acceptor (−)-13 (Scheme 34); a single diastereomer (−)-116 resulted with excellent stereocontrol, albeit in modest yield (46%). The proposed Rubottom oxidation however proved problematic, likely due to the ring oxygen. At best a 27% yield of TBS ether (−)-118 was obtained. Alternate oxidation with either methyl-(trifluoromethyl)dioxirane generated in situ,80 OsO4,81 or attempted oxidation of the enolate derived from enol acetate (+)-117 provided no improvement.
Scheme 34.

The stereogenicity of the newly generated C(9) and C(10) centers were assigned based on the observed NMR coupling constants of 8.3 Hz and 9.4 Hz (i.e., axial-axial) between H(9) and H(10) in (−)-118 and (−)-119, respectively, indicative of the trans relationships.82 The absolute stereochemistry of the C(10) center was confirmed via Kakisawa analysis29 of the corresponding Mosher esters derived from alcohol (−)-119.
To improve the efficiency of this reaction sequence, we reasoned that the sterically less demanding TES enol ether might be a better oxidation substrate compared to (−)-116. We therefore prepared TES enol ether (+)-120 (Scheme 35), via conjugate addition of the mixed higher-order cuprate [i.e., R(2-Th)Cu(CN)Li2]83 derived from vinyl bromide (−)-14 and freshly prepared (2-Th)Cu(CN)Li to dihydropyranone (−)-13; in situ capture of the enolate with triethylsilyl chloride (TESCl) furnished the somewhat hydrolytically unstable TES enol ether (−)-120 in 77% yield. Purification of (+)-120 required activity (III) basic alumina to prevent TES hydrolysis.
Scheme 35.

With (+)-120 in hand, we installed the requisite C(10) oxygen exploiting the sterically less demanding TES enol ether (Scheme 35). Optimal conditions entailed m-CPBA buffered with NaHCO3 in CH2Cl2; (−)-121 was obtained in 54% yield as a single stereoisomer. Additional screening of the Davis oxaziridines84 and magnesium bis(monoperoxyphthalate) (MMPP)85 as oxidants proved unrewarding. Utilization of TES enol ether (+)-120 in place of the TBS ether required the subsequent conversion of TES ether (−)-121 to the TBS ether (−)-118, which was achieved in two-steps (95% yield), that did not require chromatographic purification.
The requisite C(11)–C(12) unsaturation was then installed via kinetic enolate formation (LDA, THF/HMPA), followed in turn by treatment with Comins reagent [N-(5-chloro-2-pyridyl)triflimide],86 and reduction employing catalytic Pd(PPh3)4 with Et3SiH as the hydride source (Scheme 36).87 The observed nOe enhancements between the C(9) methine and the C(14) methylene protons in (−)-122 confirmed both that the C(9) and C(13) substituents remained trans and that the C(13) center had not undergone epimerization via a retro-Michael pathway upon kinetic enolization.
Scheme 36.

Oxidative removal of the PMB group in (−)-122, followed by sequential oxidations employing Dess-Martin43 and Pinnick conditions,88 and in turn protection of the resulting carboxylic acid as the tert-butyl ester employing N,N′-diisopropyl-O-tert-butylisourea (123),89 led to (−)-124 in 69% yield over the four steps (Scheme 36). Chemoselective removal of the primary TBS group was then achieved with HF•py to furnish the primary alcohol (−)-125 in 77% yield. Notably, allowing the reaction to proceed to higher conversions led to hydrolysis of the C(10) TBS ether. Dess-Martin oxidation88 then completed construction of dihydropyran fragment (−)-7. Overall, advanced fragment (−)-7 was prepared in 19 steps (longest linear sequence) and in 8% overall yield from commercially available starting materials.12b
2.8. Advanced Fragment Union: Unexpected Challenges
Having secured ample quantities of the three major subtargets (−)-5, (−)-6, and (−)-7, we turned to their union via the planned consecutive Julia–Kociénski olefinations.90 Metallation of sulfone (−)-6 with LiHMDS, followed by addition of aldehyde (−)-5 in DMF/HMPA (3:1)91 provided exclusively the desired E-isomer (−)-126, as verified by the coupling constant of H(30) (J = 15.2 Hz, Entry 1, Table 3). This transformation, although proceeding in low yield (ca. 11–24%), permitted substantial recovery of the coupling partners. Utilizing KHMDS in DME, a set of conditions traditionally employed for trans alkene production,91 led to low E/Z selectivity (2:1) with higher conversion (54%, Entry 3). After a variety of different base/solvent combinations were explored (Table 3), best results were achieved with t-BuLi in combination with polar aprotic solvents (DMF/HMPA) to furnish (−)-126 as a single E isomer in 39% yield, when run on a 300 mg scale (Entry 5). With several recycles an overall yield of 65% could be achieved.
Table 3.
Olefination Conditions to Afford (−)-126
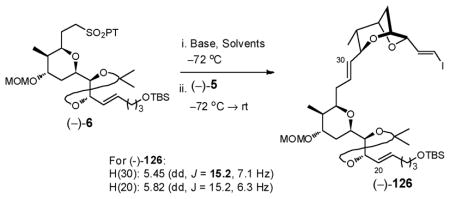 | ||||||
|---|---|---|---|---|---|---|
| Entry | Base | Solvents | Yield | E/Z Ratio | Recov’d Sulfone | Recov’d Aldehyde |
| 1 | LiHMDS | DMF/HMPA (3:1) | 24% | E-only | 63% | 36% |
| 2 | NaHMDS | DME/HMPA (3:1) | 40% | 3.6:1 | 39% | 23% |
| 3 | KHMDS | DME | 54% | 2.0:1 | 0% | 0% |
| 4 | LDA | DMF/HMPA (3:1) | 11% | E-only | 38% | 42% |
| 5 | t-BuLi | DMF/HMPA (3:1) | 39% | E-only | 39% | 13% |
Turning to the second Julia–Kociénski union (Scheme 37), removal of the TBS group in (−)-126 was achieved with Et3N•3HF to provide alcohol (−)-127, which in turn was subjected to Mitsunobu coupling with phenyltetrazole-thiol (PTSH) followed by molybdenum (VI)-catalyzed oxidation to deliver the PT-sulfone (−)-128. The Julia–Kociénski union again proved challenging. In this case, the E/Z selectivity was reversed, with KHMDS yielding the best E-selectivity in DME, with no selectivity observed with LiHMDS in DMF/HMPA. Low conversions were observed in both cases. Validation of the E-configuration at C(15)–C(16) also proved less than straightforward, as the H(15) and H(16) vinyl protons shared identical chemical shifts at 5.54 ppm. Recourse was therefore made to the method employed by Höfle and co-workers during their elucidation of (+)-sorangicin A. They assigned the E-configuration of the C(15)–C(16) olefin based on the 13C shift of the C(17) carbon at 33.4 ppm, which was in line with theoretical calculations of 33 ppm.92 In our case, the chemical shift of the C(17) carbon was 32.7 ppm, supporting the trans configuration.
Scheme 37.

