

Abstract
Latent transforming growth factor beta (TGF-β) binding proteins (LTBPs) are large extracellular glycoproteins structurally similar to fibrillins. They perform intricate and important roles in the extracellular matrix (ECM) and perturbations of their function manifest as a wide range of diseases. LTBPs are major regulators of TGF-β bioavailability and action. In addition, LTBPs interact with other ECM proteins- from cytokines to large multi-factorial aggregates like microfibrils and elastic fibers, affecting their genesis, structure, and performance. In the present paper, we review recent advancements in the field and relate the complex roles of LTBP in development and homeostasis.
Introduction
The extracellular matrix (ECM) represents a dynamic and multifarious environment that provides cells with support, anchorage, and instructive signals that guide essential cellular functions such as proliferation, migration, differentiation and death. The scaffold of the vibrant extracellular space is made primarily of fibronectin, collagenous and elastic fibers, and microfibrils, which provide tissues with resilience and elasticity and also serve as docking platforms for soluble factors involved in inter-cellular communication. Latent transforming growth factor beta (TGF-β) binding proteins (LTBPs), components of microfibrils, represent a family of matrix proteins that exert important and complex functions in the extracellular space. Below, we discuss their characteristics, functions, as well as findings that might direct future research.
Latent Transforming Growth Factor beta (TGF-β) Binding Proteins (LTBPs)
The LTBPs are large secreted glycoproteins structurally related to fibrillins. The first described LTBP (LTBP1) was identified in platelet releasate as a component of a high molecular weight complex, which also included a dimer of transforming growth factor beta (TGF-β) and a dimer of its propeptide [Miyazono et al., 1988] (Figure 1). TFG-β is a pluripotent and omnipresent growth factor. Cells secrete this cytokine as a biologically inactive aggregate comprised of a disulfide linked TFG-β dimer non-covalently associated with its propeptide homodimer. The interaction of TFG-β with its propeptide renders the cytokine inactive, hence the propeptide is dubbed the latency-associated peptide (LAP) and the complex between TFG-β and LAP is named the small latent complex (SLC). LTBP1 associates with SLC, forming a tripartite complex known as the large latent complex (LLC) (Figure 2). Since the characterization of LTBP1, three other LTBPs were identified (LTBP2-4) (reviewed in [Rifkin, 2005]). LTBP3 and LTBP4 also interact with latent TGF-B, whereas LTBP2 does not [Saharinen and Keski-Oja, 2000].
Figure 1. Schematic representation of the LTBP family domain organization.
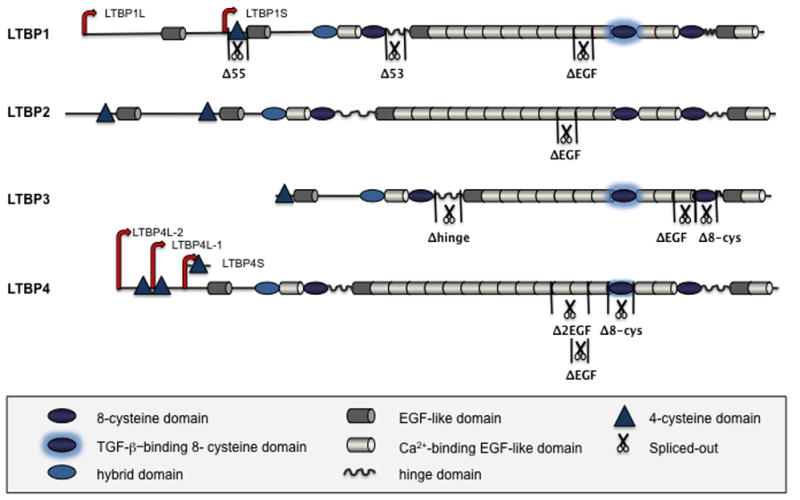
Four LTBPs are shown, with domain symbols depicted in the boxed area below the Figure. Major isoforms and known splice variants of LTBPs are also illustrated [Robertson et al., 2010].
Figure 2. TGF-β-LTBP assembly, processing, and activation.
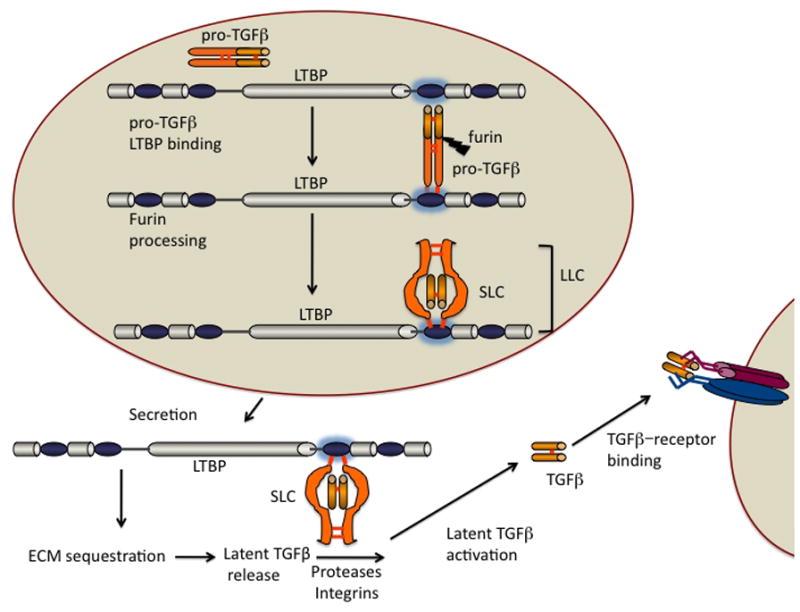
After dimerization in the endoplasmic reticulum, pro-TGFs molecules form disulfide bonds to LTBP molecules early in the secretory pathway. In the trans-Golgi, furin-like enzymes process the pro-TGF-β yielding mature TGF-β, the TGFβ propeptide (LAP), and an LTBP. This complex is called the large latent complex (LLC). When complexed with LAP, TGF-β cannot bind to its receptor. The LLC is secreted, binds to matrix components, and the latent TGF-β is activated by proteases/integrins that modify LAP or/and LTBP. The liberated TGF-β then binds to its receptor.
