

Abstract
Background. Hyperoxaluria is a major risk factor for kidney stone formation. Although urinary oxalate measurement is part of all basic stone risk assessment, there is no standardized method for this measurement.
Methods. Urine samples from 24-h urine collection covering a broad range of oxalate concentrations were aliquoted and sent, in duplicates, to six blinded international laboratories for oxalate, sodium and creatinine measurement. In a second set of experiments, ten pairs of native urine and urine spiked with 10 mg/L of oxalate were sent for oxalate measurement. Three laboratories used a commercially available oxalate oxidase kit, two laboratories used a high-performance liquid chromatography (HPLC)-based method and one laboratory used both methods.
Results. Intra-laboratory reliability for oxalate measurement expressed as intraclass correlation coefficient (ICC) varied between 0.808 [95% confidence interval (CI): 0.427–0.948] and 0.998 (95% CI: 0.994–1.000), with lower values for HPLC-based methods. Acidification of urine samples prior to analysis led to significantly higher oxalate concentrations. ICC for inter-laboratory reliability varied between 0.745 (95% CI: 0.468–0.890) and 0.986 (95% CI: 0.967–0.995). Recovery of the 10 mg/L oxalate-spiked samples varied between 8.7 ± 2.3 and 10.7 ± 0.5 mg/L. Overall, HPLC-based methods showed more variability compared to the oxalate oxidase kit-based methods.
Conclusions. Significant variability was noted in the quantification of urinary oxalate concentration by different laboratories, which may partially explain the differences of hyperoxaluria prevalence reported in the literature. Our data stress the need for a standardization of the method of oxalate measurement.
Keywords: agreement, hyperoxaluria, method, oxalate, stone
Introduction
The risk of passing a kidney stone during a lifetime is ∼10% and the risk of recurrence reaches 50% after the first episode. Metabolic evaluation of recurrent stone formers is cost-efficient and helps prevent further recurrence by guiding treatment [1]. Metabolic investigation is based on 24-h urine collections which provide the excretion rate for several identified lithogenic factors and allow the calculation of urinary saturation with respect to stone forming salts. Since ∼70% of stones contain calcium and oxalate [2], every metabolic workup includes measurement of calcium and oxalate concentrations in a 24-h urine collection, together with other key analytes. Debate about the relative contribution of hyperoxaluria and hypercalciuria to the process of calcium oxalate stone formation has been ongoing in the kidney stone community for years [3–7]. However, a direct comparison of oxalate measurement between stone laboratories has not been performed.
Several methods have been developed to measure oxalate levels [8, 9], however, two methods are currently used by most laboratories: high-performance liquid chromatography (HPLC)-based and colorimetric oxalate oxidase methods. HPLC-based methods are generally developed in-house by individual centers, each using a different setup (column, eluant, detector, retention time, etc…) while the oxalate oxidase colorimetric method has been developed, validated and largely diffused as a kit by a single vendor.
In the course of comparing stone risk profiles of stone formers recruited from two centers, one in Europe and one in the USA, we noted important variations in oxalate measurements. This motivated the current study in which we explored the intra- and inter-laboratory agreement of oxalate assessment performed by six international laboratories. We found variable agreement between laboratories, stressing the importance of correct sample handling (pre-analytic acidification) and the need for a standardization of urinary oxalate measurement.
Materials and methods
Urine samples were obtained randomly and anonymously from kidney stone formers presenting to the outpatient stone clinic of the University of Texas Southwestern Medical Center (UTSW). Twenty-four-hour urine samples were collected by carefully instructed patients and kept at 4°C without addition of any preservative or chemicals.
Experiment one
Once received by the UTSW laboratory, a regular stone risk profile was first run on the 24-h urine collection using an HPLC-based method for oxalate. The rest of the urine was aliquoted in 10 mL portions and frozen at −80°C without addition of preservative and without acidification. Twenty samples of 10 mL urine comprising urine samples from 10 patients in duplicate were sent out frozen on dry ice to six international laboratories for oxalate assessment (Lab A–F, with Laboratory D performing both oxalate oxidase and HPLC-based method receiving two separate Labels, D1 and D2, one for each method). The UTSW laboratory reran the frozen samples in duplicate in a blinded fashion similar to the other laboratories. Of note, the laboratories were blinded to sample identification and were unaware of the fact that they received duplicates to analyze. Each laboratory was instructed to thaw and acidify the samples prior to processing. Each laboratory was also asked to determine sodium and creatinine as control measurements. In addition, and in order to assess the role of acidification on oxalate measurement, two laboratories (Laboratories D2 and E, both using a HPLC-based method) were asked to compare oxalate measurement from thawed urine (‘non-acidified’) and from the same urine acidified immediately after thawing (‘acidified’).