We reasoned the observed low efficiency for both Julia–Kociénski unions resides in the nature of the THP-containing sulfones [i.e., (−)-6 and (−)-128]. The combination of difficult initial deprotonations and the presumed resultant intramolecular coordination of the sulfone anions in particular impeded efficient conversion. Reversal of the Julia–Kociénski anion and aldehyde coupling partners appeared as a viable option. For the first union however such a reversal would not be without risk, owing to potential β-elimination of the sulfone anion derived from bicycle (−)-5. Reversal of coupling partners for the second Julia–Kociénski union appeared more feasible (Scheme 38).
Scheme 38.

Towards this end, conversion of alcohol (−)-125 to PT-sulfone (+)-130 was achieved in two steps, and in high overall yield (Scheme 38). More important, use of KHMDS in DME for the Julia–Kociénski olefination with aldehyde (−)-131, derived from (−)-127, provided the coupled product (−)-129 in high yield (79%) with complete E-selectivity. With the second Julia–Kociénski olefination efficiently achieved, the three advanced fragments had thus been united. For installation of the Z,Z,E trienoate moiety, removal of the silyl group from the secondary TBS ether in (−)-129 was next readily accomplished, employing solid TBAF•3H2O, to furnish alcohol (−)-132.
2.9. The Sorangicin A Trienoate: A Model Study
Early on Höfle and co-workers demonstrated that the instability of (+)-sorangicin A was a direct result of the resident trienoate system.5 In particular, a series of products having undergone isomerization of the trienoate unit were isolated and identified. We of course fully recognized the pending challenge of the sensitive trienoate fragment. Initially we planned to mask the extended unsaturation as a dienyne; model studies thus appeared prudent.
To this end, Stille coupling93 between vinyl iodide (−)-50 and enyne 813 proceeded smoothly to furnish dienyne (−)-133 in 60% yield (Scheme 39). Subsequent semihydrogenation with 5% Pd/CaCO3, poisoned with Pb, in the presence of quinoline, as expected furnished trienoate 134, assigned based on 1H NMR of an unpurified sample. Unfortunately all attempts to remove the excess quinoline, either with NaHSO4 (1M) or by SiO2 chromatography, led to rapid isomerization. We therefore abandoned the dienyne tactic and opted to install the C(37)–C(43) fragment with the correct Z,Z,E trienoate configuration onto (−)-132, with a view to increase convergency.
Scheme 39.

With this idea in mind, we turned to the use of known stannyl dienoate 13594 bearing the desired Z,Z-configuration, which proved to be a stable compound under standard laboratory conditions, due to the electropositive tin stabilizing the electron deficient system as a σ-donor. In a second model study, Stille union between vinyl iodide (−)-70 and (Z,Z)-135, employing PdCl2(PhCN)2 (5 mol%) proceeded well in terms of yield (96%), albeit a geometric mixture of isomers was obtained, which proved difficult to separate (Entry 1, Table 4). Addition of 1.5 equivalent of Ph2PO2NBu4, an additive generally employed as a tin scavenger to improve coupling efficiency,95 did not improve the situation (Entry 2). However, when an excess of Ph2PO2NBu4 (6 equiv) was employed, trienoate (+)-136 was obtained as a single Z,Z,E-isomer in high yield (Entry 3). The requisite Z,Z,E-geometry of the extended unsaturation system was assigned based on coupling constants and nOe correlations. Presumably, Ph2PO2NBu4 inhibits Z/E isomerization by either arresting formation of detrimental iodide side products, or by accelerating dissociation of the Pd catalyst after reductive elimination. Of interest, trienoate (+)-136 proved to be stable for days in CDCl3, but quickly decomposed in the neat state.
Table 4.
Model Stille coupling to afford (+)-136
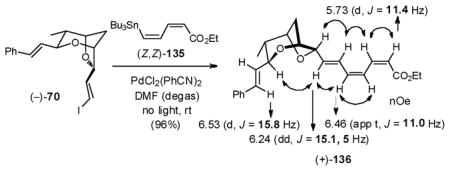 | ||
|---|---|---|
| Entry | Additive | Geometric Isomers |
| 1 | --- | mixture |
| 2 | Ph2PO2NBu4 (1.5 equiv) | mixture |
| 3 | Ph2PO2NBu4 (6 equiv) | pure Z,Z,E isomer |
2.10. Elaboration of the Complete Carbon Skeleton of (+)-Sorangicin A
Having succeeded in introducing the trienoate fragment in a simple model, we turned to the construction of the full carbon skeleton (Scheme 40). Pleasingly, the Z,Z,E-triene could be introduced in a similar fashion as the model system, albeit an even larger excess of Ph2PO2NBu4 (ca. 12 equiv) was required to furnish (+)-137 as a single Z,Z,E-isomer in 87% yield. Hydrolysis of (+)-137 was then achieved with LiOH in aqueous THF to provide acid 138 in 70% yield. Although the Z,Z,E-configuration was preserved, saponification proceeded quite slowly, with some starting material recovered even after 4 days. To improve the hydrolysis, the methyl ester, dienoate (Z,Z)-139, was prepared in analogous fashion to (Z,Z)-135 and coupled with vinyl iodide (−)-132; again the coupling proved excellent (88%). The hydrolysis now proceeded to completion in 1.5 days to furnish 138.96
Scheme 40.