The four LTBP proteins have a modular and highly repetitive structure, primarily composed of two distinct cysteine-rich motifs: one, characterized by six cysteines, is similar to the epidermal growth factor (EGF) module, and the other defined by eight intramolecularly bound cysteines (8-cys or TB domain), represents the family’s signature domain [Todorovic et al., 2005]. LTBPs can also possess an LTBP-exclusive 4-cys domain near the N-terminus. Each LTBP includes four 8-cys domains interspersed with numerous EGF-like motifs (15–20), many of which contain consensus Ca2+-binding sequences (Figure 1). The first 8-cys domain is known as the hybrid domain, since it shares similarities both with 8-cys and EGF-like motifs. Between the second 8-cys domain and the stretch of repeating EGF-like motifs, there is an unstructured proline-rich region called the hinge domain, which shows the highest degree of sequence diversity amongst the four LTBPs [Annes et al., 2003]. Another hinge region is localized between the fourth 8-cys domain and the downstream EGF-like motif of LTBP-1. The hinge sequences are sensitive to proteolytic cleavage [Dallas et al., 2002; Ge and Greenspan, 2006; Taipale et al., 1995; Unsold et al., 2001], which plays an important role in regulation of LTBP function. In addition to proteolytic processing, alternative splicing and usage of different promoters contribute to the structural variability of LTBPs (Figure 1). LTBP1 and LTBP4 exist as two major isoforms- long and short, transcribed from independent promoters [Kantola et al., 2010; Koski et al., 1999]. Transcriptional regulation of LTBP2 and LTBP3 genes might be similar, considering that Northern blot analysis of LTBP2 and LTBP3 mRNA in different tissues shows the presence of two major transcripts [Dabovic et al., 2002b; Moren et al., 1994; Shipley et al., 2000]. Different variants of each LTBP show distinct expression patterns and diverse affinities towards their binding partners [Koli et al., 2001]. The interactions of LTBPs with their intra- and extra-cellular ligands determine their localization, function and action.
LTBP Interactions
The best-studied LTBP interaction is the one that gave this protein family its name, the association with TFG-β LAP. LTBP and TFG-β proteins evolved in sea urchins and subsequently coevolved, developing into families of four LTBPs and three TFG-β isoforms [Robertson et al., 2010]. Three out of four LTBPs bind LAP, indicating that this interaction might have been the driving force for their co-evolution. LTBP1 and 3 bind all three TFG-β LAP isoforms with high affinity, whereas LTBP4 shows a weak binding capacity only for TGF-B1 LAP. LAP binds to third 8-cys domain (8-cys-3) in LTBP [Gleizes et al., 1996; Saharinen et al., 1996]. Close inspection of all 8-cys domains in fibrillins and LTBPs demonstrated that the 8-cys-3 motifs of LTBP1, 3 and 4 possess a two amino-acid insertion between the 6th and the 7th cysteine [Saharinen and Keski-Oja, 2000]. Deletion of this short insert in the 8-cys-3 motif of LTBP1 results in loss of LAP binding ability, while its inclusion in the non-TFG-β binding 8-Cys-3 repeat of LTBP2 results in TFG-β binding. This further emphasizes the importance of this short sequence in disulfide bridging with LAP. The solution structure of the LTBP1 8-cys-3 domain suggests that the dipeptide insertion just before cys7 results in exposure of the 2–6 disulfide bond, allowing covalent linkage to the pair of cys33 residues in the LAP dimer [Lack et al., 2003]. Furthermore, the 8-cys-3 solution structure suggested that a “ring” of five negatively charged amino acids surrounds the reactive disulfide bond and this electrostatic interaction facilitates covalent bonding of LAP and LTBP [Chen et al., 2005]. A recent mutational analysis showed that the crucial amino acids within LAP’s LTBP-binding epitope are four positively charged residues that surround cys33 [Walton et al., 2010]. The pentameric amino acid ring in LTBP4 8-cys-3 motif has only one negative charge, which might account for the weak and exclusive covalent bonding of LTBP4 and TFG-β1 LAP. The poor and restricted interaction between LTBP4 and TFG-β LAP implies that LTBP4 might have a role independent of TGF-B. The fact that LTBP4 is the only LAP-binding LTBP that can be presented as a variant without the LAP-binding domain (Figure 1) supports this hypothesis [Koli et al., 2001]. A recent study showed that the long form of LTBP4 binds TGF-B1 LAP much more efficiently than the short isoform, although the reason for this difference is unclear [Kantola et al., 2010].
LTBPs function as chaperones by binding LAP within the endoplasmic reticulum (Figure 2). In the absence of LTBP, the reactive cysteine (cys33) within LAP bonds with a free cysteine of the mature cytokine, causing misfolding and slow secretion of the SLC [Rifkin, 2005]. Interestingly, LTBP3 cannot be secreted from cells if not complexed to SLC [Chen et al., 2002]. Secreted LLC is either soluble or sequestered within the ECM through LTBP interactions with multiple extracellular proteins. Latent TFG-β is then stored in the extracellular space until required, and when the need arises it is liberated from its propeptide, in a process commonly referred to as “latent TFG-β activation” (Figure 2). Several different mechanisms of TFG-β activation have been described, involving diverse activators such as proteases, the integrins αvβ6, αvβ3, αvβ5 and αvβ8, thrombospondin-1, F-spondin, neuropilin-1, reactive oxygen species and low or high pH [Chandramouli et al., 2011]. The molecular targets of these divergent latent TFG-β activators can be LTBPs and/or LAPs. Binding of SLC to an LTBP is required for proper activation of latent TFG-β1 as mutation of cys33 to ser yields mice with a TFG-β1 null phenotype [Yoshinaga et al., 2008]. Moreover, activation of TFG-β1 by the integrins αvβ6 and αvβ5 requires an LLC [Annes et al., 2004; Wipff et al., 2007]. The recent x-ray structure of TFG-β1 LAP is consistent with the integrin binding to one end of the complex and thereby transmitting a distortional force through the SLC to the LTBP, which is also bound to the matrix [Shi et al., 2011]. The requirement for LLC may indicate a mechanism for force induced latent TFG-β activation in organs exposed to recurrent stretching or movement.