Experiment two
Once received by the UTSW laboratory, the regular stone risk profile was first run on the 24-h urine collection using an HPLC-based method for oxalate. Then, urine was aliquoted by 10 mL, acidified to pH < 2 with a solution of 12 N HCl and kept at 4°C. A set of samples was spiked with 10 mg/L sodium oxalate by adding 0.1 mL of a stock solution of 1 g/L of sodium oxalate to 9.9 mL of native urine. Ten native samples and ten spiked samples were sent out to five international laboratories (Laboratories A–E, as Laboratory F withdrew, recognizing a problem with its oxalate assessment method).
Oxalate levels assessment.
All participating laboratories are clinical laboratories. Four laboratories (Laboratories A, B, C and D1) used the same commercially available oxalate oxidase kit (Trinity Biotech, Co. Wicklow, Ireland), following the manufacturer’s protocol, two laboratories used locally setup HPLC-based method (Laboratories E and F) and one lab used both methods (D1 and D2) (see Table 1). HPLC-based method utilized the Dionex system, using AS22, AS4A and AS11 columns (Laboratories D2, E and F, respectively). The upper limits of linearity for the different methods were 180 mg/L for laboratories using the oxalate oxidase kit (A, B, C, D1) and 18 mg/L, 80 mg/L and 180 mg/L for Laboratories D2, E and F, respectively.
Table 1.
Analysis of the reliability within the same laboratory (analysis of the duplicates from Experiment 1)a
| Laboratories | Method for oxalate measurement | Oxalate | Sodium | Creatinine |
| Lab A | OO | 0.875 (0.599–0.967) | NA | NA |
| Lab B | OO | 0.998 (0.994–1.000) | 1.000 (0.999–1.000) | 1.000 (0.999–1.000) |
| Lab C | OO | 0.994 (0.976–0.998) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) |
| Lab D1 | OO | 0.942 (0.798–0.985) | 0.999 (0.997–1.000) | 1.000 (0.999–1.000) |
| Lab D2 | HPLC | NA | ||
| Lab E | HPLC | 0.977 (0.914–0.994) | 0.999 (0.996–1.000) | 0.995 (0.982–0.999) |
| Lab F | HPLC | 0.808 (0.427–0.948) | 1.000 (1.000–1.000) | 1.000 (0.999–1.000) |
ICC are presented with 95% upper and lower CI. NA stands for not available; OO, oxalate oxidase method.
Statistical methods
Intra-laboratory agreement was measured by comparing results of duplicate samples for oxalate, sodium and creatinine and was expressed as intraclass correlation coefficient (ICC). Urine oxalate measured in Laboratories A–E was compared in all 20 (basal and spiked) specimens using the ICC. PASW Statistics version 17 (SPSS Inc. Chicago, IL) was used. Results from Laboratories A–E were also compared among one another using Bland–Altman plots [10]. Acidified and non-acidified samples were compared by a paired Student’s t-test.
Results
Intra-laboratory comparison
We first examined the intra-laboratory variability by using sample duplicates (Experiment 1), given that the laboratories were blinded regarding sample identification. All laboratories were informed that the samples were not acidified during the collection and were asked to process them accordingly. Table 1 shows the ICC [and 95% confidence intervals (CI)] for Laboratories A–F, with the exception of Laboratory D2, from which incomplete data precluded analysis of the ICC. Important variability was observed in the analysis of the duplicates in three laboratories (Laboratories A and D1 using the oxalate oxidase method, and F using an HPLC-based method). In contrast, duplicate analysis for sodium and creatinine showed high ICC. After this initial analysis, Laboratory F (with the lowest ICC) conducted additional internal analyses and concluded that inconsistencies in their HPLC-method for oxalate determination precluded participation in Experiment 2. In summary, Experiment 1 showed important intra-laboratory variations leading even to the withdrawal of one laboratory from the rest of the study.