2.11. Macrocyclization
With the full carbon skeleton of (+)-sorangicin A assembled, attention shifted to the critical macrocyclization, a task considered nontrivial both due to the hindered C(10) 2°-hydroxyl and the labile trienoacid (Scheme 41). We first selected the conditions of Mukaiyama, as modified by Evans,97 given both the convenience of this protocol and the lack of acidic or thermal conditions commonly employed in alternative macrocyclizations that might destroy the sensitive triene. Towards this end, treatment of seco-acid 138 with Mukaiyama reagent 141 and NaHCO3 in CH2Cl2 indeed furnished macrolactone 142, albeit contaminated with minor amounts of other geometric isomers that proved difficult to remove. Recalling that the Yonemitsu modification of the Yamaguchi conditions,98 involving direct introduction of DMAP at room temperature without preformation of the mixed anhydride, had successfully been employed by the Curran group in their total synthesis of (−)-dictyostatin possessing a Z,E-dienoacid,99 we explored this tactic. Unfortunately, these conditions led to significant isomerization. We reasoned that the formation of geometric isomers might be due to a reversible Michael-type addition of DMAP to the activated trienoacid during the cyclization event. The iodide present in Mukaiyama salt 141 held a similar risk. Another risk associated with activator 141 was halogen exchange leading to 143, a reagent known to be inert toward carboxylic acid activation.100
Scheme 41.
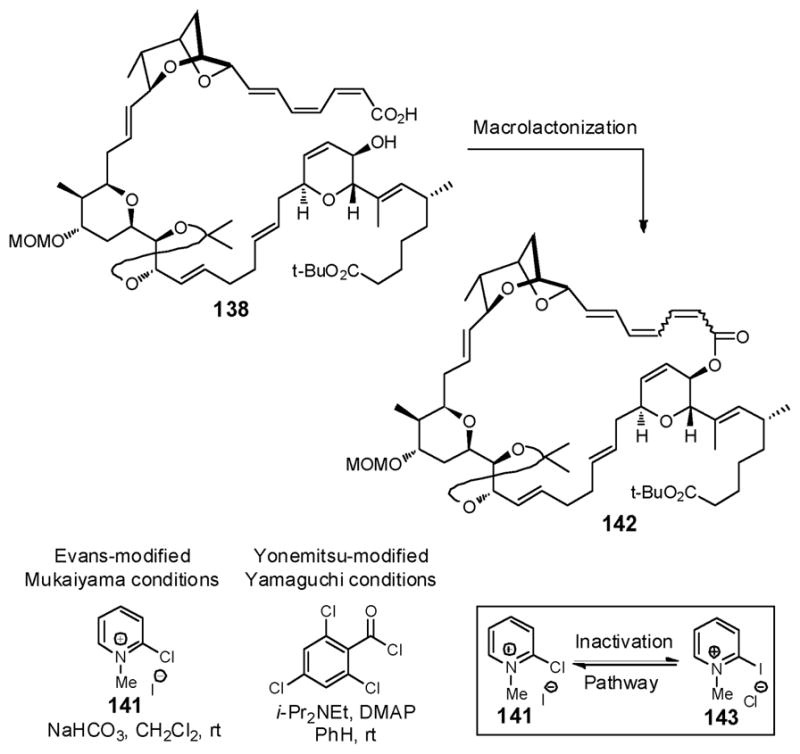
To address these issues, the modified Mukaiyama reagent 144 was adopted, which possesses a non-nucleophilic counterion, tetrafluoroborate, envisioned to mitigate both isomerization and inactivation pathways.101 Pleasingly, macrolide 142 was obtained in excellent yield (80%), with minimum isomerization (<10%) under dilute conditions (c≤1×10−3 M), employing 144 (Scheme 42). Moreover, the Z,Z,E-triene configuration was retained, as confirmed from the observed NMR coupling constants (Scheme 42). In this case, the larger 1-ethyl group in 144, may also contribute to the success, by suppressing decomposition of the pyridinium salt, a result of attack on the 1-alkyl group, while at the same time not being too sterically encumbered to hinder cyclization upon acid activation.
Scheme 42.

2.12. Global Deprotection: A Challenging Transformation
All that remained to complete the total synthesis of (+)-sorangicin A (1) was to define suitable conditions to remove the three acid labile protecting groups: the t-butyl ester, the acetonide, and the MOM ether group, while maintaining the delicate macrocycle. This proved to be a daunting task, due to the proclivity of the macrolide to undergo isomerization of the (Z,Z,E)-trienoate. As noted in the introduction, Höfle and coworkers demonstrated that natural (+)-sorangicin A (1) could be protected efficiently as acetonide 145 with TFA in acetone (ca. 98%), and that deprotection could be achieved in 70% yield employing TFA in aqueous THF at 85 °C (Scheme 43).14 Indeed, exploiting this protocol they prepared an extensive library of sorangicin analogues. The efficiency of deprotection however was highly substrate-dependent, ranging from 20% to 50%.
Scheme 43.

With this in mind, treatment of the fully protected macrolide 142 with TFA in aqueous THF, first at room temperature, led to acetonide removal, a process that was accelerated by silica gel (Scheme 44). Additional heating at 85 °C did not lead to further deprotection; only decomposition occurred.
Scheme 44.

Encouraged by our ability to remove the acetonide, we initiated a series of deprotection studies on the more readily available, advanced fragments (+)-108 and (−)-125 to preserve the valuable macrolide 142. These efforts demonstrated that aqueous TFA/THF conditions would remove the acetonide in (+)-108 at 45 °C, and when the temperature was raised to 85 °C the MOM group (Scheme 45A). Liberation of the acid from t-butyl ester (−)-125 (Scheme 45B) however could only be achieved in 42% yield at 85 °C, indicating that a more effective protocol had to be devised to hydrolyze the t-butyl ester. Anhydrous TFA in CH2Cl2 was not an option, as decomposition of the delicate trienoate linkage proved competitive. Turning to Lewis acids, B-bromocatecholborane102 removed both the MOM and acetonide groups in (+)-108, but removal of the t-butyl group of the ester remained sluggish (Scheme 45C and D).
Scheme 45.

Subsequent screening revealed that treatment of (−)-125 with TMSOTf in 2,6-lutidine liberated the acid with good overall efficiency, after an acidic workup (1M KHSO4) (88%, Scheme 46). These conditions also cleanly removed the MOM group in (+)-108. With these observations in hand, we attempted to remove both the MOM and t-butyl groups simultaneously, employing late stage intermediate 142. Only decomposition resulted!
Scheme 46.

We reasoned that TMSOTf was sufficiently powerful to liberate the acid from the t-butyl ester, but too reactive to maintain the delicate trienoate. In the end, we discovered that TBSOTf, a bulkier reagent, buffered with 2,6-lutidine, fulfilled both the requirements: selective removal of the t-butyl group without compromising the (Z,Z,E)-trienoate linkage to furnish the now TBS protected ester 153, after an acid workup (0.2 N HCl) (Scheme 47). Notably, the MOM and acetonide protecting groups in 142 were not affected by these conditions. Treatment of TBS ester 153 with B-bromocatecholborane at low temperature then furnished the fully deprotected macrolide 154.
Scheme 47.