In summarum, LTBPs orchestrate TFG-β action at multiple levels: 1) they ensure correct folding and efficient secretion 2) they direct temporal and spatial deposition in the extracellular space through their own secretion dynamics and interaction with ECM proteins and 3) they regulate activation.
Incorporation of LTBPs into the ECM is crucial for regulation of latent TFG-β storage and activation. However, significant amounts of LTBP1 and LTBP4 are secreted free of SLC, much like LTBP2, which cannot interact with SLC, suggesting that LTBPs perform roles unrelated to TGF-β. LTBPs interact with many extracellular proteins (Figure 3), but interactions with fibronectin and fibrillins seem to be crucial for proper localization and function. The initial studies of LTBP1 deposition into the ECM identified three potential ECM-interacting domains, two localized at the N-terminus and one localized at the C-terminus [Unsold et al., 2001]. N-terminal fragments were readily incorporated into the matrices and association of the binding domains with the ECM was enhanced by soluble, cell-derived factors. Similar results were obtained with binding peptides from LTBP2, LTBP3 and LTBP4, The N-terminal ECM-binding sequences of LTBP1, 2 and 4 associate with fibronectin [Chen et al., 2007; Fontana et al., 2005; Hyytiainen and Keski-Oja, 2003; Kantola et al., 2008; Taipale et al., 1996] (Figure 3) and this interaction is promoted by transglutaminase [Rifkin, 2005]. The purified N-terminal fragment of LTBP3 co-localized with fibronectin in pre-formed lung fibroblast ECM, and so did the endogenous LTBP3 at the time of its initial incorporation into mature matrices [Koli et al., 2005], suggesting an association of LTBP3 and fibronectin. However, no biochemical studies were performed thus far to investigate this possibility.
Figure 3. Protein interactions of LTBPs and ECM proteins.
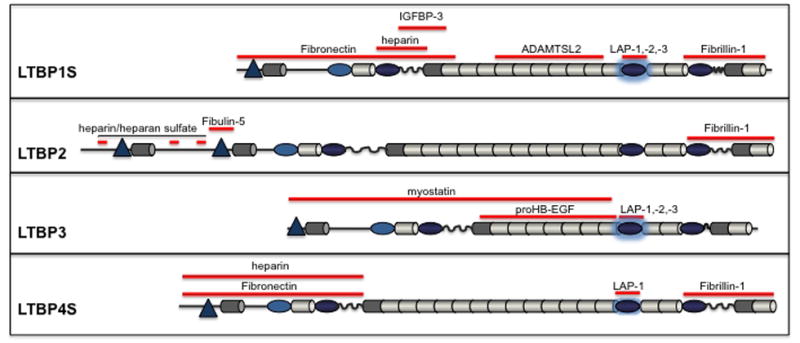
LTBP sequences that interact with given ECM proteins are depicted as red bars. Some LTBP interactions with certain ECM components are not depicted because the fine mapping of the interaction site has not been done.
LTBPs and fibrillins are highly homologous proteins, but are easily distinguished since fibrillins are much larger (~350 kDa) with almost double the number of EGF-like and 8-cys repeats compared to LTBPs. Fibrillins are major structural components of connective tissue microfibrils and elastic fibers [Olivieri et al., 2010]. Microfibril extracts from tissues do not contain LTBPs, indicating that LTBPs are not disulfide bonded to microfibrils [Isogai et al., 2003]. However, all LTBPs, with the exception of LTBP3, bind to fibrillin 1 (Figure 3), the major component of microfibrils through their C-terminus [Hirani et al., 2007; Isogai et al., 2003]. LTBP3 is unique because it cannot be secreted if not bound to latent TGF-β. Because the binding capacity of LTBP3 to fibrillin 1 was assessed only with a C-terminal peptide homologous to the fibrillin-1-binding LTBP1 and LTBP4 domains, which lack the TFG-β LAP-binding sequence (8-cys-3), this C-terminal peptide may have assumed a conformation different from the one of LTBP3-LLC. It would be advisable to use the full length LTBP3-LLC for testing its binding abilities towards fibrillins. LTBP1 and LTBP4 can also bind fibrillin 2, whereas LTBP2 cannot [Isogai et al., 2003]. Competitive binding studies showed that the binding site on fibrillin-1 for LTBP2 is the same or in close proximity to that for LTBP1 [Hirani et al., 2007], suggesting that LTBP2 might be a negative regulator of LTBP1 interaction with microfibrils. LTBP4 may also compete with its family members for binding to fibrillin-1, since its binding site lies within the same fibrillin-1 peptide as that of LTBP1 and LTBP2 (Figure 3). Competitive binding of LTBPs to microfibrils may represent an additional level of control of microfibril composition and function.
Some LTBPs (LTBP1, 2 and 4) associate with heparin and heparan sulfate, in a manner that might affect cell adhesion, migration, and micro- and elastic fiber assembly [Chen et al., 2007; Kantola et al., 2008; Parsi et al., 2010]. Elastogenesis and structural organization of elastic fibers also depend on fibulin-4 and -5. LTBP2 interacts with fibulin 5 and plays an important role in the targeting of fibulin 5 to microfibrils to form elastic fibers [Hirai et al., 2007]. LTBP1 binds to fibulin 4 [Massam-Wu et al., 2010], whose role in elastogenesis is to tether lysiloxidase (LOX) to tropoelastin to facilitate cross-linking [Horiguchi et al., 2009], whereas LTBP4 interacts with fibulin-5 (B. Dabovic, personal communication). Fibulin-2, -4, and -5 can bind to fibrillin 1 and their binding sites are within the domain responsible for interaction with LTBPs [Ono et al., 2009]. Therefore, LTBPs and fibulins may compete for interaction with fibrillin 1. However, Kielty’s group showed that fibulin-4 can simultaneously bind fibrillin-1 and LTBP1, suggesting that fibulin-4 might function as a mediator of LTBP1 association with microfibrils [Massam-Wu et al., 2010].