Pre-analytical acidification of the samples
All laboratories in the study routinely acidify urine samples at the time of analysis. We next examined the role of pre-analytical sample acidification in oxalate analysis. Two laboratories using HPLC-based methods (D2 and E) analyzed samples from native urine and urine acidified in the laboratory (to pH < 2) prior to testing. As shown in Figure 1, pre-analytical acidification of the urine samples resulted in significantly higher oxalate concentrations in both laboratories, possibly reflecting a better solubility of oxalate. These data stress the importance of pre-analytical acidification of the samples.
Fig. 1.

Comparison of urine oxalate measurement from paired samples acidified and non-acidified by the laboratories prior to analysis. Acidified samples were acidified after thawing by the receiving Laboratories (D2, open circles, and E, closed circles) and processed for oxalate measurement by an HPLC-based method. Data for individual sample pairs are connected by a dashed line for Laboratory D2 and continuous line for Laboratory E. Mean and SD is shown by the squares. P < 0.001 acidified versus non-acidified samples, n = 20 per group, with 10 samples per laboratory.
Inter-laboratory comparison
Taking into account, the limitations of the data set gathered from Experiment 1, and in view of the importance of the pre-analytical urinary acidification, we designed a second experiment in which we acidified the urine shortly after it was received in the distributing center. Acidified samples kept at 4°C were aliquoted, and duplicates were spiked with 10 mg/dL sodium oxalate and sent overnight to the participating laboratories. Figure 2 and supplementary Table 1 show the values obtained by each individual laboratory for the 20 samples. Important variations were observed between the laboratories, with systematic bias toward high values for Laboratory D1 and toward low values for Laboratory A.
Fig. 2.

Individual laboratory oxalate measurements for 20 different samples from Experiment 2. Samples are arranged in order of increasing oxalate concentration. Closed symbols represent laboratories that used HPLC-based methods, while open symbols indicate laboratories that used the oxalate-oxidase method.
Analysis of the differences between the spiked samples and the original urine was performed, and the recovery of 10 mg/L oxalate is shown in Figure 3. Important variations of recovery were identified for all laboratories, with coefficient of variation of 12% on average (minimum 5%, maximum 27%) (Table 2).
Fig. 3.

Recovery of 10 mg/L spike in oxalate in different laboratories participating in Experiment 2. Closed symbols represent laboratories that used HPLC-based methods, while open symbols indicate laboratories that used the oxalate-oxidase method.
Table 2.
Results of recovery of 10 mg/L of oxalate from Experiment 2 shown as mean, percentage of recovery and percentage of coefficient of variation for each laboratory
| Laboratory | A | B | C | D1 | D2 | E |
| Mean (mg/L) | 9.68 | 10.53 | 10.74 | 10.67 | 8.68 | 10.12 |
| SD (mg/L) | 1.38 | 0.71 | 0.53 | 0.93 | 2.34 | 0.93 |
| Recovery (%) | 96.8 | 105.3 | 107.4 | 106.7 | 86.8 | 101.2 |
| CV (%) | 14.21 | 6.71 | 4.89 | 8.76 | 26.98 | 9.17 |
To analyze in more detail the differences between the laboratories, we established ICC for all pairs of participating laboratories. Table 3 presents the data demonstrating ICC varying between 0.745 (95% CI: 0.468–0.890) and 0.986 (95% CI: 0.967–0.995). To visualize individual comparisons between pairs of laboratories, we present direct plots with the identity line drawn in dash and Bland–Altman plots (Figure 4). Illustrated are pairs of laboratories using the oxalate oxidase kit with high agreement (Panels A and B), and laboratories using the same method, but with lower correlation coefficient (Panels C and D). We performed the same comparison for the two laboratories using HPLC (Panels E and F). Finally, we compared oxalate oxidase kit-using laboratories with those using HPLC-derived methods. Examples of high and low agreement (Panels G, H and I, J, respectively) are presented.
Table 3.
ICC for urine oxalate measured in six different laboratories by two different methods (oxalate oxidase: Laboratories A, B, C, D1, clear background; HPLC-based: Laboratories D2 and E, gray background)
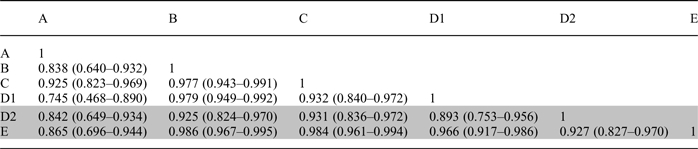 |
Fig. 4.