2.13. The Structure of (+)-Sorangicin A
With the first synthetic sample of the fully deprotected macrolide 154 in hand, careful comparison of the TLC of an authentic sample of (+)-sorangicin A (1), kindly provided by Höfle, with a sample of the synthetic macrolide was not promising, although high-resolution mass spectroscopy confirmed the correct molecular formula. The 500 MHz 1H NMR spectrum of 154, albeit not pure, clearly indicated that the trienoate remained intact during the two-step deprotection process.
Before committing additional, highly valuable advanced materials to provide access to pure 154 for detailed NMR analysis, we compared the 500 MHz 1H NMR spectrum of natural (+)-sorangicin A in CD3OD with that of the synthetic protected pure macrolide 142, having assigned the majority of the resonances in the latter via extensive 2D NMR experiments (Figure 6). This analysis revealed that: (a) the trienoate regions matched closely; (b) the C(21)–C(23) segments did not resemble each other, an understandable observation given the local conformational effects arising from the acetonide in 142; and (c) significant differences in chemical shifts and line shapes were observed for three vinyl protons: H(7), H(11) and H(12). We thereby suspected that the structure of the dihydropyran ring, and in particular the C(10) stereochemical center in 142 was not consistent with (+)-sorangicin A (1).
Fig 6.

Comparison of the 1H NMR data for natural 3 and synthetic 142
Given that single-crystal X-ray structure 1554 (Figure 7) had been employed by Höfle and coworkers to confirm their initial structure assignment of (+)-sorangicin A (1), an error involving the misassignment of backbone connectivity or relative stereochemistry was eliminated. Review of the original 1989 publication4 revealed that (+)-sorangicin A was depicted as 156 with S-C(10) configuration (Figure 7). However, in a later publication (1993),6 a publication that we had relied upon, Höfle and co-workers depicted (+)-sorangicin A as structure 3 with the C(10) R configuration. Clearly the R configuration at C(10) in synthetic 142 led to the discrepancies of the H(7), H(11) and H(12) resonances in our synthetic material.
Fig 7.

Comparison of ORTEP of (+)-sorangicin A with two reported structures
2.14. In-House X-Ray Analysis of (+)-Sorangicin A (1)
Before taking final action to complete the total synthesis of (+)-sorangicin A (1), we conducted an X-ray analysis of natural (+)-sorangicin A, kindly provided by Dr. Rolf Jansen. By collecting the X-ray intensity data at low temperature, 143 K, compared to the Höfle analysis at 293 K, we not only confirmed the S configuration at C(10), but also provided new information on the dynamic disorder identified by the Höfle team.4 In particular, the C(16)–C(17)–C(18) moiety was disordered by rotation of approximately 30° about the C(14)–C(15) and C(19)–C(20) bonds, which upon refinement of two contributing disorder fragments [C(16), C(17), C(18), and C(16′), C(17′), C(18′)] with 50% occupancies, gave rise to high resolution structures that confirmed the single bond nature of the C(17)–C(18) moiety, a matter of early concern in the initial structure analysis (Figure 8). The reader will recall that the high flexibility of the C(14)–C(20) region had been correlated with the superior adaptive ability of (+) sorangicin A (1) towards microbial mutation, leading to an advantageous cross-resistant profile over rifampicin (2), a discovery with clear implications vis-à-vis future drug design and development against fast evolving microbes.3
Fig 8.
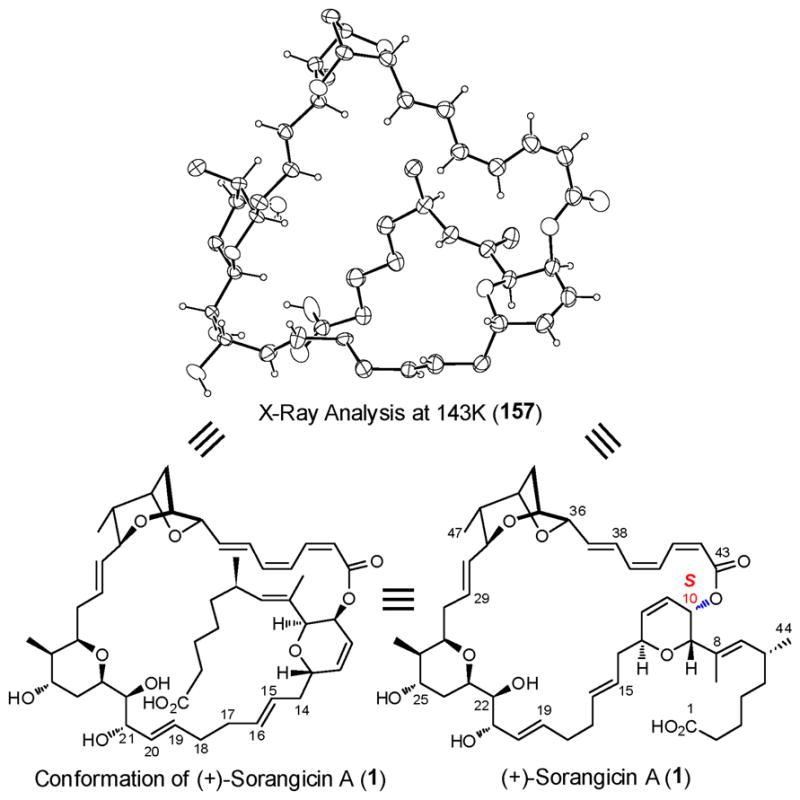
X-ray structure verification of (+)-sorangicin A
2.15. Inversion of the C(10) Stereogenicity: Access Advanced Intermediate 161
We began with global desilylation of (−)-124, followed by selective silylation of the primary hydroxyl group to furnish allylic alcohol (−)-158 in excellent yield (Scheme 48). Attempts to invert the C(10) center employing a variety of Mitsunobu conditions failed due to intervention of an SN2′ pathway, presumably due to the bulky nature of the C(1)–C(8) side chain. Recourse to a Ley oxidation103/Luche reduction104 sequence however quickly delivered the desired allylic alcohol (+)-159 as a single diastereomer. Pleasingly, the line shape of H(9) in the 1H
Scheme 48.

NMR of (+)-159 changed from a doublet (J = 7.1 Hz) as in (−)-158 to a singlet after inversion, closely resembling that of the natural product. Protecting group manipulations followed by introduction of the sulfone unit employing the conditions developed earlier (Scheme 38) completed construction of (+)-161. The eight step sequence proceeded in 46% yield.
2.16. Completion of the First Total Synthesis of (+)-Sorangicin A (1)
With the stereogenicity at C(10) now secure, Julia–Kociénski union between aldehyde (−)-131 and sulfone (+)-161 proceeded smoothly to furnish vinyl iodide (+)-162 after desilylation (Scheme 49). Additional (+)-162 was also available via an oxidation/reduction sequence employing advanced intermediate (−)-132. Conversion of (+)-162 to (+)-sorangicin A (1) now proved straight forward. Importantly the critical Mukaiyama macrolactonization was highly efficient (88%). Finally, somewhat more effective conditions to remove the MOM, acetonide, and TBS protecting group, employing 4N HCl in THF, led to synthetic (+)-sorangicin A (1). Interestingly, (+)-sorangicin A proved stable to strong aqueous protic acid in the absence of oxygen over 24 h. The overall yield for the five steps was 45%. Synthetic (+)-sorangicin A (1) proved to be indistinguishable from an authentic sample of 1 by NMR, mass spectrometric, and chromatographic comparisons (three solvent systems).12d Moreover, the chiroptic properties { : +56 (c 0.06, MeOH); lit.4 : +60.9 (c 0.7, MeOH)} confirmed that the absolute configuration of (+)-sorangicin A is as depicted in 1, consistent with the X-ray analysis achieved by Höfle and co-workers.4
Scheme 49.