Recently, a few diverse factors have been shown to bind to LTBPs. For example, LTBP1 interacts with ADAMTSL2, a secreted glycoprotein of unknown function [Le Goff et al., 2008]. ADAMTSL2 mutations in humans cause geleophysic dysplasia, an autosomal recessive disorder characterized by thick skin, short stature, fingers and toes, and cardiac valve malfunction that can lead to premature death. The functional significance of LTBP1 interaction with ADAMTSL2 is unclear, but skin fibroblasts from affected individuals secret enhanced levels of active TGF-β, suggesting that ADAMTSL2 might regulate TFG-β activity, possibly in an LTBP1-dependent fashion.
At least three LTBPs: LTBP1, 2 and 3, interact non-covalently with growth factors from the TFG-β super-family other than TGF-βs. In Xenopus embryos LTBP1 interacts with activin and modulates its action [Altmann et al., 2002], whereas LTBP2 and LTBP3 associate with a pro-form of myostatin, a TGF-β-like hormone that regulates the size of skeletal muscle [Anderson et al., 2008]. Pro-myostatin is secreted and retained in the ECM in an LTBP3-dependent manner and upon cleavage by furin, the mature growth factor is released from the complex with LTBP3. LTBP2 and LTBP3 intracellularly bind pro-GDF11 and proprotein convertase 5/6A (PC5/6A), which cleaves the GDF11 propeptide from the mature cytokine. LTBP2 and LTBP3 may act as chaperons for PC5/6A zymogen in the ECM and prevent its maturation, therefore regulating the processing of GDF11 [Sun et al., 2011]. Yeast two-hybrid system experiments revealed a potential association between LTBP1 and insulin-like growth factor binding protein-3 (IGFBP-3) (Figure 3) and LTBP3 and Heparin-Binding EGF-like growth factor (HB-EGF) precursor, but these interactions have not been confirmed biochemically [Brooke et al., 2002; Gui and Murphy, 2003].
LTBPs form multiple intra- and extra-cellular associations with divergent proteins. These interactions affect the bioavailability of TGF-βs as well as other TFG-β super-family members like activin, myostatin and GDF11. In addition, LTBPs play an important structural role in microfibril and elastic fibril assembly and organization.
Dynamic integration of LTBPs into the ECM
The backbone of the ECM is composed of fibrilar proteins, such as collagen, fibronectin, and elastin to which many adaptor proteins and proteoglycans associate, together forming a complex and dynamic 3-D structure. The building blocks of elastic matrices are fibrillins and tropoelastin, but elastogenesis and maintenance of elastic fibers would be impossible without a number of other factors, including specific LTBPs and fibulins.
LTBPs initially incorporate into the ECM via interaction with fibronectin. Fibronectin is one of the first molecules to be deposited in the extracellular space, and regulates the initial and continued assembly and stability of several other ECM components (reviewed in [Dallas et al., 2006]). Without a pre-laid fibronectin matrix, fibrillins, LTBPs and tropoelastin are not integrated into the ECM.
Cultured human lung fibroblasts deposit LTBP1 immediately after formation of fibronectin matrices; LTBP1 is clearly detectable after 2 days of culture and it co-localizes with fibronectin meshwork [Koli et al., 2005]. LTBP2 forms extracellular fibers on day 4 [Vehvilainen et al., 2009], whereas an LTBP4 network is not visible until day 7, and both proteins initially co-localized with fibronectin. LTBP3 is the last LTBP to be deposited into fibroblast matrices at about 10–14 days of culture [Koli et al., 2005]. After an extended period of culture, the localization of LTBPs startes to diverge from that of fibronectin, with the LTBPs being organized into fibrilar arrays in a layer above the fibronectin fibers. This phenomenon was previously described for fibrillin deposition in the ECM and, indeed, in mature matrices LTBPs co-localized with fibrillin-1. Experiments following the dynamics of ECM formation and reorganization explained this change in localization of fibronectin and LTBPs/fibrillins upon long culture periods, since LTBP matrices are much more stable and less prone to cell-mediated reorganization than the fibronectin meshwork, which is constantly assembled and turned over [Dallas et al., 2006].
Fibrillin-1 and -2 self-associate both longitudinally and through lateral binding, giving rise to microfibrils with an average diameter of 10 nm (reviewed in [Olivieri et al., 2010]). Lack of fibrillin-1 microfibrils affects ECM deposition of all LTBPs ([Massam-Wu et al., 2010; Ono et al., 2009; Vehvilainen et al., 2009]; L. Zilberberg, personal communication). Abnormal LTBP-fibrillin interactions in mice and humans yield promiscuous TFG-β signaling and changes in p38 MAPK, ERK1/2 and JNK1 signaling pathways [Carta et al., 2009; Holm et al., 2011].
LTBP2 and LTBP4 have been implicated in the formation of elastic fibers. Elastogenesis requires coaecervation of tropoelastin and is modulated by fibulin-4 and -5, cell-surface glycosaminoglycans, and cross-linking enzymes lysiloxidase (LOX) and LOX-like (LOXL) (Figure 4). Fibulin-5 and elastin deposition in cell culture is dependent on fibrillin-1 [Hirai et al., 2007]. However, in the absence of LTBP2, fibulin-5 targets tropoelastin mainly to fibrillin-2. Therefore, LTBP2 might function as a molecular switch that regulates the differential usage of microfibrils during assembly of elastic matrices.
Figure 4. Organization of microfibrils and elastic assemblies.