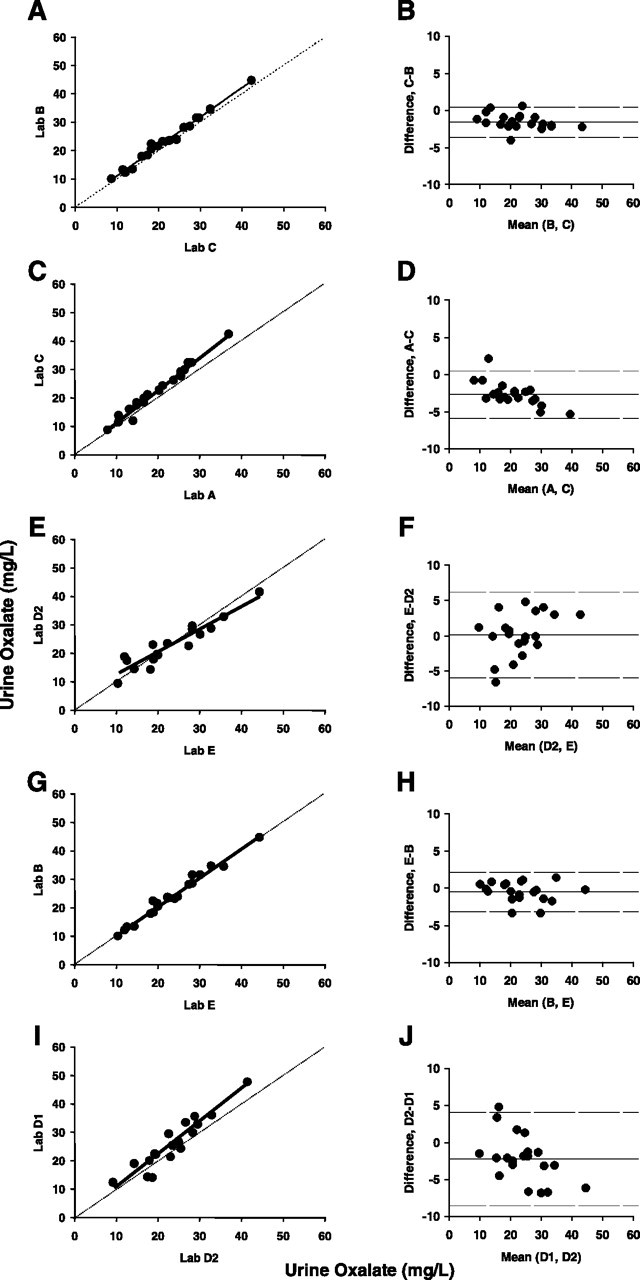
(A–J) Between laboratory agreement from Experiment 2. (A, C, E, G, I) Direct comparison between indicated pairs of laboratories is shown with dashed identity line and trend line in solid. (B, D, F, H, J) Bland–Altman plots for the same pairs of laboratories. Solid line represents the mean bias and the dashed lines indicate upper and lower limits of agreement. Panels A and B show pairs of highly correlated laboratories using the oxalate oxidase kit, while Panels C and D compare laboratories with lower correlation coefficients using the same method. Panels E and F show comparison between the two laboratories using HPLC-based methods. Panels G, H and I, J show pairs in which one laboratory uses the oxalate oxidase method, with the other using an HPLC-derived method, and presenting high (Panels G and H) or weak agreement (Panels I and J).
Discussion
The goal of this study was to assess the degree of agreement in urine oxalate measurements from various laboratories. While an excellent consistency was noted in the measurement of urine sodium and creatinine, we observed substantial variation in intra-laboratory (Table 1) and inter-laboratory (Table 3) agreement in urine oxalate measurement. Overall, there was some suggestion of systematic bias for two of the laboratories (Figure 2), and HPLC-based methods showed more variability compared to the oxalate oxidase kit-based methods. The presence of systematic bias between laboratories suggests that the use of a standardized calibrator(s) could enhance between-laboratory agreement.