3. Conclusion
The first and to date only total synthesis of (+)-sorangicin A (1) has been achieved. The longest linear sequence was 30 steps that proceeded in 3.2% overall yield. The enantioselective route features high convergency, as well as a number of key features and/or tactics, including: a first- and second-generation synthesis of the dioxabicyclo[3.2.1]octane core, effective constructions of the advanced di- and tetrahydropyran ring systems, consecutive Julia–Kociénski olefinations to unite three advanced fragments with high E-stereoselectivity, a modified Stille union to introduce the sensitive Z,Z,E triene functionality, a highly efficient Mukaiyama macrolactonization, and carefully defined Lewis and protic acid-promoted deprotection protocols to achieve global deprotection in the final stage of the synthetic venture.
Supplementary Material
Acknowledgments
This article is dedicated to Professor Gilbert Stork (Columbia University), outstanding scientist, scholar and friend, on the occasion of his 90th birthday. Over nearly 70 years, Professor Stork and his group have transformed the field of Synthetic Organic Chemistry that now make possible total syntheses such as (+)-sorangicin A. Happy Birthday Gilbert!!! Support was provided by the National Institutes of Health (National Institute of General Medical Sciences) through Grant GM-29028. We also thank Drs. George Furst, Rakesh Kohli and Patrick Carroll (University of Pennsylvania) for assistance in obtaining NMR spectra, high resolution mass spectra, and X-ray crystallographic data, respectively. We also thank Dr. Rolf Jansen (Helmholtz Center for Infection Research, Braunschweig, Germany) for an authentic sample of (+)-sorangicin A.
Footnotes
Experimental procedures, crystallographic information files, complete spectroscopic and analytical data for new compounds can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Jansen R, Wray V, Irschik H, Reichenbach H, Höfle G. Tetrahedron Lett. 1985;26:6031. [Google Scholar]
- 2.Irschik H, Jansen R, Gerth K, Höfle G, Reichenbach H. J Antibiot. 1987;40:7. doi: 10.7164/antibiotics.40.7. [DOI] [PubMed] [Google Scholar]
- 3.Campbell EA, Pavlova O, Zenkin N, Leon F, Irschik H, Jansen R, Severinov K, Darst SA. The EMBO Journal. 2005;24:674. doi: 10.1038/sj.emboj.7600499. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansen R, Irschik H, Reichenbach H, Schomburg D, Wray V, Höfle G. Liebigs Ann Chem. 1989;111 [Google Scholar]
- 5.Jansen R, Irschik H, Reichenbach H, Wray V, Höfle G. Liebigs Ann Chem. 1989;213 [Google Scholar]
- 6.Schummer D, Irschik H, Höfle G. Liebigs Ann Chem. 1993;293 [Google Scholar]
- 7.Danek SC. PhD Thesis. Morken Group, University of North Carolina; Chapel Hill: 2003. [Google Scholar]
- 8.Schinzer D, Schulz C, Krug O. Synlett. 2004;15:2689. [Google Scholar]
- 9.Park SH, Lee HW. Bull Korean Chem Soc. 2008;29:1661. [Google Scholar]
- 10.(a) Srihari P, Kumaraswamy B, Yadav JS. Tetrahedron. 2009;65:6304. [Google Scholar]; (b) Mohapatra DK, Das PP, Pattanayak MR, Yadav JS. Chem Eur J. 2010;16:2072. doi: 10.1002/chem.200902999. [DOI] [PubMed] [Google Scholar]
- 11.(a) Crimmins MT, Haley MW. Org Lett. 2006;8:4223. doi: 10.1021/ol061339e. [DOI] [PubMed] [Google Scholar]; (b) Crimmins MT, Haley MW, O’Bryan EA. Org Lett ASAP. doi: 10.1021/ol201920j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Smith AB, III, Fox RJ. Org Lett. 2004;6:1477. doi: 10.1021/ol049644s. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Fox RJ, Vanecko JA. Org Lett. 2005;7:3099. doi: 10.1021/ol051119l. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, III, Dong S. Org Lett. 2009;11:1099. doi: 10.1021/ol802942j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith AB, III, Dong S, Brenneman JB, Fox RJ. J Am Chem Soc. 2009;131:12109. doi: 10.1021/ja906115a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rossi R, Bellina F, Catanese A, Mannina L, Valensin D. Tetrahedron. 2000;56:479. [Google Scholar]
- 14.Jansen R, Schummer D, Irschik H, Höfle G. Liebigs Ann Chem. 1990;975 [Google Scholar]
- 15.Ferrier RJ, Middleton S. Chem Rev. 1993;93:2779.Petasis NA, Lu SP. Tetrahedron Lett. 1996;37:141.Smith AB, III, Fox RJ, Razler TM. Acc Chem Res. 2008;41:675. doi: 10.1021/ar700234r.For examples exploiting the Petasis-Ferrier union/rearrangement tactic in natural product synthesis, see: Smith AB, III, Minbiole KP, Verhoest PR, Schelhaas M. J Am Chem Soc. 2001;123:10942. doi: 10.1021/ja011604l.Smith AB, III, Verhoest PR, Minbiole KP, Lim JJ. Org Lett. 1999;1:909. doi: 10.1021/ol990830l.Smith AB, III, Minbiole KP, Verhoest PR, Beauchamp TJ. Org Lett. 1999;1:913. doi: 10.1021/ol990829m.Smith AB, III, Sfouggatakis C, Gotchev DB, Shirakami S, Bauer D, Zhu W, Doughty VA. Org Lett. 2004;6:3637. doi: 10.1021/ol048418f.Smith AB, III, Safonov IG, Corbett RM. J Am Chem Soc. 2002;124:11102. doi: 10.1021/ja020635t.Smith AB, III, Safonov IG, Corbett RM. J Am Chem Soc. 2001;123:12426. doi: 10.1021/ja012220y.For more recent modifications, see: O’Neil KE, Kingree SV, Minbiole KPC. Org Lett. 2005;7:515. doi: 10.1021/ol047426t.