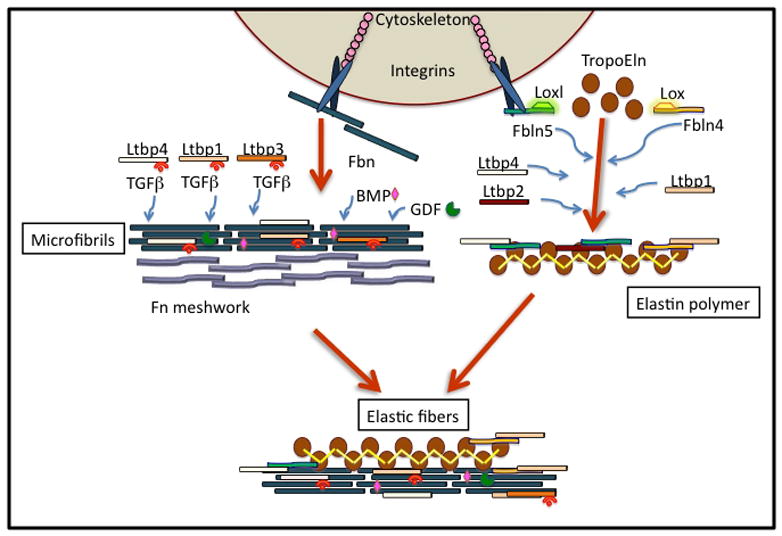
The initial step in the progressive and hierarchical building of microfibrils and elastic fibers is the deposition of the fibronectin (Fn) meshwork. Fibrillin (Fbn) -1 and -2 self-associate on head-to-tail principle resulting in longitudinal polymerization, but also through lateral binding, altogether giving rise to microfibrils. BMPs, GDFs and TGF-β are probably incorporated at this point because of their direct binding to Fbn and association with LTBPs. As the initial microfibrils form, a second series of reactions in which tropoelastin (TropoEln) binds to complexes of fibulins (Fbln-4 and Fbln-5), each of which is associated with lyslyoxidase (Lox) and LOX-like (Loxl) molecules. Fbln-4-Lox and Fbln-5-Loxl promote TropoELN conversion to elastin polymers. The microfibrils and elastin polymers combine to form elastic fibers in a manner that is Fbln- and LTBP4-dependent. LTBPs, with their associated TGFβs, bind to the microfibrils independently of Fbln incorporation. However, LTBPs might associate with Fbln-4 and -5 during elastin polymerization. LTBPs are illustrated within the microfibrils as both with and without TGFβ because a significant amount of LTBPs is found without any bound TGFβ.
Ltbp4S−/− mice show impaired elastogenesis because elastin coacervates cannot integrate into microfibril bundles. LTBP4 colocalizes with both fibrillins and fibulin-5 (B. Dabovic, personal communication) and might operate as a tethering factor between the microfibrils and tropo-elastin aggregates. In the absence of LTBP4, elastin coacervates and microfibril bundles do not interact to form functional elastic fibers. Studies of Ltbp4S null cells and mice showed that the absence of LTBP4 causes both impaired elastin and fibulin 5 deposition, further emphasizing the importance of LTBP4-fibulin-5 interaction in elastogenesis.
LTBP-related pathologies in mice and humans
Genetic disorders arising as a result of mutations in LTBP genes in mice and humans underline the importance of the LTBP family. Deficiency of the long form of Ltbp1 (Ltbp1L) in mice causes serious disruption of great vessels and cardiac valve development, resulting in perinatal death [Todorovic et al., 2011; Todorovic et al., 2007]. These defects stem from decreased TFG-β activity that occurs at specific times during heart and great vessel formation and within defined cellular populations such as cardiac neural crest cells and valvular mesenchymal cells. However, mice with deletion of the first exon of the Ltbp1 gene common for both the long and the short form display only mild cranio-facial abnormalities and live a normal lifespan [Drews et al., 2008]. The sequence deleted in these mice codes for a 4-cys domain that can be spliced out giving rise to an Ltbp1L splice variant called Ltbp1LΔ55 (Figure 1). It is possible that mice with deletion of the first exon shared by Ltbp1L and Ltbp1S make Ltbp1LΔ55, which might explain the discrepancy between phenotypes of these mutants and Ltbp1L−/− mice. The Ltbp1L isoform is expressed mostly during embryonic development, whereas Ltbp1S expression initiates late during organogenesis and becomes the major isoform in adults. The lack of the long form at the time when it represents the only Ltbp1 isoform expressed might have grave consequences on development, whereas the lack of the short form later in life when a splice variant of Ltbp1L is still expressed might result only in mild developmental defects. Currently, no LTBP1 mutation has been linked to any human pathology.
Null mutations in LTBP2 have been associated with ocular malformations, such as development of a small and dislocated lens and glaucoma (reviewed in [Robertson et al., 2010]. An earlier report indicating that Ltbp2 null mice died at the time of implantation has not been reproduced.
Homozygous mutation within the 7th EGF-like domain in LTBP3 caused a dramatic decrease in LTBP3 expression and was manifested as oligodontia (agenesis of ≥six teeth), short stature and scoliosis [Noor et al., 2009]. Ltbp3 null mice are also shorter than their littermates and have multiple skeletal and craniofacial abnormalities, including kyphosis, extension of the mandible beyond the maxilla and cranial doming. As the animals age, their long bones and vertebrae become osteoarthritic and osteopetrotic and they develop pulmonary emphysema [Colarossi et al., 2005; Dabovic et al., 2002a]. Similar to Ltbp1L−/− mice, developmental defects and pathologies that arise in the adulthood of Ltbp3−/− mice are consistent with decreased TFG-β levels.
Autosomal recessive mutations in LTBP4 cause Urban-Rifkin-Davis syndrome (URDS), which is characterized by multi-organ developmental defects, such as aberrant lung, gastrointestinal, genitourinary, musculoskeletal and dermal organogenesis [Urban et al., 2009]. Human patients with a mutation in LTBP4 were identified based upon comparative electron microscopy studies of patients’ tissues and tissue samples from mice lacking the short form of Ltbp4 (Ltbp4S). Tissues from both sources showed strikingly similar elastic fiber pathology-elastin appeared as big globules positioned next to the microfibrils with only a few elastin polymers integrated within the microfibril bundle, instead of having the microfibrils enmeshed into elastin [Dabovic et al., 2009]. Lack of Ltbp4S in mice results in defective differentiation of terminal air sacs, cardiomyopathy and colon carcinomas [Sterner-Kock et al., 2002]. The lung abnormalities in Ltbp4S−/− mice are rescued when Tgfb2 expression is genetically abolished, implicating excess TFG-β signaling in the pathogenesis of the phenotype. Similarly, skin fibroblasts from URDS patients produce more active TGF-β than controls. However, genetic ablation of Tgfb2 expression in Ltbp4S−/− mice did not improve the organization of elastic matrices, indicating a role for LTBP4 in elastogenesis independent of its role in modulating TFG-β activity. Collectively, the human and mouse findings imply that LTBP4 may perform two roles: 1) a structural role in promoting elastogenesis and 2) a functional role in regulating TGF-β activity. However, it is possible that LTBP4 plays exclusively a structural role and that increased TFG-β activity is actually a consequence of abnormal elastic fibers and perturbed extracellular milieu, which, in turn, leads to a promiscuous activation of latent TGF-β, similarly to what is seen in human and mouse mutants for fibrillin-1 (reviewed in[Olivieri et al., 2010]). The fact that attenuation of TFG-β2 activity, a TFG-β isoform that does not interact directly with LTBP4, partially rescues abnormal lung development in Ltbp4S−/− mice, favors the latter hypothesis.