A number of pre-analytical factors are known to influence urine oxalate measurement [11], including the conversion of ascorbic acid to oxalate which falsely increases measured oxalate. This conversion occurs with longer duration of sample storage and more rapidly at alkaline pH and higher storage temperature. It can be minimized by acidifying urine and/or maintaining it at temperatures ≤4°C if stored for prolonged durations [11]. The time at which the sample is acidified—upon collection of the urine or right before oxalate measurement—has been shown not to affect the measured oxalate concentration [12]. In our experiments, non-acidified samples were frozen at −80°C, shipped on dry ice and acidified before analysis for Experiment 1, while samples from Experiment 2 were acidified shortly after the end of collection, kept at 4°C, and shipped with ice packs. In both experiments, samples were analyzed within 10 days of collection. We therefore feel that pre-analytical handling is unlikely to have contributed to the variance in urine oxalate measurement between laboratories.
We did note a significant difference in HPLC-measured oxalate concentrations when samples were run acidified versus non-acidified (Figure 1). This might reflect a better solubility of oxalate in acidified urine or a direct effect of the pH on the chromatography.
Urine oxalate measurement is routinely performed in the evaluation of patients with nephrolithiasis. Initially introduced three decades ago [13], the enzymatic method for determining oxalate concentration in urine using oxalate oxidase is now available through a commercially available kit (Trinity Biotech). This kit was used by four laboratories participating in our studies, Laboratories A, B, C and D1. Among themselves, Laboratories B, C and D1 showed excellent intra- and inter-laboratory reliability (ICC for analysis of duplicates 0.942–0.998 and for inter-laboratory comparison 0.932–0.979—Tables 1 and 3 and Figure 4A and B). On the other hand, Laboratory A, using the same kit, displayed lower intra-laboratory reliability (ICC: 0.875) and less agreement with the other laboratories, with a tendency for returning lower values (Figures 2 and 4C and D). This could reflect handling problems of the samples or any aspect of assay performance. HPLC was adapted for the measurement of urine oxalate a few years after the oxalate oxidase method [14–16] and is less expensive once the method is established in the laboratory. Laboratories utilize different columns, solvents, detectors and retention times for oxalate detection. Although laboratories periodically perform internal validation studies, external validation may not be performed on a regular basis. In our study, Laboratory F utilizing HPLC displayed very low intra-laboratory reliability and eventually identified a problem with its method to isolate oxalate (it was measuring an additional compound besides oxalate, hidden in the same peak) and withdrew from the rest of the study. Laboratory D2 utilizing HPLC displayed lower inter-laboratory ICCs (0.842–0.931), while Laboratory E, also utilizing HPLC, displayed better agreement with Laboratories B, C and D1 which utilizes the oxalate oxidase kit (ICC: 0.966–0.986).
Our findings suggest that direct comparison of urinary oxalate concentrations measured by different laboratories is not always possible. This is clinically important as discrepancies in the prevalence of hyperoxaluria between various stone centers could be due to differences in oxalate measurement rather than to dietary or genetic factors. Furthermore, a number of investigators have debated the relative roles of urinary calcium and oxalate in the formation of calcium oxalate stones [3–7]. Some of the arguments provided are based on results of metabolic studies, in which urinary oxalate was measured by various techniques. We propose that variations of the measurement of oxalate in the urine may have led to overestimation or underestimation of the real prevalence of hyperoxaluria in the population of stone formers.
Limitations of this study include the small sample size and the fact that not all methodologies available for oxalate measurement were evaluated. However, samples were analyzed by the two most commonly used techniques. Furthermore, oxalate concentrations evaluated were within the reference range, but lower than what one may see in patients with primary hyperoxaluria.
In this study assessing the degree of agreement in urine oxalate measurements from various laboratories, significant variability was noted between laboratories. Our findings stress the need for a standardization of the method of measurement in order to allow accurate comparison of urinary oxalate values between laboratories. Use of an international calibrator might help in improving some systematic bias noted for some laboratories.
Supplementary data
Supplementary Table 1 is available online at http://ndt.oxfordjournals.org.
Acknowledgments
The authors would like to acknowledge the assistance of Ms Kathy Hill and Ms Janice Koska from the University of Texas Southwestern Medical Center in the preparation of the urine samples prior to shipping to various laboratories.