- 16.Redlich H, Bruns W, Francke W, Schuring V, Payne TL, Vite JP. Tetrahedron. 1987;2029 [Google Scholar]
- 17.Keck GE, Li X-Y, Krishnamurthy D. J Org Chem. 1995;60:5998. [Google Scholar]
- 18.Mukai C, Sugimoto Y-i, Ikeda Y, Hanaoka M. J Chem Soc Chem Commun. 1994;1161 [Google Scholar]
- 19.Nicholas KM. Acc Chem Res. 1987;20:207–214. [Google Scholar]
-
20.Upon generation of the activated complex 163, an anchimeric assisted SN2 epoxide ring-opening, via Co2(CO)6-alkyne complex, initially furnishes intermediate 164. See Schreiber SL, Sammakia T, Crowe WE. J Am Chem Soc. 1986;108:3128.Schreiber SL, Klimas MT, Sammakia T. J Am Chem Soc. 1987;109:5749.Intramolecular SN2 capture of cation 164 by the hydroxy group, from the same face as the epoxide, then leads to the Co2(CO)6-alkyne complex (165) with overall retention of stereochemistry. See: Mukai C, Sugimoto Y-i, Ikeda Y, Hanaoka M. Tetrahedron. 1998;54:823.
- 21.Seng F, Negishi E-I. Org Lett. 2001;3:719. doi: 10.1021/ol000384y.For related compounds and applications, see Negishi E-I, Alimardanov A, Xu C. Org Lett. 2000;2:65. doi: 10.1021/ol990336h.Negishi E-I, Hata M, Xu C. J Am Chem Soc. 2003;125:13636. doi: 10.1021/ja0304392.
- 22.For the preparation of the enantiomer of (+)-28, see Zheng W, DeMattei JA, Wu J–P, Duan JJ–W, Cook LR, Oinuma H, Kishi Y. J Am Chem Soc. 1996;118:7946.
- 23.(a) England P, Chun KH, Moran EJ, Armstrong RW. Tetrahedron Lett. 1990;31:2669. [Google Scholar]; (b) Ishihara J, Mikakawa J, Tsujimoto T, Murai A. Synlett. 1997:1417. [Google Scholar]; (c) Chênevert R, Dasser M. J Org Chem. 2000;65:4529. doi: 10.1021/jo991989e. [DOI] [PubMed] [Google Scholar]; (d) Keck GE, Wager CA, Wager TT, Savin KA, Covel JA, McLaws MD, Krishnamurthy D, Cee VJ. Angew Chem Int Ed. 2001;40:231. [PubMed] [Google Scholar]; (e) Piggott MJ, Wege D. Aust J Chem. 2003;56:691. [Google Scholar]; (f) Marshall JA, Schaaf GM. J Org Chem. 2003;68:7428. doi: 10.1021/jo0348930. [DOI] [PubMed] [Google Scholar]
- 24.Bailey S, Harnden MR, Jarvest RL, Parkin A, Boyd MR. J Med Chem. 1991;34:57. doi: 10.1021/jm00105a010. [DOI] [PubMed] [Google Scholar]
- 25.Veysoglu T, Mitscher LA, Swayze JK. Synthesis. 1980;807 [Google Scholar]
- 26.Kjell DP, Slattery BJ, Semo MJ. J Org Chem. 1999;64:5722. doi: 10.1021/jo990543v. [DOI] [PubMed] [Google Scholar]
- 27.(a) Brown HC, Bhat KS. J Am Chem Soc. 1986;108:293. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]; (b) Brown HC, Bhat KS. J Am Chem Soc. 1986;108:5919. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 28.Crispino GA, Jeong K-S, Kolb HC, Wang Z-M, Xu D, Sharpless KB. J Org Chem. 1993;58:3785.and references cited therein. Kolb HC, VanNiewenhze MS, Sharpless KB. Chem Rev. 1994;94:2483.
- 29.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092. [Google Scholar]
- 30.(a) Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945. [Google Scholar]; (b) Evans DA, Rieger DL, Gage JR. Tetrahedron Lett. 1990;31:7099. [Google Scholar]
- 31.Hicks DR, Fraser-Reid B. Synthesis. 1974;203 [Google Scholar]
- 32.Dounay AB, Florence GJ, Saito A, Forsyth CJ. Tetrahedron. 2002;58:1865. [Google Scholar]
- 33.Whereas vinyl bromide 24 has been reported to be a competent component in cross-coupling reactions, anions derived from 24 have not been described.
- 34.Although commercially available, 1,4-bis(trimethylsilyl)-1,3-butadiyne was prepared on large-scale according to Jones GE, Kendrick DA, Holmes AB. Organic Syntheses, Coll. 865:63. 52.For the initial preparation of 1,4-bis(trimethylsilyl)-1,3-butadiyne, see Walton DRM, Waugh F. J Organometallic Chem. 1972;37:45.See Supporting Information for details.
- 35.(a) Bohlmann F, Enkelmann R, Plettner W. Chem Ber. 1964;97:2118. [Google Scholar]; (b) Doolittle RE. Synthesis. 1984:730. [Google Scholar]
- 36.(a) While Holmes and co-workers reported the generation of lithium trimethylsilylbutadiyne from 1,4-bis(trimethylsilyl)-1,3-butadiyne (33), followed by reaction with a wide range of alkyl halides and carbonyl compounds, examples of epoxide ring-opening by lithium trimethylsilylbutadiyne have not been recorded Holmes AB, Jennings-Whie CLD, Schulthess AH. J Chem Soc Chem Comm. 1979:840.Holmes AB, Jones GE. Tetrahedron Lett. 1980;21:3111.
- 37.Crousse B, Alami M, Linstrumelle G. Synlett. 1997;992 [Google Scholar]
- 38.Studies on the LiAlH4 reduction of propargylic alcohols and 4-substituted-3-butyne-1-ols suggest that the mechanism may be more complicated. See Molloy BB, Hauser KL. J Chem Soc Chem Commun. 1968:1017.Borden WT. J Am Chem Soc. 1970;92:4898.Grant B, Djerassi C. J Org Chem. 1974;39:968.Kang MJ, Jang J-S, Lee S-G. Tetrahedron Lett. 1995;36:8829.
- 39.Wang Z-X, Tu Y, Frohn M, Zhang J-R, Shi Y. J Am Chem Soc. 1997;119:11224.Wang Z-X, Shi Y. J Org Chem. 1998;63:3099. doi: 10.1021/jo980604+.For epoxidation of enyne systems, see Wang Z-X, Cao G-A, Shi Y. J Org Chem. 1999;64:7646.For a recent review, see Shi Y. Acc Chem Res. 2004;37:488. doi: 10.1021/ar030063x.