Taken together, the human and mouse pathologies arising from mutations in LTBP genes underscore the importance of TFG-β interactions in the biology of these ECM proteins. However, LTBPs (especially LTBP4) are also important structural elements of the ECM.
Conclusions and perspectives
Amongst ECM proteins, LTBPs occupy a special place warranted by their interaction with the omnipresent latent TGF-β. LTBPs participate and regulate every step in TGF-β’s biology- from its folding and secretion, through its localization, to, finally, its activation. To rephrase an old proverb: Behind every successful TGF-β there is an LTBP. Yet, the LTBPs are more than just TGF-β chaperones: they interact with other proteins- from potent growth factors to structural proteins of the ECM and in doing so, they play a vital role in organ formation and tissue homeostasis. The biological importance of LTBPs is emphasized by the pathology of mouse and human LTBP mutants. Though much work has been done to understand the mechanism(s) of LTBP action, these efforts have been primarily related to studies of TGF-B activity. However, not all pathologies that arise from LTBP mutations and, consequently, lack or change of this protein’s function, can be explained by changes in TGF-β activity. LTBPs also play important roles in elastogenesis and in maintenance of ECM structure. Further investigations are required to determine the sequence and molecular players that yield LTBP-related pathologies in order not only to better understand LTBP function, but also to find suitable tools to prevent or meliorate the devastating consequences caused by LTBP malfunction.
Acknowledgments
The authors apologize to those of our colleagues whose work was not cited because of space restrictions. We know you did it.
D.B.R. is funded by NIH R01: CA034282-26, GM083220 and AR054817-01A2; and P01 AR49698.
References
- Altmann CR, Chang C, Munoz-Sanjuan I, Bell E, Heke M, Rifkin DB, Brivanlou AH. The latent-TGFbeta-binding-protein-1 (LTBP-1) is expressed in the organizer and regulates nodal and activin signaling. Dev Biol. 2002;248:118–27. doi: 10.1006/dbio.2002.0716. [DOI] [PubMed] [Google Scholar]
- Anderson SB, Goldberg AL, Whitman M. Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J Biol Chem. 2008;283:7027–35. doi: 10.1074/jbc.M706678200. [DOI] [PubMed] [Google Scholar]
- Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol. 2004;165:723–34. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Brooke JS, Cha JH, Eidels L. Latent transforming growth factor beta-binding protein-3 and fibulin-1C interact with the extracellular domain of the heparin-binding EGF-like growth factor precursor. BMC Cell Biol. 2002;3:2. doi: 10.1186/1471-2121-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB, Ramirez F. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. 2009;284:5630–6. doi: 10.1074/jbc.M806962200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouli A, Simundza J, Pinderhughes A, Cowin P. Choreographing metastasis to the tune of LTBP. J Mammary Gland Biol Neoplasia. 2011;16:67–80. doi: 10.1007/s10911-011-9215-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sivakumar P, Barley C, Peters DM, Gomes RR, Farach-Carson MC, Dallas SL. Potential role for heparan sulfate proteoglycans in regulation of transforming growth factor-beta (TGF-beta) by modulating assembly of latent TGF-beta-binding protein-1. J Biol Chem. 2007;282:26418–30. doi: 10.1074/jbc.M703341200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Ali T, Todorovic V, O'Leary JM, Kristina Downing A, Rifkin DB. Amino acid requirements for formation of the TGF-beta-latent TGF-beta binding protein complexes. J Mol Biol. 2005;345:175–86. doi: 10.1016/j.jmb.2004.10.039. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dabovic B, Annes JP, Rifkin DB. Latent TGF-beta binding protein-3 (LTBP-3) requires binding to TGF-beta for secretion. FEBS Lett. 2002;517:277–80. doi: 10.1016/s0014-5793(02)02648-0. [DOI] [PubMed] [Google Scholar]
- Colarossi C, Chen Y, Obata H, Jurukovski V, Fontana L, Dabovic B, Rifkin DB. Lung alveolar septation defects in Ltbp-3-null mice. Am J Pathol. 2005;167:419–28. doi: 10.1016/S0002-9440(10)62986-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabovic B, Chen Y, Choi J, Vassallo M, Dietz HC, Ramirez F, von Melchner H, Davis EC, Rifkin DB. Dual functions for LTBP in lung development: LTBP-4 independently modulates elastogenesis and TGF-beta activity. J Cell Physiol. 2009;219:14–22. doi: 10.1002/jcp.21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabovic B, Chen Y, Colarossi C, Obata H, Zambuto L, Perle MA, Rifkin DB. Bone abnormalities in latent TGF-[beta] binding protein (Ltbp)-3-null mice indicate a role for Ltbp-3 in modulating TGF-[beta] bioavailability. J Cell Biol. 2002a;156:227–32. doi: 10.1083/jcb.200111080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabovic B, Chen Y, Colarossi C, Zambuto L, Obata H, Rifkin DB. Bone defects in latent TGF-beta binding protein (Ltbp)-3 null mice; a role for Ltbp in TGF-beta presentation. J Endocrinol. 2002b;175:129–41. doi: 10.1677/joe.0.1750129. [DOI] [PubMed] [Google Scholar]
- Dallas SL, Chen Q, Sivakumar P. Dynamics of assembly and reorganization of extracellular matrix proteins. Curr Top Dev Biol. 2006;75:1–24. doi: 10.1016/S0070-2153(06)75001-3. [DOI] [PubMed] [Google Scholar]
- Dallas SL, Rosser JL, Mundy GR, Bonewald LF. Proteolysis of latent transforming growth factor-beta (TGF-beta )-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J Biol Chem. 2002;277:21352–60. doi: 10.1074/jbc.M111663200. [DOI] [PubMed] [Google Scholar]