Funding. N.M.M. was supported by the National Institutes of Health (grants K23-RR21710). O.B. was supported by a bridge grant of the Faculté de Biologie et Médecine of the University of Lausanne and by the Foundation Prof. Placide Nicod.
Conflict of interest statement. None declared.
References
- 1.Lotan Y, Cadeddu JA, Pearle MS. International comparison of cost effectiveness of medical management strategies for nephrolithiasis. Urol Res. 2005;33:223–230. doi: 10.1007/s00240-005-0463-9. [DOI] [PubMed] [Google Scholar]
- 2.Daudon M, Dore JC, Jungers P, et al. Changes in stone composition according to age and gender of patients: a multivariate epidemiological approach. Urol Res. 2004;32:241–247. doi: 10.1007/s00240-004-0421-y. [DOI] [PubMed] [Google Scholar]
- 3.Pak CY, Adams-Huet B, Poindexter JR, et al. Relative effect of urinary calcium and oxalate on saturation of calcium oxalate. Kidney Int. 2004;66:2032–2037. doi: 10.1111/j.1523-1755.2004.00975.x. [DOI] [PubMed] [Google Scholar]
- 4.Robertson WG, Peacock M. The cause of idiopathic calcium stone disease: hypercalciuria or hyperoxaluria? Nephron. 1980;26:105–110. doi: 10.1159/000181963. [DOI] [PubMed] [Google Scholar]
- 5.Taylor EN, Curhan GC. Determinants of 24-hour urinary oxalate excretion. Clin J Am Soc Nephrol. 2008;3:1453–1460. doi: 10.2215/CJN.01410308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zimmermann DJ, Hesse A, von Unruh GE. Influence of a high-oxalate diet on intestinal oxalate absorption. World J Urol. 2005;23:324–329. doi: 10.1007/s00345-005-0028-0. [DOI] [PubMed] [Google Scholar]
- 7.Curhan GC, Willett WC, Speizer FE, et al. Twenty-four-hour urine chemistries and the risk of kidney stones among women and men. Kidney Int. 2001;59:2290–2298. doi: 10.1046/j.1523-1755.2001.00746.x. [DOI] [PubMed] [Google Scholar]
- 8.Zerwekh J, Drake E, Gregory J, et al. Assay of urinary oxalate: six methodologies compared. Clin Chem. 1983;29:1977–1980. [PubMed] [Google Scholar]
- 9.Hesse A, Bongartz D, Heynck H, et al. Measurement of urinary oxalic acid: a comparison of five methods. Clin Biochem. 1996;29:467–472. doi: 10.1016/0009-9120(96)00067-7. [DOI] [PubMed] [Google Scholar]
- 10.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1:307–310. [PubMed] [Google Scholar]
- 11.Mazzachi B, Teubner J, Ryall R. Factors affecting measurement of urinary oxalate. Clin Chem. 1984;30:1339–1343. [PubMed] [Google Scholar]
- 12.Ferraz RR, Baxmann AC, Ferreira LG, et al. Preservation of urine samples for metabolic evaluation of stone-forming patients. Urol Res. 2006;34:329–337. doi: 10.1007/s00240-006-0064-2. [DOI] [PubMed] [Google Scholar]
- 13.Kohlbecker G, Richter L, Butz M. Determination of oxalate in urine using oxalate oxidase: comparison with oxalate decarboxylase. J Clin Chem Clin Biochem. 1979;17:309–313. doi: 10.1515/cclm.1979.17.5.309. [DOI] [PubMed] [Google Scholar]
- 14.Hughes H, Hagen L, Sutton RA. Determination of urinary oxalate by high-performance liquid chromatography. Anal Biochem. 1982;119:1–3. doi: 10.1016/0003-2697(82)90656-x. [DOI] [PubMed] [Google Scholar]
- 15.Larsson L, Libert B, Asperud M. Determination of urinary oxalate by reversed-phase ion-pair “high-performance” liquid chromatography. Clin Chem. 1982;28:2272–2274. [PubMed] [Google Scholar]
- 16.Honow R, Bongartz D, Hesse A. An improved HPLC-enzyme-reactor method for the determination of oxalic acid in complex matrices. Clin Chim Acta. 1997;261:131–139. doi: 10.1016/s0009-8981(97)06521-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.