- 40.The reaction was also executed in the extremely polar LiClO4•OEt2 solvent system developed by the Dailey and Grieco groups, with the idea of increasing carbocationic character in the ring-opening transition state. Although the reaction rate was enhanced dramatically, the ratio of (−)-21 to (−)-45 remained nearly 1:1. See: Grieco PA, Nunes JJ, Gaul MD. J Am Chem Soc. 1990;112:4595.Grieco PA. Organic Chemistry in Lithium Perchlorate/Diethyl Ether. In: Kisakürek V, editor. Organic Chemistry: Its Language and Its State of the Art. VCH; Basel: 1993. p. 133.Forman MA, Dailey WP. J Am Chem Soc. 1991;113:2761.
- 41.It is important to note that in the conversion of (+)-49 to (−)-46, degassing of all solvents (i.e., CH2Cl2 and MeOH) was imperative to achieve high yields reproducibly, indicating the high air sensitivity of the reaction.
- 42.Burchat AF, Chong JM, Nielson N. J Organometallic Chem. 1997;542:281. [Google Scholar]
- 43.Dess DB, Martin JC. J Org Chem. 1983;48:4155. [Google Scholar]
- 44.For an example of an alkyne possessing both α- and β-ether substituents proceeding with excellent regioselectivity to furnish the β-isomer, see Mukai C, Miyakoshi N, Hanaoka M. J Org Chem. 2001;66:5875. doi: 10.1021/jo0104532.
- 45.Although reproducible, the synthetic yield and enantioselectivity for (−)-13 was highly dependent on the degree of molecular sieve activation based on Keck’s protocol as in ref. 17.
- 46.Danishefsky S, Kitahara T. J Am Chem Soc. 1974;96:7807. [Google Scholar]
- 47.Cardani S, Toma CD, Gennari C, Scolastico C. Tetrahedron. 1992;48:5557. [Google Scholar]
- 48.(a) Dossetter AG, Jamison TF, Jacobsen EN. Angew Chem Int Ed. 1999;38:2398. doi: 10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; (b) Gademann K, Chavez DE, Jacobsen EN. Angew Chem Int Ed. 2002;41:3059. doi: 10.1002/1521-3773(20020816)41:16<3059::AID-ANIE3059>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 49.Paterson I, Steven A, Luckhurst CA. Org Biomol Chem. 2004;2:3026. doi: 10.1039/B407240E. [DOI] [PubMed] [Google Scholar]
- 50.(a) Alexakis A, Vastra J, Mangeney P. Tetrahedron Lett. 1997;38:7745. [Google Scholar]; (b) Agapiou K, Cauble DF, Krische MJ. J Am Chem Soc. 2004;126:4528. doi: 10.1021/ja030603l. [DOI] [PubMed] [Google Scholar]
- 51.Hutchinson DK, Fuchs PL. J Am Chem Soc. 1987;109:4930. [Google Scholar]
- 52.Suzuki M, Morita Y, Koyano H, Koga M, Noyori R. Tetrahedron. 1990;46:4809. [Google Scholar]
- 53.Fürstner A, Grela K, Mathes C, Lehmann CW. J Am Chem Soc. 2000;122:11799. [Google Scholar]
- 54.Joly GD, Jacobsen EN. Org Lett. 2002;4:1795. doi: 10.1021/ol0258785. [DOI] [PubMed] [Google Scholar]
- 55.Aldehyde (−)-58, although commercially available, was prepared in two steps from L-gulonic acid γ-lactone; see Hubschwerlen C, Specklin JL, Higelin J. Organic Syntheses. 1995;72:1.
- 56.That is, during the enolate alkylation, which is a very slow process, the Zn-methyl group of 60 is sufficiently basic, in the presence of excess HMPA, to deprotonate (+)-61; the resulting secondary kinetic enolate then reacts with MeI to yield side product (+)-62, see: Rathgeb X, March S, Alexakis A. J Org Chem. 2006;71:5737. doi: 10.1021/jo060814j.Lowering the alkylation temperature from −40 °C to −60 °C led to a slower reaction and in turn an increase of (+)-62 (38%). Higher temperatures (ca. −20 °C) however had a beneficial effect on the yield of (+)-61, but did not rectify the problem completely. Application of the more active alkylation reagent MeOTf proved no better.
- 57.Kojima N, Maezaki N, Tominaga H, Asai M, Yanai M, Tanaka T. Chem Eur J. 2003;9:4980. doi: 10.1002/chem.200305185. [DOI] [PubMed] [Google Scholar]
- 58.Parikh J, Doering W. J Am Chem Soc. 1967;89:5505. [Google Scholar]
- 59.Takai K, Nitta K, Utimoto K. J Am Chem Soc. 1986;108:7408. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]
- 60.Kende AS, DeVita RJ. Tetrahedron Lett. 1990;31:307. [Google Scholar]
- 61.Evans DA, Black WC. J Am Chem Soc. 1993;115:4497. [Google Scholar]
- 62.Seebach D, Imwinkelried R, Stucky G. Helv Chim Acta. 1987;70:448. [Google Scholar]
- 63.Petasis NA, Bzowej EI. J Am Chem Soc. 1990;112:6392. [Google Scholar]
- 64.Although dioxanone formation and methylenation proceeded smoothly, attempts to perform the rearrangement with an acetonide at C(21,22) led to decomposition.
- 65.Cossey KN, Funk RL. J Am Chem Soc. 2004;126:12216. doi: 10.1021/ja046940r. [DOI] [PubMed] [Google Scholar]
- 66.This highly selective aldol strategy has been successfully applied by the Crimmins group in the synthesis of (+)-spongistatin 1, in which the α and β oxygen substituents of a methyl ketone acted in synergy to deliver a single diastereomeric aldol adduct, see: Crimmins MT, Katz JD, McAtee LC, Tabet EA, Kirincich SJ. Org Lett. 2001;3:949. doi: 10.1021/ol015652m.
- 67.Marshall JA, Seletsky JA, Boris M, Luke GP. J Org Chem. 1994;59:3413. [Google Scholar]
- 68.(a) Miwa K, Aoyama T, Shioiri T. Synlett. 1994:107. [Google Scholar]; (b) Myers AG, Goldberg SD. Angew Chem, Int Ed. 2000;29:2732. doi: 10.1002/1521-3773(20000804)39:15<2732::aid-anie2732>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 69.Hart DH, Blackburn TF, Schwartz J. J Am Chem Soc. 1975;97:679. [Google Scholar]
- 70.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 71.Alkyl boronate 76 was prepared from 1,3-propanediol; see Supporting Information