- Drews F, Knobel S, Moser M, Muhlack KG, Mohren S, Stoll C, Bosio A, Gressner AM, Weiskirchen R. Disruption of the latent transforming growth factor-beta binding protein-1 gene causes alteration in facial structure and influences TGF-beta bioavailability. Biochim Biophys Acta. 2008;1783:34–48. doi: 10.1016/j.bbamcr.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Fontana L, Chen Y, Prijatelj P, Sakai T, Fassler R, Sakai LY, Rifkin DB. Fibronectin is required for integrin alphavbeta6-mediated activation of latent TGF-beta complexes containing LTBP-1. FASEB J. 2005;19:1798–808. doi: 10.1096/fj.05-4134com. [DOI] [PubMed] [Google Scholar]
- Ge G, Greenspan DS. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J Cell Biol. 2006;175:111–20. doi: 10.1083/jcb.200606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleizes PE, Beavis RC, Mazzieri R, Shen B, Rifkin DB. Identification and characterization of an eight-cysteine repeat of the latent transforming growth factor-beta binding protein-1 that mediates bonding to the latent transforming growth factor-beta1. J Biol Chem. 1996;271:29891–6. doi: 10.1074/jbc.271.47.29891. [DOI] [PubMed] [Google Scholar]
- Gui Y, Murphy LJ. Interaction of insulin-like growth factor binding protein-3 with latent transforming growth factor-beta binding protein-1. Mol Cell Biochem. 2003;250:189–95. doi: 10.1023/a:1024990409102. [DOI] [PubMed] [Google Scholar]
- Hirai M, Horiguchi M, Ohbayashi T, Kita T, Chien KR, Nakamura T. Latent TGF-beta-binding protein 2 binds to DANCE/fibulin-5 and regulates elastic fiber assembly. EMBO J. 2007;26:3283–95. doi: 10.1038/sj.emboj.7601768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirani R, Hanssen E, Gibson MA. LTBP-2 specifically interacts with the amino-terminal region of fibrillin-1 and competes with LTBP-1 for binding to this microfibrillar protein. Matrix Biol. 2007;26:213–23. doi: 10.1016/j.matbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, Loeys BL, Thomas CJ, Patnaik S, Marugan JJ, Judge DP, Dietz HC. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–61. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi M, Inoue T, Ohbayashi T, Hirai M, Noda K, Marmorstein LY, Yabe D, Takagi K, Akama TO, Kita T, Kimura T, Nakamura T. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc Natl Acad Sci U S A. 2009;106:19029–34. doi: 10.1073/pnas.0908268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyytiainen M, Keski-Oja J. Latent TGF-beta binding protein LTBP-2 decreases fibroblast adhesion to fibronectin. J Cell Biol. 2003;163:1363–74. doi: 10.1083/jcb.200309105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, Charbonneau NL, Reinhardt DP, Rifkin DB, Sakai LY. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278:2750–7. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- Kantola AK, Keski-Oja J, Koli K. Fibronectin and heparin binding domains of latent TGF-beta binding protein (LTBP)-4 mediate matrix targeting and cell adhesion. Exp Cell Res. 2008;314:2488–500. doi: 10.1016/j.yexcr.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Kantola AK, Ryynanen MJ, Lhota F, Keski-Oja J, Koli K. Independent regulation of short and long forms of latent TGF-beta binding protein (LTBP)-4 in cultured fibroblasts and human tissues. J Cell Physiol. 2010;223:727–36. doi: 10.1002/jcp.22082. [DOI] [PubMed] [Google Scholar]
- Koli K, Hyytiainen M, Ryynanen MJ, Keski-Oja J. Sequential deposition of latent TGF-beta binding proteins (LTBPs) during formation of the extracellular matrix in human lung fibroblasts. Exp Cell Res. 2005;310:370–82. doi: 10.1016/j.yexcr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Koli K, Saharinen J, Karkkainen M, Keski-Oja J. Novel non-TGF-beta-binding splice variant of LTBP-4 in human cells and tissues provides means to decrease TGF-beta deposition. J Cell Sci. 2001;114:2869–78. doi: 10.1242/jcs.114.15.2869. [DOI] [PubMed] [Google Scholar]
- Koski C, Saharinen J, Keski-Oja J. Independent promoters regulate the expression of two amino terminally distinct forms of latent transforming growth factor-beta binding protein-1 (LTBP-1) in a cell type-specific manner. J Biol Chem. 1999;274:32619–30. doi: 10.1074/jbc.274.46.32619. [DOI] [PubMed] [Google Scholar]
- Lack J, O'Leary JM, Knott V, Yuan X, Rifkin DB, Handford PA, Downing AK. Solution structure of the third TB domain from LTBP1 provides insight into assembly of the large latent complex that sequesters latent TGF-beta. J Mol Biol. 2003;334:281–91. doi: 10.1016/j.jmb.2003.09.053. [DOI] [PubMed] [Google Scholar]
- Le Goff C, Morice-Picard F, Dagoneau N, Wang LW, Perrot C, Crow YJ, Bauer F, Flori E, Prost-Squarcioni C, Krakow D, Ge G, Greenspan DS, Bonnet D, Le Merrer M, Munnich A, Apte SS, Cormier-Daire V. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat Genet. 2008;40:1119–23. doi: 10.1038/ng.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massam-Wu T, Chiu M, Choudhury R, Chaudhry SS, Baldwin AK, McGovern A, Baldock C, Shuttleworth CA, Kielty CM. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF beta. J Cell Sci. 2010;123:3006–18. doi: 10.1242/jcs.073437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono K, Hellman U, Wernstedt C, Heldin CH. Latent high molecular weight complex of transforming growth factor beta 1. Purification from human platelets and structural characterization. J Biol Chem. 1988;263:6407–15. [PubMed] [Google Scholar]
- Moren A, Olofsson A, Stenman G, Sahlin P, Kanzaki T, Claesson-Welsh L, ten Dijke P, Miyazono K, Heldin CH. Identification and characterization of LTBP-2, a novel latent transforming growth factor-beta-binding protein. J Biol Chem. 1994;269:32469–78. [PubMed] [Google Scholar]
- Noor A, Windpassinger C, Vitcu I, Orlic M, Rafiq MA, Khalid M, Malik MN, Ayub M, Alman B, Vincent JB. Oligodontia is caused by mutation in LTBP3, the gene encoding latent TGF-beta binding protein 3. Am J Hum Genet. 2009;84:519–23. doi: 10.1016/j.ajhg.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri J, Smaldone S, Ramirez F. Fibrillin assemblies: extracellular determinants of tissue formation and fibrosis. Fibrogenesis Tissue Repair. 2010;3:24. doi: 10.1186/1755-1536-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono RN, Sengle G, Charbonneau NL, Carlberg V, Bachinger HP, Sasaki T, Lee-Arteaga S, Zilberberg L, Rifkin DB, Ramirez F, Chu ML, Sakai LY. Latent transforming growth factor beta-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J Biol Chem. 2009;284:16872–81. doi: 10.1074/jbc.M809348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsi MK, Adams JR, Whitelock J, Gibson MA. LTBP-2 has multiple heparin/heparan sulfate binding sites. Matrix Biol. 2010;29:393–401. doi: 10.1016/j.matbio.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005;280:7409–12. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- Robertson I, Jensen S, Handford P. TB domain proteins: evolutionary insights into the multifaceted roles of fibrillins and LTBPs. Biochem J. 2010;433:263–76. doi: 10.1042/BJ20101320. [DOI] [PubMed] [Google Scholar]
- Saharinen J, Keski-Oja J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol Biol Cell. 2000;11:2691–704. doi: 10.1091/mbc.11.8.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen J, Taipale J, Keski-Oja J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 1996;15:245–53. [PMC free article] [PubMed] [Google Scholar]
- Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-beta structure and activation. Nature. 2011;474:343–9. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley JM, Mecham RP, Maus E, Bonadio J, Rosenbloom J, McCarthy RT, Baumann ML, Frankfater C, Segade F, Shapiro SD. Developmental expression of latent transforming growth factor beta binding protein 2 and its requirement early in mouse development. Mol Cell Biol. 2000;20:4879–87. doi: 10.1128/mcb.20.13.4879-4887.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner-Kock A, Thorey IS, Koli K, Wempe F, Otte J, Bangsow T, Kuhlmeier K, Kirchner T, Jin S, Keski-Oja J, von Melchner H. Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 2002;16:2264–73. doi: 10.1101/gad.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Essalmani R, Susan-Resiga D, Prat A, Seidah NG. Latent Transforming Growth Factor {beta}-Binding Proteins-2 and -3 Inhibit the Proprotein Convertase 5/6A. J Biol Chem. 2011;286:29063–73. doi: 10.1074/jbc.M111.242479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem. 1995;270:4689–96. doi: 10.1074/jbc.270.9.4689. [DOI] [PubMed] [Google Scholar]
- Taipale J, Saharinen J, Hedman K, Keski-Oja J. Latent transforming growth factor-beta 1 and its binding protein are components of extracellular matrix microfibrils. J Histochem Cytochem. 1996;44:875–89. doi: 10.1177/44.8.8756760. [DOI] [PubMed] [Google Scholar]
- Todorovic V, Finnegan E, Freyer L, Zilberberg L, Ota M, Rifkin DB. Long form of latent TGF-beta binding protein 1 (Ltbp1L) regulates cardiac valve development. Dev Dyn. 2011;240:176–87. doi: 10.1002/dvdy.22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic V, Frendewey D, Gutstein DE, Chen Y, Freyer L, Finnegan E, Liu F, Murphy A, Valenzuela D, Yancopoulos G, Rifkin DB. Long form of latent TGF-beta binding protein 1 (Ltbp1L) is essential for cardiac outflow tract septation and remodeling. Development. 2007;134:3723–32. doi: 10.1242/dev.008599. [DOI] [PubMed] [Google Scholar]
- Todorovic V, Jurukovski V, Chen Y, Fontana L, Dabovic B, Rifkin DB. Latent TGF-beta binding proteins. Int J Biochem Cell Biol. 2005;37:38–41. doi: 10.1016/j.biocel.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Unsold C, Hyytiainen M, Bruckner-Tuderman L, Keski-Oja J. Latent TGF-beta binding protein LTBP-1 contains three potential extracellular matrix interacting domains. J Cell Sci. 2001;114:187–197. doi: 10.1242/jcs.114.1.187. [DOI] [PubMed] [Google Scholar]
- Urban Z, Hucthagowder V, Schurmann N, Todorovic V, Zilberberg L, Choi J, Sens C, Brown CW, Clark RD, Holland KE, Marble M, Sakai LY, Dabovic B, Rifkin DB, Davis EC. Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am J Hum Genet. 2009;85:593–605. doi: 10.1016/j.ajhg.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehvilainen P, Hyytiainen M, Keski-Oja J. Matrix association of latent TGF-beta binding protein-2 (LTBP-2) is dependent on fibrillin-1. J Cell Physiol. 2009;221:586–93. doi: 10.1002/jcp.21888. [DOI] [PubMed] [Google Scholar]
- Walton KL, Makanji Y, Chen J, Wilce MC, Chan KL, Robertson DM, Harrison CA. Two distinct regions of latency-associated peptide coordinate stability of the latent transforming growth factor-beta1 complex. J Biol Chem. 2010;285:17029–37. doi: 10.1074/jbc.M110.110288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–23. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaga K, Obata H, Jurukovski V, Mazzieri R, Chen Y, Zilberberg L, Huso D, Melamed J, Prijatelj P, Todorovic V, Dabovic B, Rifkin DB. Perturbation of transforming growth factor (TGF)-beta1 association with latent TGF-beta binding protein yields inflammation and tumors. Proc Natl Acad Sci U S A. 2008;105:18758–63. doi: 10.1073/pnas.0805411105. [DOI] [PMC free article] [PubMed] [Google Scholar]