- 72.Shekhani MS, Khan KM, Mahwood K, Shah PM, Malik S. Tetrahedron Lett. 1990;31:1669. [Google Scholar]
- 73.Rauhala V, Nevalainen M, Koskinen AMP. Tetrahedron. 2004;60:9199. [Google Scholar]
- 74.Paterson I, Coster MJ, Chen DY-K, Aceña JL, Bach J, Keown LE, Trieselmann T. Org Biomol Chem. 2005;3:2420. doi: 10.1039/b504149j. [DOI] [PubMed] [Google Scholar]
- 75.One possible explanation for the observed decrease in C(25) OH elimination upon changing the reaction solvent from CH2Cl2 to CH3CN is that the Lewis basic CH3CN attenuated the Lewis acidity of the BF3•OEt2.
- 76.Schultz HS, Freyermuth HB, Buc SR. J Org Chem. 1963;28:1140. [Google Scholar]
- 77.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J Am Chem Soc. 1997;119:6496. [Google Scholar]
- 78.The stereochemical outcome for the Myers alkylation and reduction protocol was confirmed via reduction of aldehyde (−)-114 to the corresponding alcohol, removal of the PMB ether and conversion to the known bis-Mosher esters, see Kubota T, Tsuda M, Kobayashi J. J Org Chem. 2002;67:1651. doi: 10.1021/jo016326n. and references cited therein.
- 79.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;36:3769. [Google Scholar]
- 80.(a) Yang DY, Wong M-K, Yip Y-C. J Org Chem. 1995;60:3887. [Google Scholar]; (b) Thompson CF, Jamison TF, Jacobsen EN. J Am Chem Soc. 2001;123:9974. doi: 10.1021/ja016615t. [DOI] [PubMed] [Google Scholar]
- 81.(a) McCormick JP, Tomasik W, Johnson MW. Tetrahedron Lett. 1981;22:607. [Google Scholar]; (b) Boeckman RK, Jr, Starrett JE, Jr, Nickell DG, Sum P-E. J Am Chem Soc. 1986;108:5549. [Google Scholar]
- 82.This assignment is based on the precedent that 236-trisubstituted tetrahydropyranones approximate chair conformations see: Goodwin TE, Crowder CM, White RB, Swanson JS. J Org Chem. 1983;48:376.
- 83.(a) Lipshutz BH, Kozlowski JA, Parker DA, Nguyen SL, McCarthy KE. J Organometallic Chem. 1985;285:437. [Google Scholar]; (b) Lipshutz BH, Koerner M, Parker DA. Tetrahedron Lett. 1987;28:945. [Google Scholar]
- 84.Davis FA, Sheppard AC. J Org Chem. 1987;52:955.Davis FA, Lal SG. J Org Chem. 1988;53:5004.Davis FA, Chattopadhyay S, Towson JC, Lal S, Reddy T. J Org Chem. 1988;53:2087.and Vishwakarma LC, Stringer OD, Davis FA. Organic Syntheses, Coll. 866:546. 203.
- 85.Brougham P, Cooper MS, Cummerson DA, Heaney H, Thompson N. Synthesis. 1987;1015 [Google Scholar]
- 86.(a) Comins DL, Dehghani A. Tetrahedron Lett. 1992;33:6299. [Google Scholar]; (b) Comins DL, Dehghani A, Foti CJ, Joseph SP. Organic Syntheses, Coll. 974:165. 77. [Google Scholar]
- 87.Scott WJ, Stille JK. J Am Chem Soc. 1986;108:3033. doi: 10.1021/ja00263a015. [DOI] [PubMed] [Google Scholar]
- 88.Bal BS, Childers WE, Jr, Pinnick HW. Tetrahedron. 1981;37:2091. [Google Scholar]
- 89.For a review on isourea reagent see Mathias LJ. Synthesis. 1979;561
- 90.Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998;26 [Google Scholar]
- 91.Liu P, Jacobsen EN. J Am Chem Soc. 2001;123:10772. doi: 10.1021/ja016893s. [DOI] [PubMed] [Google Scholar]
- 92.In the structural elucidation paper of sorangicin A by Höfle et al., ref. 1, based on assumption of additivity of effects and the known carbon chemical shifts of hexa-1,5-diene, E- and Z-hex-3-ene and but-1-ene, the authors calculated the carbon chemical shifts of C(17) in the E- and Z-configuration to be ca. 33 and 27 ppm, respectively.
- 93.(a) Milstein D, Stille JK. J Am Chem Soc. 1978;100:3636. [Google Scholar]; (b) Stille JK. Angew Chem Int Ed. 1986;25:508. [Google Scholar]
- 94.Franci X, Martina SLX, McGrady JE, Webb MR, Donald C, Taylor RJK. Tetrahedron Lett. 2003;44:7735. [Google Scholar]
- 95.Srogl J, Allred GD, Liebeskind LS. J Am Chem Soc. 1997;119:12376. [Google Scholar]
- 96.Acid 138 proved to be extremely unstable, and therefore was employed in the next step without full characterization.
- 97.(a) Mukaiyama T, Usui M, Saigo K. Chem Lett. 1976:49. [Google Scholar]; (b) Evans DA, Starr JT. J Am Chem Soc. 2003;125:13531. doi: 10.1021/ja037643+. [DOI] [PubMed] [Google Scholar]
- 98.(a) Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989. [Google Scholar]; (b) Hikota M, Sakurai Y, Horita K, Yonemitsu O. Tetrahedron Lett. 1990;31:6367. [Google Scholar]
- 99.Shin Y, Fournier J-H, Brückner A, Madiraju C, Balachandran R, Raccor BS, Edler MC, Hamel E, Sikorski RP, Vogt A, Day BW, Curran DP. Tetrahedron. 2007;63:8537. doi: 10.1016/j.tet.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.(a) Oh SH, Cortez GS, Romo D. J Org Chem. 2005;70:2835. doi: 10.1021/jo050024u. [DOI] [PubMed] [Google Scholar]; (b) Bradlow HL, Vanderwerf CA. J Org Chem. 1951;16:1143. [Google Scholar]
- 101.Xu JC, Li P. Tetrahedron. 2000;56:8119.For advantages of using modified Mukaiyama reagents, see: Folmer JJ, Acero C, Thai DL, Rapoport H. J Org Chem. 1998;63:8170.
- 102.Boeckman RK, Jr, Potenza JC. Tetrahedron Lett. 1985;26:1411. [Google Scholar]
- 103.Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994;639 [Google Scholar]
- 104.Luche JL. J Am Chem Soc. 1978;100:2226. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.